Hematological Diseases and Osteoporosis

 ,
,  ,
, 
Abstract
1. Introduction
2. Data Source and Search
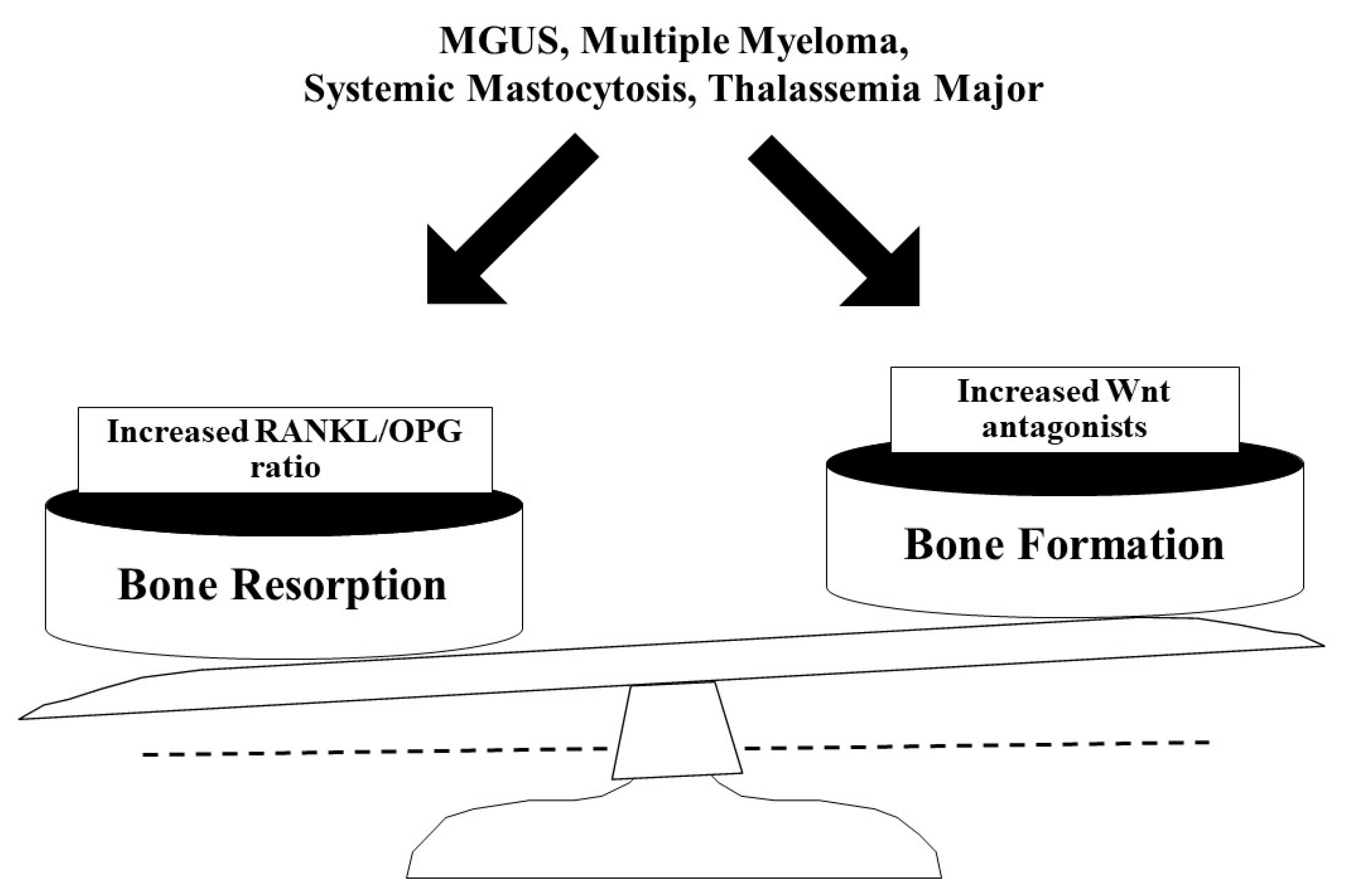
3. Monoclonal Gammopathy of Undetermined Significance
3.1. Bone Involvement
3.2. Treatment
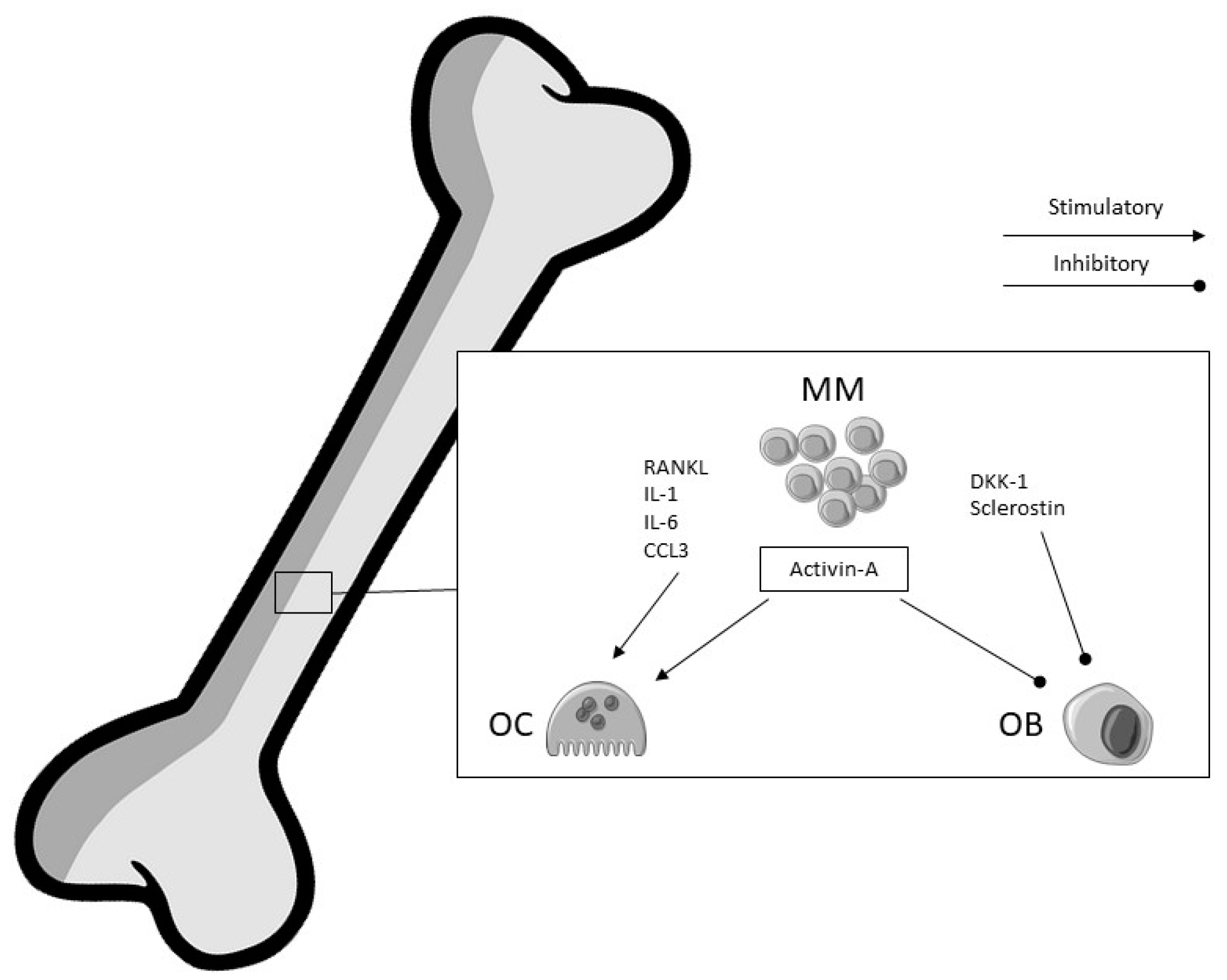
4. Multiple Myeloma
4.1. Bone Involvement
4.2. Treatment
5. Systemic Mastocytosis
5.1. Bone Involvement
5.2. Treatment
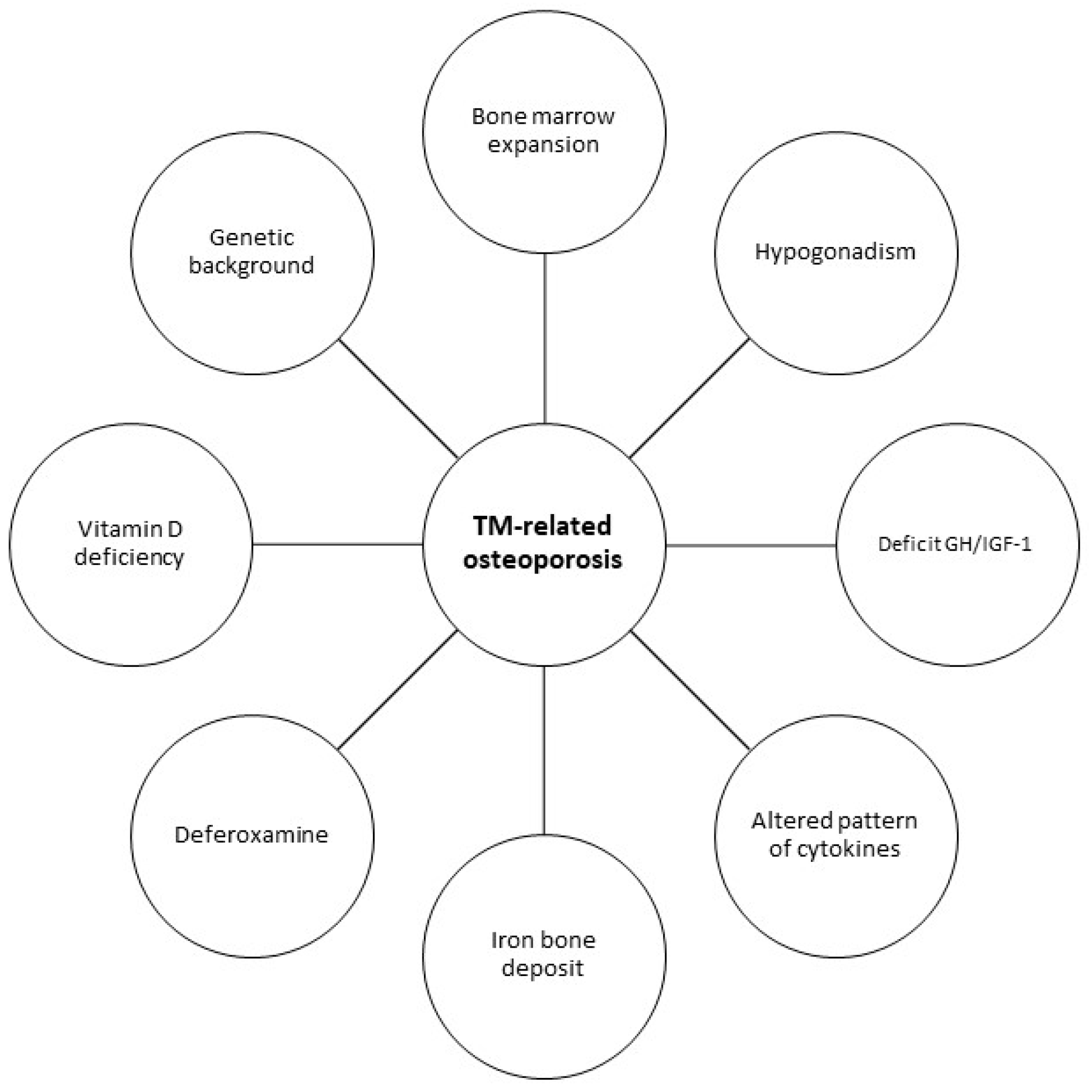
6. Thalassemia Major
6.1. Bone Involvement
6.2. Treatment
7. Sickle Cell Disease
7.1. Bone Involvement
7.2. Treatment
8. Hemophilia
8.1. Bone Involvement
8.2. Treatment
9. Conclusions
Funding
Conflicts of Interest
References
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Corrado, A.; Cantatore, F.P. Osteocytes: Central conductors of bone biology in normal and pathological conditions. Acta Physiol. 2012, 204, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, A.; Pennisi, P.; Bratengeier, C.; Torrisi, V.; Lindner, B.; Mangiafico, R.A.; Pulvirenti, I.; Hawa, G.; Tringali, G.; Fiore, C.E. Increased sclerostin serum levels associated with bone formation and resorption markers in patients with immobilization-induced bone loss. J. Clin. Endocrinol. Metab. 2010, 95, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Intemann, J.; De Gorter, D.J.J.; Naylor, A.J.; Dankbar, B.; Wehmeyer, C. Importance of osteocyte-mediated regulation of bone remodelling in inflammatory bone disease. Swiss Med. Wkly. 2020, 150, 20187. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, A.; Privitera, F.; Pulvirenti, I.; Canzonieri, E.; Rapisarda, R.; Fiore, C.E. Relationships between osteoprotegerin, receptor activator of the nuclear factor kB ligand and serum levels and carotid intima-media thickness in patients with type 2 diabetes mellitus. Panminerva Med. 2014, 56, 221–225. [Google Scholar]
- Gaudio, A.; Fiore, V.; Rapisarda, R.; Sidoti, M.H.; Xourafa, A.; Catalano, A.; Tringali, G.; Zanoli, L.; Signorelli, S.S.; Fiore, C.E. Sclerostin is a possible candidate marker of arterial stiffness: Results from a cohort study in Catania. Mol. Med. Rep. 2017, 15, 3420–3424. [Google Scholar] [CrossRef]
- Lorentzon, M.; Cummings, S.R. Osteoporosis: The evolution of a diagnosis. J. Intern. Med. 2015, 277, 650–661. [Google Scholar] [CrossRef]
- NIH. Consensus Development Panel on Osteoporosis Prevention D Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001, 285, 785–795. [Google Scholar] [CrossRef]
- Mirza, F.; Canalis, E. Management of endocrine disease: Secondary osteoporosis: Pathophysiology and management. Eur. J. Endocrinol. 2015, 173, R131–R151. [Google Scholar] [CrossRef]
- Valderrábano, R.J.; Wu, J.Y. Bone and blood interactions in human health and disease. Bone 2019, 119, 65–70. [Google Scholar] [CrossRef]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, 29, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Toxqui, L.; Vaquero, M.P. Chronic iron deficiency as an emerging risk factor for osteoporosis: A hypothesis. Nutrients 2015, 7, 2324–2344. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Durie, B.G.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kröger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Melton, L.J., 3rd; Rajkumar, S.V.; Khosla, S.; Achenbach, S.J.; Oberg, A.L.; Kyle, R.A. Fracture risk in monoclonal gammopathy of undetermined significance. J. Bone Miner. Res. 2004, 19, 25–30. [Google Scholar] [CrossRef]
- Bida, J.P.; Kyle, R.A.; Therneau, T.M.; Melton, L.J., 3rd; Plevak, M.F.; Larson, D.R.; Dispenzieri, A.; Katzmann, J.A.; Rajkumar, S.V. Disease associations with monoclonal gammopathy of undetermined significance: A population-based study of 17,398 patients. Mayo Clin. Proc. 2009, 84, 685–693. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Tang, M.; Pfeiffer, R.M.; Bjorkholm, M.; Blimark, C.; Mellqvist, U.H.; Wahlin, A.; Turesson, I.; Landgren, O. Monoclonal gammopathy of undetermined significance and risk of skeletal fractures: A population-based study. Blood 2010, 116, 2651–2655. [Google Scholar] [CrossRef]
- Piot, J.M.; Royer, M.; Schmidt-Tanguy, A.; Hoppé, E.; Gardembas, M.; Bourrée, T.; Hunault, M.; François, S.; Boyer, F.; Ifrah, N.; et al. Factors associated with an increased risk of vertebral fracture in monoclonal gammopathies of undetermined significance. Blood Cancer J. 2015, 5, e345. [Google Scholar] [CrossRef][Green Version]
- Pepe, J.; Petrucci, M.T.; Nofroni, I.; Fassino, V.; Diacinti, D.; Romagnoli, E.; Minisola, S. Lumbar bone mineral density as the major factor determining increased prevalence of vertebral fractures in monoclonal gammopathy of undetermined significance. Br. J. Haematol. 2006, 134, 485–490. [Google Scholar] [CrossRef]
- Ng, A.C.; Khosla, S.; Charatcharoenwitthaya, N.; Kumar, S.K.; Achenbach, S.J.; Holets, M.F.; McCready, L.K.; Melton, L.J., 3rd; Kyle, R.A.; Rajkumar, S.V.; et al. Bone microstructural changes revealed by high-resolution peripheral quantitative computed tomography imaging and elevated DKK1 and MIP-1α levels in patients with MGUS. Blood 2011, 118, 6529–6534. [Google Scholar] [CrossRef]
- Farr, J.N.; Zhang, W.; Kumar, S.K.; Jacques, R.M.; Ng, A.C.; McCready, L.K.; Rajkumar, S.V.; Drake, M.T. Altered cortical microarchitecture in patients with monoclonal gammopathy of undetermined significance. Blood 2014, 123, 647–649. [Google Scholar] [CrossRef]
- Woitge, H.W.; Horn, E.; Keck, A.V.; Auler, B.; Seibel, M.J.; Pecherstorfer, M. Biochemical markers of bone formation in patients with plasma cell dyscrasias and benign osteoporosis. Clin. Chem. 2001, 47, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Laroche, M.; Attal, M.; Dromer, C. Bone remodelling in monoclonal gammopathies of uncertain significance, symptomatic and non symptomatic myeloma. Clin. Rheumatol. 1996, 15, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsdottir, S.; Lund, S.H.; Lindqvist, E.K.; Thordardottir, M.; Sigurdsson, G.; Costello, R.; Burton, D.; Steingrimsdottir, H.; Gudnason, V.; Eiriksdottir, G.; et al. Bone disease in monoclonal gammopathy of undetermined significance: Results from a screened population-based study. Blood Adv. 2017, 21, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Veronese, N.; Luchini, C.; Solmi, M.; Sergi, G.; Manzato, E.; Stubbs, B. Monoclonal gammopathy of undetermined significance and bone health outcomes: A systematic review and exploratory meta-analysis. J. Bone Miner. Metab. 2018, 36, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Pepe, J.; Petrucci, M.T.; Mascia, M.L.; Piemonte, S.; Fassino, V.; Romagnoli, E.; Minisola, S. The effects of alendronate treatment in osteoporotic patients affected by monoclonal gammopathy of undetermined significance. Calcif. Tissue Int. 2008, 82, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R.; Yellin, O.; Boccia, R.V.; Flam, M.; Wong, S.F.; Batuman, O.; Moezi, M.M.; Woytowitz, D.; Duvivier, H.; Nassir, Y.; et al. Zoledronic acid markedly improves bone mineral density for patients with monoclonal gammopathy of undetermined significance and bone loss. Clin. Cancer Res. 2008, 14, 6289–6295. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Palumbo, A.; Johnsen, H.E.; Engelhardt, M.; Gay, F.; Gregersen, H.; Hajek, R.; Kleber, M.; Ludwig, H.; Morgan, G.; et al. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: Recommendations from the European Myeloma Network. Haematologica 2014, 99, 984–996. [Google Scholar] [CrossRef]
- Moreau, P.; San Miguel, J.; Sonneveld, P.; Mateos, M.V.; Zamagni, E.; Avet-Loiseau, H.; Hajek, R.; Dimopoulos, M.A.; Ludwig, H.; Einsele, H.; et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28 (Suppl. 4), iv52–iv61. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma: Diagnosis and Treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef]
- Silbermann, R.; Roodman, G.D. Myeloma bone disease: Pathophysiology and management. J. Bone Oncol. 2013, 2, 59–69. [Google Scholar] [CrossRef]
- Greipp, P.R.; San Miguel, J.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Kastritis, E.; Zomas, A.; Terpos, E.; Katodritou, E.; Symeonidis, A.; Delimpasi, S.; Pouli, A.; Vassilakopoulos, T.P.; Michalis, E.; et al. Hypercalcemia remains an adverse prognostic factor for newly diagnosed multiple myeloma patients in the era of novel antimyeloma therapies. Eur. J. Haematol. 2017, 99, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Filzmoser, J.M.; Pecherstorfer, M.; Podar, K. Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies. Pharmaceutics 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Sezer, O.; Heider, U.; Zavrski, I.; Kühne, C.A.; Hofbauer, L.C. RANK ligand and osteoprotegerin in myeloma bone disease. Blood 2003, 101, 2094–2098. [Google Scholar] [CrossRef]
- Giuliani, N.; Lisignoli, G.; Colla, S.; Lazzaretti, M.; Storti, P.; Mancini, C.; Bonomini, S.; Manferdini, C.; Codeluppi, K.; Facchini, A.; et al. CC-chemokine ligand 20/macrophage inflammatory protein-3α and CC-chemokine receptor 6 are overexpressed in myeloma microenvironment related to osteolytic bone lesions. Cancer Res. 2008, 68, 6840–6850. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Katagiri, T.; Tamura, T.; Wada, S.; Findlay, D.M.; Martin, T.J.; Hirota, H.; Taga, T.; Kishimoto, T.; et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J. Exp. Med. 1995, 182, 1461–1468. [Google Scholar] [CrossRef]
- Spaan, I.; Raymakers, R.A.; van de Stolpe, A.; Peperzak, V. Wnt signaling in multiple myeloma: A central player in disease with therapeutic potential. J. Hematol. Oncol. 2018, 11, 67. [Google Scholar] [CrossRef]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009, 23, 10–24. [Google Scholar] [CrossRef]
- Michigami, T.; Shimizu, N.; Williams, P.J.; Niewolna, M.; Dallas, S.L.; Mundy, G.R.; Yoneda, T. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast-stimulating activity. Blood 2000, 96, 1953–1960. [Google Scholar] [CrossRef]
- Hideshima, T.; Podar, K.; Chauhan, D.; Anderson, K.C. Cytokines and signal transduction. Best Pract. Res. Clin. Haematol. 2005, 18, 509–524. [Google Scholar] [CrossRef]
- Gupta, D.; Treon, S.P.; Shima, Y.; Hideshima, T.; Podar, K.; Tai, Y.T.; Lin, B.; Lentzsch, S.; Davies, F.E.; Chauhan, D.; et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia 2001, 15, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Storti, P.; Bolzoni, M.; Palma, B.D.; Bonomini, S. Angiogenesis and multiple myeloma. Cancer Microenviron. 2011, 4, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Kastritis, E.; Christoulas, D.; Gkotzamanidou, M.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Papatheodorou, A.; Dimopoulos, M.A. Circulating activin-A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post-lenalidomide and dexamethasone therapy. Ann. Oncol. 2012, 23, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Mukherjee, S.; Vaghela, N.; Hideshima, T.; Fulciniti, M.; Pozzi, S.; Santo, L.; Cirstea, D.; Patel, K.; Sohani, A.R.; et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc. Natl. Acad. Sci. USA 2010, 107, 5124–5129. [Google Scholar] [CrossRef]
- Berenson, J.R.; Lichtenstein, A.; Porter, L.; Dimopoulos, M.A.; Bordoni, R.; George, S.; Lipton, A.; Keller, A.; Ballester, O.; Kovacs, M.J.; et al. Efficacy of pamidronate in reducing skeletal events in patients with advanced multiple myeloma. Myeloma Aredia Study Group. New Engl. J. Med. 1996, 334, 488–493. [Google Scholar] [CrossRef]
- Terpos, E.; Sezer, O.; Croucher, P.I.; García-Sanz, R.; Boccadoro, M.; San Miguel, J.; Ashcroft, J.; Bladé, J.; Cavo, M.; Delforge, M.; et al. The use of bisphosphonates in multiple myeloma: Recommendations of an expert panel on behalf of the European Myeloma Network. Ann. Oncol. 2009, 20, 1303–1317. [Google Scholar] [CrossRef]
- Raje, N.; Anderson, K.C. Introduction: The evolving role of bisphosphonate therapy in multiple myeloma. Blood 2000, 96, 381–383. [Google Scholar] [CrossRef]
- Mhaskar, R.; Kumar, A.; Miladinovic, B.; Djulbegovic, B. Bisphosphonates in multiple myeloma: An updated network meta-analysis. Cochrane. Database Syst. Rev. 2017, 12, CD003188. [Google Scholar] [CrossRef]
- Major, P.; Lortholary, A.; Hon, J.; Abdi, E.; Mills, G.; Menssen, H.D.; Yunus, F.; Bell, R.; Body, J.; Quebe-Fehling, E.; et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: A pooled analysis of two randomized, controlled clinical trials. J. Clin. Oncol. 2001, 19, 558–567. [Google Scholar] [CrossRef]
- Kostenuik, P.J.; Nguyen, H.Q.; McCabe, J.; Warmington, K.S.; Kurahara, C.; Sun, N.; Chen, C.; Li, L.; Cattley, R.C.; Van, G.; et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J. Bone Miner. Res. 2009, 24, 182–195. [Google Scholar] [CrossRef]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; García-Sanz, R.; Durie, B.; Legieć, W.; Krejčí, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Tsourdi, E.; Langdahl, B.; Cohen-Solal, M.; Aubry-Rozier, B.; Eriksen, E.F.; Guañabens, N.; Obermayer-Pietsch, B.; Ralston, S.H.; Eastell, R.; Zillikens, M.C. Discontinuation of Denosumab therapy for osteoporosis: A systematic review and position statement by ECTS. Bone 2017, 105, 11–17. [Google Scholar] [CrossRef] [PubMed]
- von Metzler, I.; Krebbel, H.; Hecht, M.; Manz, R.A.; Fleissner, C.; Mieth, M.; Kaiser, M.; Jakob, C.; Sterz, J.; Kleeberg, L.; et al. Bortezomib inhibits human osteoclastogenesis. Leukemia 2007, 21, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Toscani, D.; Palumbo, C.; Dalla Palma, B.; Ferretti, M.; Bolzoni, M.; Marchica, V.; Sena, P.; Martella, E.; Mancini, C.; Ferri, V.; et al. The Proteasome Inhibitor Bortezomib Maintains Osteocyte Viability in Multiple Myeloma Patients by Reducing Both Apoptosis and Autophagy: A New Function for Proteasome Inhibitors. J. Bone Miner. Res. 2016, 31, 815–827. [Google Scholar] [CrossRef]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef]
- Heath, D.J.; Chantry, A.D.; Buckle, C.H.; Coulton, L.; Shaughnessy, J.D., Jr.; Evans, H.R.; Snowden, J.A.; Stover, D.R.; Vanderkerken, K.; Croucher, P.I. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J. Bone Miner. Res. 2009, 24, 425–436. [Google Scholar] [CrossRef]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef]
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016, pdated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [PubMed]
- Uzzaman, A.; Maric, I.; Noel, P.; Kettelhut, B.V.; Metcalfe, D.; Carter, M.C. Pediatric-onset mastocytosis: A long term clinical follow-up and correlation with bone marrow histopathology. Pediatr. Blood Cancer 2009, 53, 629–634. [Google Scholar] [CrossRef] [PubMed]
- von Bubnoff, N.; Gorantla, S.H.; Kancha, R.K.; Lordick, F.; Peschel, C.; Duyster, J. The systemic mastocytosis-specific activating cKit mutation D816V can be inhibited by the tyrosine kinase inhibitor AMN107. Leukemia 2005, 19, 1670–1671. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Zanotti, R.; Bonadonna, P.; Artuso, A.; Caruso, B.; Schena, D.; Vecchiato, D.; Bonifacio, M.; Viapiana, O.; Gatti, D.; et al. Bone mineral density, bone turnover markers and fractures in patients with indolent systemic mastocytosis. Bone 2011, 49, 880–885. [Google Scholar] [CrossRef] [PubMed]
- van der Veer, E.; van der Goot, W.; de Monchy, J.G.; Kluin-Nelemans, H.C.; van Doormaal, J.J. High prevalence of fractures and osteoporosis in patients with indolent systemic mastocytosis. Allergy 2012, 67, 431–438. [Google Scholar] [CrossRef]
- Seitz, S.; Barvencik, F.; Koehne, T.; Priemel, M.; Pogoda, P.; Semler, J.; Minne, H.; Pfeiffer, M.; Zustin, J.; Püschel, K.; et al. Increased osteoblast and osteoclast indices in individuals with systemic mastocytosis. Osteoporos. Int. 2013, 24, 2325–2334. [Google Scholar] [CrossRef]
- Greene, L.W.; Asadipooya, K.; Corradi, P.F.; Akin, C. Endocrine manifestations of systemic mastocytosis in bone. Rev. Endocr. Metab. Disord. 2016, 17, 419–431. [Google Scholar] [CrossRef]
- Rossini, M.; Viapiana, O.; Adami, S.; Idolazzi, L.; Zanotti, R.; Gatti, D. Rapid skeletal turnover in radiographic mimic of osteopetrosis might be secondary to systemic mastocytosis [corrected]. J. Bone Miner. Res. 2015, 30, 945. [Google Scholar] [CrossRef]
- Kushnir-Sukhov, N.M.; Brittain, E.; Reynolds, J.C.; Akin, C.; Metcalfe, D.D. Elevated tryptase levels are associated with greater bone density in a cohort of patients with mastocytosis. Int. Arch. Allergy Immunol. 2006, 139, 265–270. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Boucher, W.; Spear, K. Serum interleukin-6 reflects disease severity and osteoporosis in mastocytosis patients. Lnt. Arch. Allergy lmmunol. 2002, 128, 344–350. [Google Scholar] [CrossRef]
- Dobigny, C.; Saffar, J.L. H1 and H2 histamine receptors modulate osteoclastic resorption by different pathways: Evidence obtained by using receptor antago nists in a rat synchronized resorption model. J. Cell Physiol. 1997, 173, 10–18. [Google Scholar] [CrossRef]
- Biosse-Duplan, M.; Baroukh, B.; Dy, M.; de Vernejoul, M.C.; Saffar, J.L. Histamine promotes osteoclastogenesis through the differential expression of histamine receptors on osteoclasts and osteoblasts. Am. J. Pathol. 2009, 174, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Zanotti, R.; Viapiana, O.; Tripi, G.; Orsolini, G.; Idolazzi, L.; Bonadonna, P.; Schena, D.; Escribano, L.; Adami, S.; et al. Bone involvement and osteoporosis in mastocytosis. Immunol. Allergy Clin. North Am. 2014, 34, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Viapiana, O.; Zanotti, R.; Tripi, G.; Perbellini, O.; Idolazzi, L.; Bonifacio, M.; Adami, S.; Gatti, D. Dickkopf-1 and sclerostin serum levels in patients with systemic mastocytosis. Calcif. Tissue Int. 2015, 96, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, A.; Christopeit, B.; Leja, S.; Gerbaulet, A.; Kleiner, S.; Förster, A.; Raap, U.; Wickenhauser, C.; Hartmann, K. Serum levels of bone cytokines are increased in indolent systemic mastocytosis associated with osteopenia or osteoporosis. J. Allergy Clin. Immunol. 2013, 132, 1234–1237.e7. [Google Scholar] [CrossRef]
- Laroche, M.; Livideanu, C.; Paul, C.; Cantagrel, A. Interferon alpha and pamidronate in osteoporosis with fracture secondary to mastocytosis. Am. J. Med. 2011, 124, 776–778. [Google Scholar] [CrossRef]
- Guillaume, N.; Desoutter, J.; Chandesris, O.; Merlusca, L.; Henry, I.; Georgin-Lavialle, S.; Barete, S.; Hirsch, I.; Bouredji, D.; Royer, B.; et al. Bone complications of mastocytosis: A link between clinical and biological characteristics. Am. J. Med. 2013, 126, 75.e1–e7. [Google Scholar] [CrossRef]
- Barete, S.; Assous, N.; de Gennes, C.; Grandpeix, C.; Feger, F.; Palmerini, F.; Dubreuil, P.; Arock, M.; Roux, C.; Launay, J.M.; et al. Systemic mastocytosis and bone involvement in a cohort of 75 patients. Ann. Rheum. Dis. 2010, 69, 1838–1841. [Google Scholar] [CrossRef]
- Cundy, T.; Beneton, M.N.; Darby, A.J.; Marshall, W.J.; Kanis, J.A. Osteopenia in systemic mastocytosis: Natural history and responses to treatment with inhibitors of bone resorption. Bone 1987, 8, 149–155. [Google Scholar] [CrossRef]
- Rossini, M.; Zanotti, R.; Viapiana, O.; Tripi, G.; Idolazzi, L.; Biondan, M.; Orsolini, G.; Bonadonna, P.; Adami, S.; Gatti, D. Zoledronic acid in osteoporosis secondary to mastocytosis. Am. J. Med. 2014, 127, 1127.e1–e4. [Google Scholar] [CrossRef]
- Ali, A.S.; Lax, A.S.; Liljeström, M.; Paakkari, I.; Ashammakhi, N.; Kovanen, P.T.; Konttinen, Y.T. Mast cells in atherosclerosis as a source of the cytokine RANKL. Clin. Chem. Lab Med. 2006, 44, 672–674. [Google Scholar] [PubMed]
- Orsolini, G.; Gavioli, I.; Tripi, G.; Viapiana, O.; Gatti, D.; Idolazzi, L.; Zanotti, R.; Rossini, M. Denosumab for the Treatment of Mastocytosis-Related Osteoporosis: A Case Series. Calcif. Tissue Int. 2017, 100, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, P.; Hungsprenges, S.; Fucharoen, S.; Baylink, D.; Thompson, E.; English, E.; Lee, M.; Burnell, J.; Finch, C. Relation between erythropoiesis and bone metabolism in thalassemia. New Engl. J. Med. 1981, 304, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, L.; De Sanctis, V. Multicentre study on prevalence of fractures in transfusion dependent thalassemic patients. J. Pediatr. Endocrinol. Metab. 1998, 11, 773–778. [Google Scholar] [PubMed]
- Poggi, M.; Sorrentino, F.; Pugliese, P.; Smacchia, M.P.; Daniele, C.; Equitani, F.; Terlizzi, F.; Guitarrini, M.R.; Monti, S.; Maffei, L.; et al. Longitudinal changes of endocrine and bone disease in adults with β-thalassemia major receiving different iron chelators over 5 years. Ann. Hematol. 2016, 95, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.E.; Tuck, S.M.; Agnew, J.E.; Koneru, S.; Morris, R.W.; Yardumian, A.; Prescott, E.; Hoffbrand, A.V.; Wonke, B. High prevalence of low bone mass in thalassaemia major. Br. J. Haematol. 1998, 103, 911–915. [Google Scholar] [CrossRef]
- Gaudio, A.; Morabito, N.; Catalano, A.; Rapisarda, R.; Xourafa, A.; Lasco, A. Pathogenesis of Thalassemia Major-associated Osteoporosis: A Review with Insights from Clinical Experience. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 110–117. [Google Scholar] [CrossRef]
- Fung, E.B.; Harmatz, P.R.; Milet, M.; Coates, T.D.; Thompson, A.A.; Ranalli, M.; Mignaca, R.; Scher, C.; Giardina, P.; Robertson, S.; et al. Fracture prevalence and relationship to endocrinopathy in iron overloaded patients with sickle cell disease and thalassemia. Bone 2008, 43, 162–168. [Google Scholar] [CrossRef]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Vichinsky, E.; Olivieri, N.; Kwiatkowski, J.; Cohen, A.; Neufeld, E.; Giardina, P.J. Prevalence of fractures among the Thalassemia syndromes in North America. Bone 2006, 38, 571–575. [Google Scholar] [CrossRef]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Cheung, A.M.; Vichinsky, E.; Olivieri, N.; Kirby, M.; Kwiatkowski, J.L.; Cunningham, M.; Holm, I.A.; et al. Bone disease in thalassemia: A frequent and still unresolved problem. J. Bone Miner. Res. 2009, 24, 543–557. [Google Scholar] [CrossRef]
- Engkakul, P.; Mahachoklertwattana, P.; Jaovisidha, S.; Chuansumrit, A.; Poomthavorn, P.; Chitrapazt, N.; Chuncharunee, S. Unrecognized vertebral fractures in adolescents and young adults with thalassemia syndromes. J. Pediatr. Hematol. Oncol. 2013, 35, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Anapliotou, M.L.; Kastanias, I.T.; Psara, P.; Evangelou, E.A.; Liparaki, M.; Dimitriou, P. The contribution of hypogonadism to the development of osteoporosis in thalassemia major: New therapeutic approaches. Clin. Endocrinol. 1995, 42, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.T.; El Banna, N.; Abdel Fattah, M.; El Zalabani, M.M.; Ansari, B.M. Bone mineral density in prepubertal children with β-thalassemia: Correlation with growth and hormonal data. Metabolism 1998, 47, 541–548. [Google Scholar] [CrossRef]
- Scacchi, M.; Danesi, L.; Cattaneo, A.; Valassi, E.; Pecori Giraldi, F.; Argento, C.; D’Angelo, E.; Mirra, N.; Carnelli, V.; Zanaboni, L.; et al. Bone demineralization in adult thalassaemic patients: Contribution of GH and IGF-I at different skeletal sites. Clin. Endocrinol. 2008, 69, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Lasco, A.; Morabito, N.; Gaudio, A.; Crisafulli, A.; Meo, A.; Denuzzo, G.; Frisina, N. Osteoporosis and beta-thalassemia major: Role of the IGF-I/IGFBP-III axis. J. Endocrinol. Invest. 2002, 25, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Morabito, N.; Russo, G.T.; Gaudio, A.; Lasco, A.; Catalano, A.; Morini, E.; Franchina, F.; Maisano, D.; La Rosa, M.; Plota, M.; et al. The “lively” cytokines network in beta-Thalassemia Major-related osteoporosis. Bone 2007, 40, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Bordat, C.; Constans, A.; Bouet, O.; Blanc, I.; Trubert, C.L.; Girot, R.; Cournot, G. Iron distribution in thalassemic bone by energy-loss spectroscopy and electron spectroscopic imaging. Calcif. Tissue Int. 1993, 53, 29–37. [Google Scholar] [CrossRef]
- Chan, Y.L.; Pang, L.M.; Chik, K.W.; Cheng, J.C.; Li, C.K. Patterns of bone diseases in transfusion-dependent homozygous thalassaemia major: Predominance of osteoporosis and desferrioxamine-induced bone dysplasia. Pediatr. Radiol. 2002, 32, 492–497. [Google Scholar] [CrossRef]
- Dandona, P.; Menon, R.K.; Houlder, S.; Thomas, M.; Hoffbrand, A.V.; Flynn, D.M. Serum 1,25 dihydroxyvitamin D and osteocalcin concentrations in thalassemia major. Arch. Dis. Child. 1987, 62, 474–477. [Google Scholar] [CrossRef]
- Gaudio, A.; Morabito, N.; Xourafa, A.; Currò, M.; Caccamo, D.; Ferlazzo, N.; Macrì, I.; La Rosa, M.A.; Meo, A.; Ientile, R. Role of genetic pattern on bone mineral density in thalassemic patients. Clin. Biochem. 2010, 43, 805–807. [Google Scholar] [CrossRef]
- Morabito, N.; Gaudio, A.; Lasco, A.; Atteritano, M.; Pizzoleo, M.A.; Cincotta, M.; La Rosa, M.; Guarino, R.; Meo, A.; Frisina, N. Osteoprotegerin and RANKL in the pathogenesis of thalassemia-induced osteoporosis: New pieces of the puzzle. J. Bone Miner. Res. 2004, 19, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Christoulas, D.; Plata, E.; Bratengeier, C.; Anastasilakis, A.D.; Komninaka, V.; Kaliontzi, D.; Gkotzamanidou, M.; Polyzos, S.A.; Dimopoulou, M.; et al. High circulating sclerostin is present in patients with thalassemia-associated osteoporosis and correlates with bone mineral density. Horm. Metab. Res. 2012, 44, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Marsella, M. Iron Chelation in Thalassemia Major. Clin. Ther. 2015, 37, 2866–2877. [Google Scholar] [CrossRef] [PubMed]
- Dede, A.D.; Trovas, G.; Chronopoulos, E.; Triantafyllopoulos, I.K.; Dontas, I.; Papaioannou, N.; Tournis, S. Thalassemia-associated osteoporosis: A systematic review on treatment and brief overview of the disease. Osteoporos. Int. 2016, 27, 3409–3425. [Google Scholar] [CrossRef]
- Lasco, A.; Morabito, N.; Gaudio, A.; Buemi, M.; Wasniewska, M.; Frisina, N. Effects of hormonal replacement therapy on bone metabolism in young adults with beta-thalassemia major. Osteoporos. Int. 2001, 12, 570–575. [Google Scholar] [CrossRef]
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef]
- Giusti, A.; Pinto, V.; Forni, G.L.; Pilotto, A. Management of beta-thalassemia-associated osteoporosis. Ann. NY Acad. Sci. 2016, 1368, 73–81. [Google Scholar] [CrossRef]
- Tsartsalis, A.N.; Lambrou, G.I.; Tsartsalis, D.; Savvidis, C.; Karantza, M.; Terpos, E.; Kanaka-Gantenbein, C.; Chrousos, G.P.; Kattamis, A. The role of biphosphonates in the management of thalassemia-induced osteoporosis: A systematic review and meta-analysis. Hormones 2018, 17, 153–166. [Google Scholar] [CrossRef]
- Gaudio, A.; Morabito, N.; Xourafa, A.; Macrì, I.; Meo, A.; Morgante, S.; Trifiletti, A.; Lasco, A.; Frisina, N. Bisphosphonates in the treatment of thalassemia-associated osteoporosis. J. Endocrinol. Invest. 2008, 31, 181–184. [Google Scholar] [CrossRef]
- Voskaridou, E.; Ntanasis-Stathopoulos, I.; Papaefstathiou, A.; Christoulas, D.; Dimopoulou, M.; Repa, K.; Papatheodorou, A.; Peppa, M.; Terpos, E. Denosumab in transfusion-dependent thalassemia osteoporosis: A randomized, placebo-controlled, double-blind phase 2b clinical trial. Blood Adv. 2018, 2, 2837–2847. [Google Scholar] [CrossRef]
- Morabito, N.; Catalano, A.; Gaudio, A.; Morini, E.; Bruno, L.M.; Basile, G.; Tsiantouli, E.; Bellone, F.; Agostino, R.M.; Piraino, B.; et al. Effects of strontium ranelate on bone mass and bone turnover in women with thalassemia major-related osteoporosis. J. Bone Miner. Metab. 2016, 34, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Tournis, S.; Dede, A.D.; Savvidis, C.; Triantafyllopoulos, I.K.; Kattamis, A.; Papaioannou, N. Effects of teriparatide retreatment in a patient with β-thalassemia major. Transfusion 2015, 55, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Pinto, V.M.; Balocco, M.; Quintino, S.; Forni, G.L. Sickle cell disease: A review for the internist. Int. Emerg. Med. 2019, 14, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Segal, J.B.; Ashar, B.H.; Leung, S.; Ahmed, S.; Siddique, S.; Rice, T.; Lanzkron, S. High prevalence and correlates of low bone mineral density in young adults with sickle cell disease. Am. J. Hematol. 2006, 81, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Garadah, T.S.; Hassan, A.B.; Jaradat, A.A.; Diab, D.E.; Kalafalla, H.O.; Kalifa, A.K.; Sequeira, R.P.; Alawadi, A.H. Predictors of abnormal bone mass density in adult patients with homozygous sickle-cell disease. Clin. Med. Insights Endocrinol. Diabetes 2015, 8, 35–40. [Google Scholar] [CrossRef]
- Bordbar, M.R.; Haghpanah, S.; Zarei, T.; Dabbaghmanesh, M.H.; Omrani, G.R.; Saki, F. Evaluation of bone mineral density in children with sickle-cell anemia and its associated factors in the south of Iran: A case-control study. Arch. Osteoporos. 2017, 12, 70. [Google Scholar] [CrossRef]
- De Luna, G.; Ranque, B.; Courbebaisse, M.; Ribeil, J.A.; Khimoud, D.; Dupeux, S.; Silvera, J.; Offredo, L.; Pouchot, J.; Arlet, J.B. High bone mineral density in sickle cell disease: Prevalence and characteristics. Bone 2018, 110, 199–203. [Google Scholar] [CrossRef]
- Voskaridou, E.; Stoupa, E.; Antoniadou, L.; Premetis, E.; Konstantopoulos, K.; Papassotiriou, I.; Terpos, E. Osteoporosis and osteosclerosis in sickle cell/beta-thalassemia: The role of the RANKL/osteoprotegerin axis. Haematologica 2006, 91, 813–816. [Google Scholar]
- Sadat-Ali, M.; Al-Elq, A.; Sultan, O.; Al-Turki, H. Secondary osteoporosis due to sickle cell anemia: Do sex steroids play a role? Indian J. Med. Sci. 2008, 62, 193–198. [Google Scholar] [CrossRef]
- Boettger, P.C.; Knupp, C.L.; Liles, D.K.; Walker, K. Vitamin D Deficiency in Adult Sickle Cell Patients. J. Natl. Med. Assoc. 2017, 109, 36–43. [Google Scholar] [CrossRef]
- Paschou, S.A.; Anagnostis, P.; Karras, S.; Annweiler, C.; Vakalopoulou, S.; Garipidou, V.; Goulis, D.G. Bone mineral density in men and children with haemophilia A and B: A systematic review and meta-analysis. Osteoporos. Int. 2014, 25, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Merchan, E.C.; Valentino, L.A. Increased bone resorption in hemophilia. Blood Rev. 2019, 33, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Norris, E.; Petrucci, J.; James, P.D.; Lee, A.; Poon, M.C.; Floros, G.; Boma-Fischer, L.; Teitel, J.; Nisenbaum, R.; et al. Bone health in symptomatic carriers of haemophilia A: A protocol for a multicentre prospective matched-cohort study. BMJ Open 2019, 9, e032891. [Google Scholar] [CrossRef] [PubMed]
- Mansouritorghabeh, H.; Rezaieyazdi, Z. Bone Density Status in Bleeding Disorders: Where Are We and What Needs to be Done? J. Bone Metab. 2017, 24, 201–206. [Google Scholar] [CrossRef]
- Gerstner, G.; Damiano, M.L.; Tom, A.; Worman, C.; Schultz, W.; Recht, M.; Stopeck, A.T. Prevalence and risk factors associated with decreased bone mineral density in patients with haemophilia. Haemophilia 2009, 15, 559–565. [Google Scholar] [CrossRef]
- Iorio, A.; Fabbriciani, G.; Marcucci, M.; Brozzetti, M.; Filipponi, P. Bone mineral density in haemophilia patients. A meta-analysis. Thromb. Haemost. 2010, 103, 596–603. [Google Scholar]
- Hoots, W.K.; Rodriguez, N.; Boggio, L.; Valentino, L.A. Pathogenesis of haemophilic synovitis: Clinical aspects. Haemophilia 2007, 13 (Suppl. 3), 4–9. [Google Scholar] [CrossRef]
- Forsyth, A.L.; Quon, D.V.; Konkle, B.A. Role of exercise and physical activity on haemophilic arthropathy, fall prevention and osteoporosis. Haemophilia 2011, 17, e870–e876. [Google Scholar] [CrossRef]
- Anagnostis, P.; Vakalopoulou, S.; Slavakis, A.; Charizopoulou, M.; Kazantzidou, E.; Chrysopoulou, T.; Vyzantiadis, T.A.; Moka, E.; Agapidou, A.; Garipidou, V. Reduced bone mineral density in patients with haemophilia A and B in Northern Greece. Thromb. Haemost. 2012, 107, 545–551. [Google Scholar]
- Kempton, C.L.; Antoniucci, D.M.; Rodriguez-Merchan, E.C. Bone health in persons with haemophilia. Haemophilia 2015, 21, 568–577. [Google Scholar] [CrossRef]
- Anagnostis, P.; Vyzantiadis, T.A.; Charizopoulou, M.; Adamidou, F.; Karras, S.; Goulis, D.G.; Karagiannis, A.; Garipidou, V.; Vakalopoulou, S. The effect of monthly ibandronate on bone mineral density and bone turnover markers in patients with haemophilia A and B and increased risk for fracture. Thromb. Haemost. 2013, 110, 257–263. [Google Scholar] [PubMed]
- Ghosh, K.; Shetty, S. Bone health in persons with haemophilia: A review. Eur. J. Haematol. 2012, 89, 95–102. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Major |
| Multifocal, dense aggregates of at least 15 mast cells in the bone marrow (BM) and/or other extracutaneous organ(s) |
| Minor |
| >25% of mastcells (MC) in the infiltrate of biopsy sections are spindle-shaped or have atypical morphology or, of all MC in the BM aspirate smears, >25% are immature or atypical |
| Activating point mutation at codon 816 of c-KIT in BM, blood, or another extracutaneous organ |
| MC in BM, blood, or extracutaneous organs express CD2 and/or CD25 in addition to normal MC markers |
| Total serum tryptase persistently exceeds 20 ng/mL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaudio, A.; Xourafa, A.; Rapisarda, R.; Zanoli, L.; Signorelli, S.S.; Castellino, P. Hematological Diseases and Osteoporosis. Int. J. Mol. Sci. 2020, 21, 3538. https://doi.org/10.3390/ijms21103538
Gaudio A, Xourafa A, Rapisarda R, Zanoli L, Signorelli SS, Castellino P. Hematological Diseases and Osteoporosis. International Journal of Molecular Sciences. 2020; 21(10):3538. https://doi.org/10.3390/ijms21103538
Chicago/Turabian StyleGaudio, Agostino, Anastasia Xourafa, Rosario Rapisarda, Luca Zanoli, Salvatore Santo Signorelli, and Pietro Castellino. 2020. "Hematological Diseases and Osteoporosis" International Journal of Molecular Sciences 21, no. 10: 3538. https://doi.org/10.3390/ijms21103538
APA StyleGaudio, A., Xourafa, A., Rapisarda, R., Zanoli, L., Signorelli, S. S., & Castellino, P. (2020). Hematological Diseases and Osteoporosis. International Journal of Molecular Sciences, 21(10), 3538. https://doi.org/10.3390/ijms21103538