Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1

Abstract
1. Introduction: Complex Molecular Pathophysiology of DM1
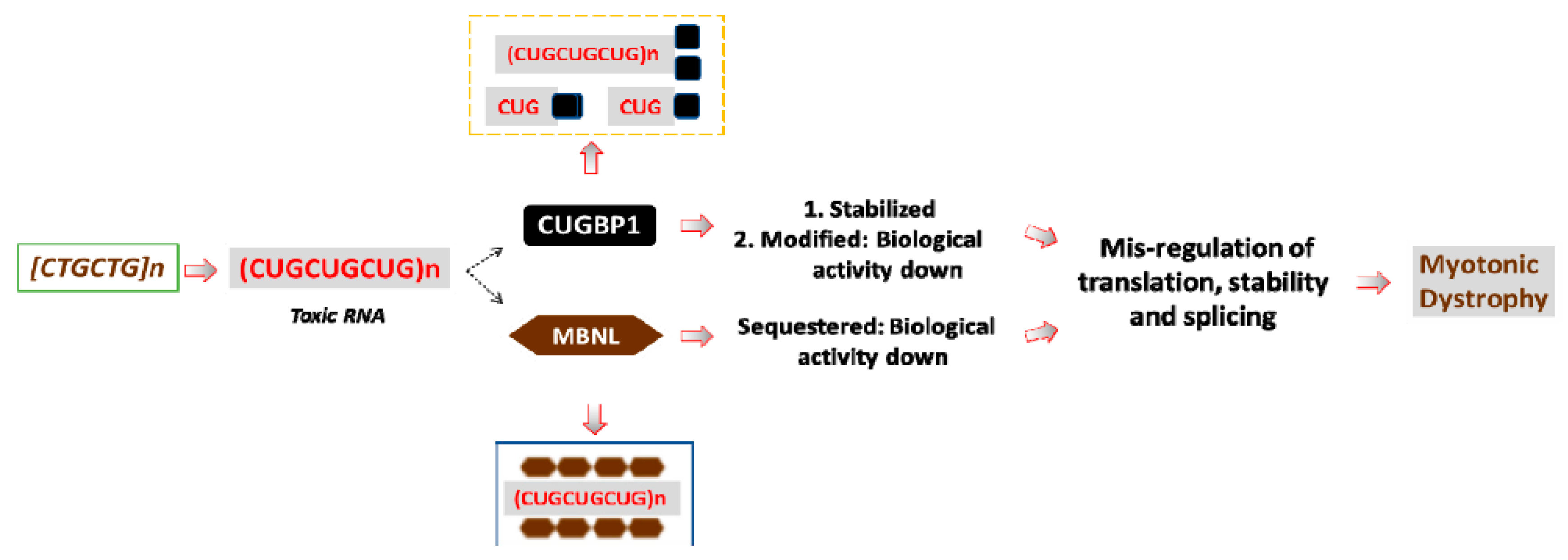
2. Why Are Expanded RNA CUG Repeats Toxic?
3. The Misregulation of CUGBP1 in DM1 Leads to the Abnormal RNA Processing at Multiple Levels
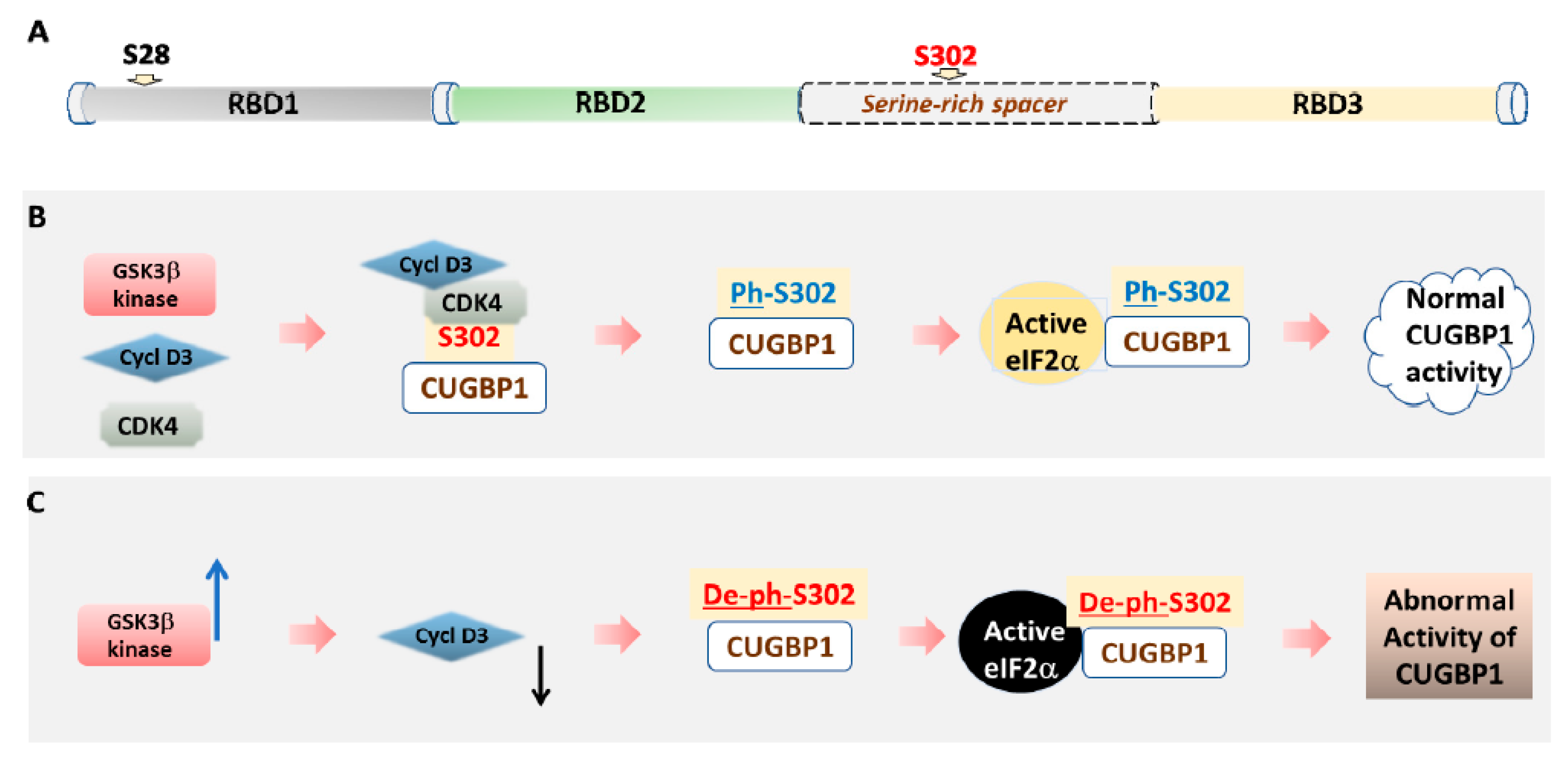
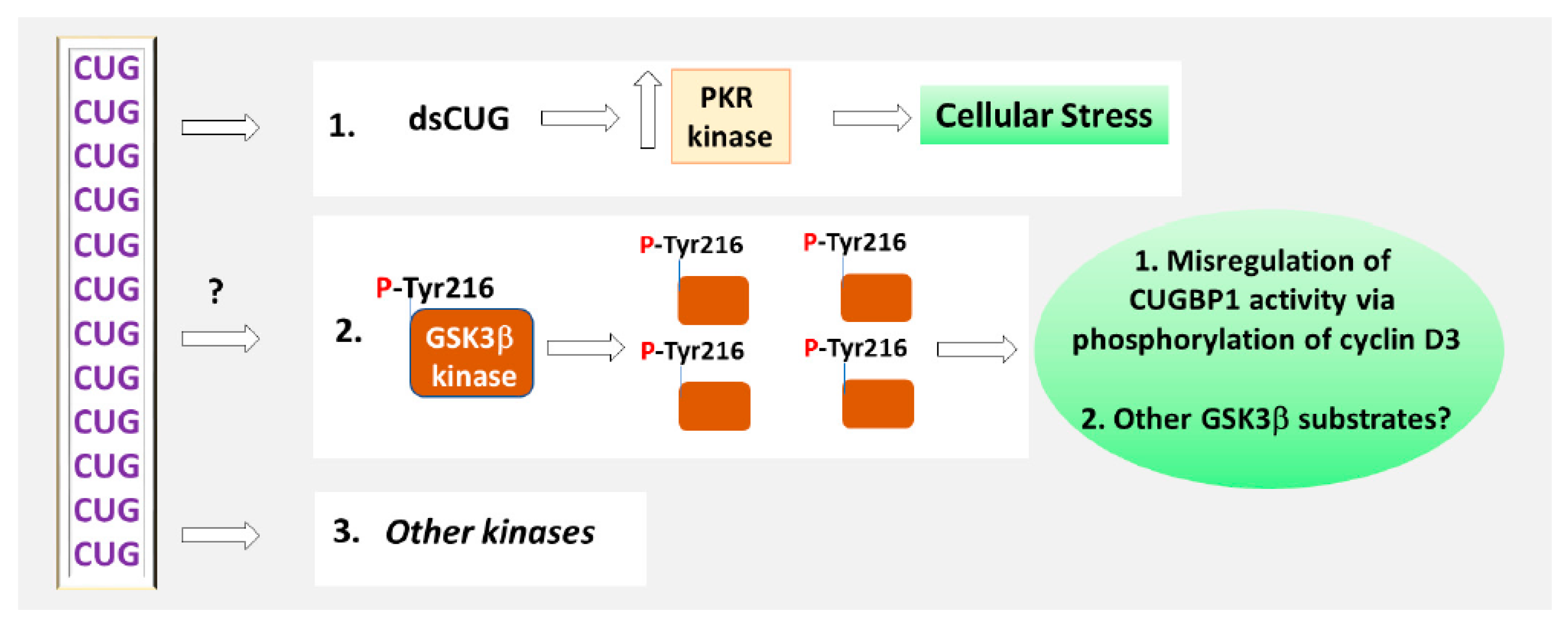
3.1. The Role of Phosphorylation in the Regulation of CUGBP1 Intracellular Localization and Translational Functions of CUGBP1
3.2. The Role of Splicing Activity of CUGBP1 in DM1
3.3. CUGBP1 Is a Regulator of mRNA Stability
4. Inhibitors of GSK3 in DM1 and CDM1 Therapeutic Approaches
5. Conclusions: What Is Next for Development of CUGBP1-GSK3-Based Therapy in CDM1 and DM1?
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Serine/threonine AKT or Protein kinase B |
| Ank-2 | Ankyrin 2 |
| ARE | AU-rich element |
| BIN1 | Bridging Integrator 1 gene |
| BIO | 6-bromoindirubin-3-oxime |
| Capzb | F actin capping proteinβ subunit |
| CDK | Cyclin dependent kinase |
| CDM | Congenital Myotonic Dystrophy |
| C/EBPβ | CCAAT-enhancer-binding proteinβ |
| c-fos | Proto-oncogene |
| CNS | Central Nervous System |
| Col | Collagen |
| CUGBP1/CELF1 | CUGBP1 elav-like factor 1 |
| Cx43 | Connexin 43 |
| Cypher | Striated Z-line protein |
| DCX | Doublecortin protein |
| DM1 | Myotonic Dystrophy type 1 |
| DDX | DEAD-box protein |
| DMPK | Myotonin protein kinase |
| DMSXL | Mutant mice expressing human DMPK gene with more than 1000 CTG repeats |
| dsCUG | double-stranded CUG RNA |
| eIF2 | Eukaryotic initiation factor 2 |
| FISH | Fluorescence in situ hybridization |
| FGF-2 | Basic fibroblast growth factor 2 |
| Fxr1 | Fragile X mental retardation-related protein 1 |
| Fyn | Protein-tyrosine kinase |
| GRE | GU-rich element |
| GSK3β | Glycogen synthase kinase 3β |
| hnRNPs | Heterogeneous nuclear ribonucleoproteins |
| HO-1 | Heme oxygenase 1 |
| HSALR | mutant mice expressing ~250 CTG repeats driven by human skeletal actin promoter |
| HuR | RNA-binding protein human antigen R |
| LEF1 | Lymphoid enhancer-binding factor 1 |
| MBNL | Muscleblind-like protein |
| MEF2 | Myocyte enhancer factor 2 |
| MYC | Proto-oncogene |
| MyoD | Myoblast determination protein |
| PARN | Poly (A)-specific ribonuclease |
| PAX-7 | Paired box protein Pax-7 |
| PB | Processing bodies |
| PKC | Protein kinase C |
| PKR | Protein kinase R |
| PYK2 | Protein-tyrosine kinase 2 |
| Rb | Retinoblastoma protein |
| RBM45 | RNA binding motif 45 protein |
| RyR1 | Ryanodine receptor 1 |
| SERCA1 | Skeletal muscle sarcoplasmic reticulum Ca2+ ATPase 1 |
| Smn1 | Spinal motor neuron 1 protein |
| TDZD-8 | 4-benzyl-2-methyl-1:2,4-thiadiazolidine-3,5-dione |
| TG | Tideglusib |
| TIA1 | RNA-binding protein T-cell intracellular antigen-1 |
| TIAR | T-cell intracellular antigen-related protein |
| TNF | Tumor necrosis factor |
| TnT | troponin T |
| Ub | ubiquitin |
| UTR | untranslated region |
References
- Harper, P.S. Myotonic Dystrophy; WB Saunders: London, UK, 2001. [Google Scholar]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G., Jr.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; De Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, L. Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int. J. Biochem. Cell Biol. 2013, 45, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Wei, C.; Schoser, B.; Meola, G.; Timchenko, N.; Timchenko, L. Reduction of toxic RNAs in myotonic dystrophies type 1 and type 2 by the RNA helicase p68/DDX5. Proc. Natl. Acad. Sci. USA 2015, 112, 8041–8045. [Google Scholar] [CrossRef] [PubMed]
- Laurent, F.X.; Sureau, A.; Klein, A.F.; Trouslard, F.; Gasnier, E.; Furling, D.; Marie, J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 2012, 40, 3159–3171. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, O.J.; Aagaard, L.; Andrejeva, D.; Thomsen, R.; Jensen, T.G.; Damgaard, C.K. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res. 2014, 42, 7186–7200. [Google Scholar] [CrossRef] [PubMed]
- Ravel-Chapuis, A.; Bélanger, G.; Yadava, R.S.; Mahadevan, M.S.; DesGroseillers, L.; Côté, J.; Jasmin, B.J. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J. Cell Biol. 2012, 196, 699–712. [Google Scholar] [CrossRef]
- Ravel-Chapuis, A.; Gunnewiek, A.K.; Bélanger, G.; Parks, T.E.C.; Côté, J.; Jasmin, B.J. Staufen1 impairs stress granule formation in skeletal muscle cells from myotonic dystrophy type 1 patients. Mol. Biol. Cell 2016, 27, 1728–1739. [Google Scholar] [CrossRef]
- Parks, T.E.C.; Ravel-Chapuis, A.; Bondy-Chorney, E.; Renaud, J.M.; Côté, J.; Jasmin, B.J. Muscle-specific expression of the RNA-binding protein Staufen1 induces progressive skeletal muscle atrophy via regulation of phosphatase tensin homolog. Hum. Mol. Genet. 2017, 26, 1821–1838. [Google Scholar] [CrossRef]
- Bondy-Chorney, E.; Parks, T.E.C.; Ravel-Chapuis, A.; Klinck, R.; Rocheleau, L.; Pelchat, M.; Chabot, B.; Jasmin, B.J.; Côté, J. Staufen1 regulates multiple alternative splicing events either positively or negatively in DM1 indicating its role as a disease modifier. PLoS Genet. 2016, 12, e1005827. [Google Scholar] [CrossRef]
- Paul, S.; Dansithong, W.; Kim, D.; Rossi, J.; Webster, N.J.; Comai, L.; Reddy, S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006, 25, 4271–4283. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Ranum, L.P. Repeat associated non-ATG (RAN) translation: New starts in microsatellite expansion disorders. Curr. Opin. Genet. Dev. 2014, 26, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Groenen, P.; Wieringa, B. Expanding complexity in myotonic dystrophy. Bioessays 1998, 20, 901–912. [Google Scholar] [CrossRef]
- Rastinejad, F.; Blau, H.M. Genetic complementation reveals a novel regulatory role for 3′ untranslated regions in growth and differentiation. Cell 1993, 72, 903–917. [Google Scholar] [CrossRef]
- Wang, J.; Pegoraro, E.; Menegazzo, E.; Gennarelli, M.; Hoop, R.C.; Angelini, C.; Hoffman, E.P. Myotonic dystrophy: Evidence for a possible dominant-negative RNA mutation. Hum. Mol. Genet. 1995, 4, 599–606. [Google Scholar] [CrossRef]
- Krahe, R.; Ashizawa, T.; Abbruzzese, C.; Roeder, E.; Carango, P.; Giacanelli, M.; Funanage, V.L.; Siciliano, M.J. Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics 1995, 28, 1–14. [Google Scholar] [CrossRef]
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef]
- Jansen, G.; Groenen, P.J.; Bächner, D.; Jap, P.H.; Coerwinkel, M.; Oerlemans, F.; Van Den Broek, W.; Gohlsch, B.; Pette, D.; Plomp, J.J.; et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 1996, 13, 316–324. [Google Scholar] [CrossRef]
- Timchenko, L.T.; Timchenko, N.A.; Caskey, C.T.; Roberts, R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: Implications for myotonic dystrophy. Hum. Mol. Genet. 1996, 5, 115–121. [Google Scholar] [CrossRef][Green Version]
- Timchenko, L.T.; Miller, J.W.; Timchenko, N.A.; DeVore, D.R.; Datar, K.V.; Lin, L.; Roberts, R.; Caskey, C.T.; Swanson, M.S. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996, 24, 4407–4414. [Google Scholar] [CrossRef]
- Timchenko, L.T. Myotonic dystrophy: The role of RNA CUG triplet repeats. Am. J. Hum. Genet. 1999, 64, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.M.; McCurrach, M.E.; Taneja, K.L.; Singer, R.H.; Housman, D.E. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA 1997, 94, 7388–7393. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Timchenko, N.A.; Timchenko, L.T. Cardiac elav-type RNA-binding protein (ETR-3) binds to RNA CUG repeats expanded in myotonic dystrophy. Hum. Mol. Genet. 1999, 8, 53–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Antic, D.; Keene, J.D. Embryonic lethal abnormal visual RNA-binding proteins involved in growth, differentiation, and posttranscriptional gene expression. Am. J. Hum. Genet. 1997, 61, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, N.A.; Cai, Z.-J.; Welm, A.L.; Reddy, S.; Ashizawa, T.; Timchenko, L.T. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 2001, 276, 7820–7826. [Google Scholar] [CrossRef]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef]
- Michalowski, S.; Miller, J.W.; Urbinati, C.R.; Paliouras, M.; Swanson, M.S.; Griffith, J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res. 1999, 27, 3534–3542. [Google Scholar] [CrossRef]
- Roberts, R.; Timchenko, N.A.; Miller, J.W.; Reddy, S.; Caskey, C.T.; Swanson, M.S.; Timchenko, L.T. Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13221–13226. [Google Scholar] [CrossRef]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 2012, 122, 4461–4472. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.-S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Amack, J.D.; Reagan, S.R.; Mahadevan, M.S. Mutant DMPK 3′-UTR transcripts disrupt C2C12 myogenic differentiation by compromising MyoD. J. Cell Biol. 2002, 159, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-McCardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006, 38, 1066–1070. [Google Scholar] [CrossRef]
- Orengo, J.P.; Chambon, P.; Metzger, D.; Mosier, D.R.; Snipes, G.J.; Cooper, T.A. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2008, 105, 2646–2651. [Google Scholar] [CrossRef]
- Huichalaf, C.; Sakai, K.; Jin, B.; Jones, K.; Wang, G.-L.; Schoser, B.; Schneider-Gold, C.; Sarkar, P.; Pereira-Smith, O.M.; Timchenko, N.; et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in Myotonic Dystrophy 1 (DM1) cells. FASEB J. 2010, 24, 3706–3719. [Google Scholar] [CrossRef]
- Salisbury, E.; Sakai, K.; Schoser, B.; Huichalaf, C.; Schneider-Gold, C.; Nguyen, H.; Wang, G.-L.; Albrecht, J.H.; Timchenko, L.T. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp. Cell Res. 2008, 314, 2266–2278. [Google Scholar] [CrossRef]
- Bohjanen, P.R.; Moua, M.L.; Guo, L.; Taye, A.; Vlasova-St-Louis, I.A. Altered CELF1 binding to target transcripts in malignant T cells. RNA 2015, 21, 1757–1769. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Wang, G.-L.; Timchenko, L.T. RNA CUG-binding protein 1 increases translation of 20-kDa isoform of CCAAT/Enhancer-binding protein by interacting with the and subunits of eukaryotic initiation translation factor 2. J. Biol. Chem. 2005, 280, 20549–20557. [Google Scholar] [CrossRef]
- Timchenko, L.T.; Salisbury, E.; Wang, G.-L.; Nguyen, H.; Albrecht, J.H.; Hershey, J.W.; Timchenko, N.A. Age-specific CUGBP1-eIF2 complex increases translation of CCAAT/enhancer-binding protein beta in old liver. J. Biol. Chem. 2006, 281, 32806–32819. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Iakova, P.; Cai, Z.J.; Smith, J.R.; Timchenko, L.T. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol. Cell. Biol. 2001, 21, 6927–6938. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, N.A.; Welm, A.L.; Lu, X.; Timchenko, L.T. CUG repeat binding protein (CUGBP1) interacts with the 5′ region of C/EBPbeta mRNA and regulates translation of C/EBPbeta isoforms. Nucleic Acids Res. 1999, 27, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, N.A.; Patel, R.; Iakova, P.; Cai, Z.J.; Quan, L.; Timchenko, L.T. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J. Biol. Chem. 2004, 279, 13129–13139. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Dulić, V. Molecular mechanisms for the senescent cell cycle arrest. In Journal of Investigative Dermatology Symposium Proceedings; Elsevier: Amsterdam, The Netherlands, 1998; Volume 3, pp. 14–18. [Google Scholar]
- Baker, G.L.; Landis, M.W.; Hinds, P.W. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 2005, 4, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Naderi, S.; Gutzkow, K.B.; Lahne, H.U.; Lefdal, S.; Ryves, W.J.; Harwood, A.J.; Blomhoff, H.K. cAMP induced degradation of cyclin D3 through association with GSK-3 beta. J. Cell Sci. 2004, 117, 3769–3783. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying > 1000 CTG repeats from the human DM1 locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Valanejad, L.; Zalewski, Z.A.; Karns, R.; Puymirat, J.; Nelson, D.; Witte, D.; Woodgett, J.; Timchenko, N.A.; et al. Correction of GSK3β at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J. 2018, 32, 2073–2085. [Google Scholar] [CrossRef]
- Wang, M.; Weng, W.C.; Stock, L.; Lindquist, D.; Martinez, A.; Gourdon, G.; Timchenko, N.; Snape, M.; Timchenko, L. Correction of Glycogen Synthase Kinase 3β in DM1 reduces the mutant RNA and improves postnatal survival of DMSXL mice. Mol. Cell. Biol. 2019, 39, e00155-19. [Google Scholar] [CrossRef]
- Doble, B.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef]
- Tian, B.; White, R.J.; Xia, T.; Welle, S.; Turner, D.H.; Mathews, M.B.; Thornton, C.A. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 2000, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lesort, M.; Jope, R.S.; Johnson, G.V. Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 1999, 72, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A.; Xiong, W.C.; Johnson, G.V. Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem. Biophys. Res. Commun. 2001, 284, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Ariaens, A.; Ponsioen, B.; Moolenaar, W.H. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol. Biol. Cell 2006, 17, 1834–1844. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Philips, A.V.; Timchenko, L.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Wang, G.S.; Kearney, D.L.; De Biasi, M.; Taffet, G.; Cooper, T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Investig. 2007, 117, 2802–2811. [Google Scholar] [CrossRef]
- Charlet-B, N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef]
- Wang, E.T.; Ward, A.G.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, H.; Wei, B.; Guo, Y.; Gu, L.; Yang, Z.; Zhang, Q.; Wu, Y.; Yuan, Q.; Zhao, G.; et al. CUG-BP1 regulates RyR1 ASI alternative splicing in skeletal muscle atrophy. Sci. Rep. 2015, 5, 16083. [Google Scholar] [CrossRef] [PubMed]
- Moraes, K.C.; Wilusz, C.J.; Wilusz, J. CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA 2006, 12, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Bareau, C.; Paillard, L.; Mereau, A.; Osborne, H.B. Mammalian CELF/Bruno-like RNA-binding proteins: Molecular characteristics and biological functions. Biochimie 2006, 88, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Beisang, D.; Reilly, C.; Bohjanen, P.R. Alternative polyadenylation regulates CELF1/CUGBP1 target transcripts following T cell activation. Gene 2014, 550, 93–100. [Google Scholar] [CrossRef]
- Marquis, J.; Paillard, L.; Audic, Y.; Cosson, B.; Danos, O.; Le Bec, C.; Osborne, H.B. CUGBP1/CELF1 requires UGU-rich sequences for high-affinity binding. Biochem. J. 2006, 400, 291–301. [Google Scholar] [CrossRef]
- Mori, D.; Sasagawa, N.; Kino, Y.; Ishiura, S. Quantitative analysis of CUG-BP1 binding to RNA repeats. J. Biochem. 2008, 143, 377–383. [Google Scholar] [CrossRef]
- Takahashi, N.; Sasagawa, N.; Suzuki, K.; Ishiura, S. The CUG-binding protein binds specifically to UG dinucleotide repeats in a yeast three-hybrid system. Biochem. Biophys. Res. Commun. 2000, 277, 518–523. [Google Scholar] [CrossRef]
- Tsuda, K.; Kuwasako, K.; Takahashi, M.; Someya, T.; Inoue, M.; Terada, T.; Kobayashi, N.; Shirouzu, M.; Kigawa, T.; Tanaka, A.; et al. Structural basis for the sequence-specific RNA-recognition mechanism of human CUG-BP1 RRM3. Nucleic Acids Res. 2009, 37, 5151–5166. [Google Scholar] [CrossRef]
- Vlasova, I.A.; Bohjanen, P.R. Posttranscriptional regulation of gene networks by GU-rich elements and CELF proteins. RNA Biol. 2008, 5, 201–207. [Google Scholar] [CrossRef]
- Vlasova, I.A.; Tahoe, N.M.; Fan, D.; Larsson, O.; Rattenbacher, B.; Sternjohn, J.R.; Vasdewani, J.; Karypis, G.; Reilly, C.S.; Bitterman, P.B.; et al. Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol. Cell 2008, 29, 263–270. [Google Scholar] [CrossRef]
- Rattenbacher, B.; Beisang, D.; Wiesner, D.L.; Jeschke, J.C.; Von Hohenberg, M.; St Louis-Vlasova, I.A.; Bohjanen, P.R. Analysis of CUGBP1 targets identifies GU-repeat sequences that mediate rapid mRNA decay. Mol. Cell. Biol. 2010, 30, 3970–3980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lee, J.Y.; Wilusz, J.; Tian, B.; Wilusz, C.J. Systematic analysis of cis elements in unstable mRNAs demonstrates that CUGBP1 is a key regulator of mRNA decay in muscle cells. PLoS ONE 2010, 5, e11201. [Google Scholar] [CrossRef] [PubMed]
- Vlasova-St Louis, I.; Bohjanen, P.R. Coordinate regulation of mRNA decay networks by GU-rich elements and CELF1. Curr. Opin. Genet. Dev. 2011, 21, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Blech-Hermoni, Y.; Dasgupta, T.; Coram, R.J.; Ladd, A.N. Identification of targets of CUG-BP, Elav-like family member 1 (CELF1) regulation in embryonic heart muscle. PLoS ONE 2016, 11, e0149061. [Google Scholar] [CrossRef]
- Liu, L.; Ouyang, M.; Rao, J.N.; Zou, T.; Xiao, L.; Chung, H.K.; Wu, J.; Donahue, J.M.; Gorospe, M.; Wang, J.Y. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol. Biol. Cell 2015, 26, 1797–1810. [Google Scholar] [CrossRef]
- Xiao, L.; Cui, Y.H.; Rao, J.N.; Zou, T.; Liu, L.; Smith, A.; Turner, D.J.; Gorospe, M.; Wang, J.Y. Regulation of cyclin-dependent kinase 4 translation through CUG-binding protein 1 and microRNA-222 by polyamines. Mol. Biol. Cell 2011, 22, 3055–3069. [Google Scholar] [CrossRef]
- Yu, T.X.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Ouyang, M.; Cao, S.; Gorospe, M.; Wang, J.Y. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol. Biol. Cell 2013, 24, 85–99. [Google Scholar] [CrossRef]
- Yu, T.X.; Gu, B.L.; Yan, J.K.; Zhu, J.; Yan, W.H.; Chen, J.; Qian, L.X.; Cai, W. CUGBP1 and HuR regulate E-cadherin translation by altering recruitment of E-cadherin mRNA to processing bodies and modulate epithelial barrier function. Am. J. Physiol. Cell Physiol. 2016, 310, C54–C65. [Google Scholar] [CrossRef]
- Chang, K.T.; Cheng, C.F.; King, P.C.; Liu, S.Y.; Wang, G.S. CELF1 mediates connexin 43 mRNA degradation in dilated cardiomyopathy. Circ. Res. 2017, 121, 1140–1152. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Wang, J.; Wei, B.; Zhang, X.; Zhang, M.; Cao, D.; Dai, J.; Wang, Z.; Nyirimigabo, E.; et al. A positive feedback regulation of Heme oxygenase 1 by CELF1 in cardiac myoblast cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 209–218. [Google Scholar] [CrossRef]
- Beisang, D.; Rattenbacher, B.; Vlasova-St Louis, I.A.; Bohjanen, P.R. Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J. Biol. Chem. 2012, 287, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Tamada, H.; Sakashita, E.; Shimazaki, K.; Ueno, E.; Hamamoto, T.; Kagawa, Y.; Endo, H. cDNA cloning and characterization of Drb1, a new member of RRM-type neural RNA-binding protein. Biochem. Biophys. Res. Commun. 2002, 297, 96–104. [Google Scholar] [CrossRef]
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef] [PubMed]
- Poulos, M.G.; Batra, R.; Li, M.; Yuan, Y.; Zhang, C.; Darnell, R.B.; Swanson, M.S. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum. Mol. Genet. 2013, 22, 3547–3558. [Google Scholar] [CrossRef]
- Choi, J.; Dixon, D.M.; Dansithong, W.; Abdallah, W.F.; Roos, K.P.; Jordan, M.C.; Trac, B.; Lee, H.S.; Comai, L.; Reddy, S. Muscleblind-like 3 deficit results in a spectrum of age-associated pathologies observed in myotonic dystrophy. Sci. Rep. 2016, 6, 30999. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Urbinati, C.R.; Crusselle, V.J.; Luo, D.; Lee, Y.J.; Harrison, J.K.; Oh, S.P.; Swanson, M.S. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr. Patterns 2003, 3, 459–462. [Google Scholar] [CrossRef]
- Ho, T.H.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547. [Google Scholar] [CrossRef]
- Koshelev, M.; Sarma, S.; Price, R.E.; Wehrens, X.H.; Cooper, T.A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 1066–1075. [Google Scholar] [CrossRef]
- Ward, A.J.; Rimer, M.; Killian, J.M.; Dowling, J.J.; Cooper, T.A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 3614–3622. [Google Scholar] [CrossRef]
- Kress, C.; Gautier-Courteille, C.; Osborne, H.B.; Babinet, C.; Paillard, L. Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol. Cell. Biol. 2007, 27, 1146–1157. [Google Scholar] [CrossRef]
- Berger, D.S.; Moyer, M.; Kliment, G.M.; Van Lunteren, E.; Ladd, A.N. Expression of a dominant negative CELF protein in vivo leads to altered muscle organization, fiber size, and subtype. PLoS ONE 2011, 6, e19274. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Kuyumcu-Martinez, M.N.; Sarma, S.; Mathur, N.; Wehrens, X.H.; Cooper, T.A. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Investig. 2009, 119, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Morato, M.; Brook, J.D.; Wojciechowska, M. Small molecules which improve pathogenesis of Myotonic Dystrophy type 1. Front. Neurol. 2018, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- Study of Tideglusib in Adolescent and Adult Patients with Myotonic Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT02858908 (accessed on 10 November 2019).
- Hernández-Hernández, O.; Guiraud-Dogan, C.; Sicot, G.; Huguet, A.; Luilier, S.; Steidl, E.; Saenger, S.; Marciniak, E.; Obriot, H.; Chevarin, C.; et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behavior. Brain 2013, 136, 957–970. [Google Scholar] [CrossRef]
- Morriss, G.R.; Rajapakshe, K.; Huang, S.; Coarfa, C.; Cooper, T.A. Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1. Hum. Mol. Genet. 2018, 27, 2789–2804. [Google Scholar] [CrossRef] [PubMed]
- Botta, A.; Malena, A.; Tibaldi, E.; Rocchi, L.; Loro, E.; Pena, E.; Cenci, L.; Ambrosi, E.; Bellocchi, M.C.; Pagano, M.A.; et al. MBNL142 and MBNL143 gene isoforms, overexpressed in DM1-patient muscle, encode for nuclear proteins interacting with Src family kinases. Cell Death Dis. 2013, 4, e770. [Google Scholar] [CrossRef][Green Version]
- Brockhoff, M.; Rion, N.; Chojnowska, K.; Wiktorowicz, T.; Eickhorst, C.; Erne, B.; Frank, S.; Angelini, C.; Furling, D.; Rüegg, M.A.; et al. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J. Clin. Investig. 2017, 127, 549–563. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| GSK3 Inhibitor | Effect on the HSALR Phenotype | Molecular Effects |
|---|---|---|
| Lithium [31] |
|
|
| TDZD-8 [31] |
|
|
| BIO [49] |
|
|
| Tideglusib [50] |
|
|
| GSK3 Inhibitor | Effect on the Phenotype of DMSXL Mice | Molecular Effects |
|---|---|---|
| Tideglusib [50] |
|
|
| GSK3 Inhibitor | Effects on Human CDM1 and DM1 Myoblasts |
|---|---|
| BIO [49] |
|
| Tideglusib [50] |
|
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timchenko, L. Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2020, 21, 94. https://doi.org/10.3390/ijms21010094
Timchenko L. Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1. International Journal of Molecular Sciences. 2020; 21(1):94. https://doi.org/10.3390/ijms21010094
Chicago/Turabian StyleTimchenko, Lubov. 2020. "Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1" International Journal of Molecular Sciences 21, no. 1: 94. https://doi.org/10.3390/ijms21010094
APA StyleTimchenko, L. (2020). Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1. International Journal of Molecular Sciences, 21(1), 94. https://doi.org/10.3390/ijms21010094