A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases

 ,
,  ,
,  , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Biological Characteristics of VIP
2.1. VIP Discovery, Cellular Location, and Structure
2.2. General Biological Functions
3. VIP Receptors, Ligands, and Signaling Pathways
3.1. VIP Receptors
3.1.1. VPAC1 Receptor
3.1.2. VPAC2 Receptor
3.2. Ligands
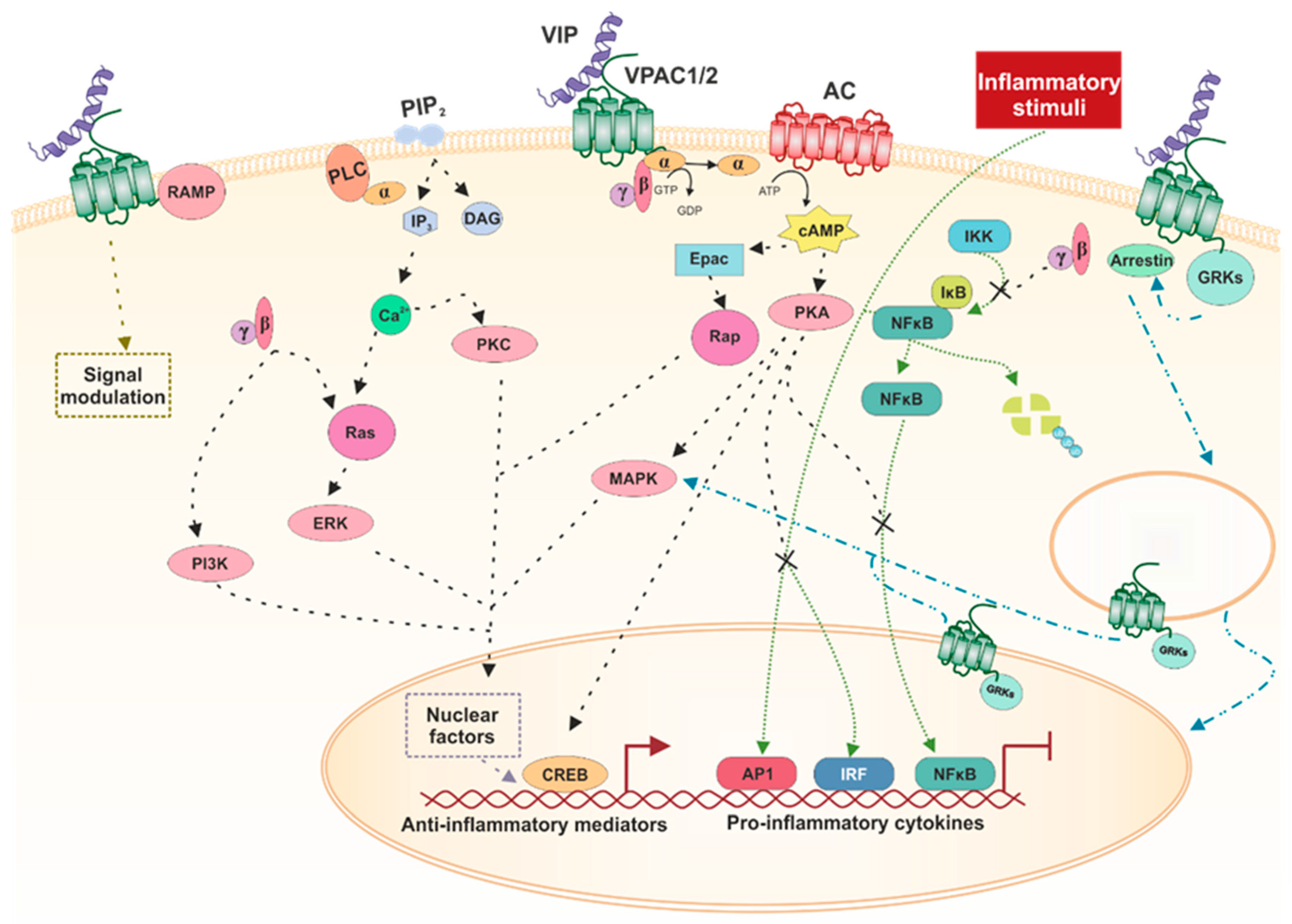
3.3. Signaling Pathways
4. A Very Important Peptide in Inflammation and Autoimmunity
4.1. Targeting Balance of Inflammatory Factors
4.2. Modulating the Expression of TLRs
4.3. Regulating Th Cells
4.4. Inducing Tolerogenic Dendritic Cells
5. Protective Effects of VIP in Inflammatory/Autoimmune Diseases
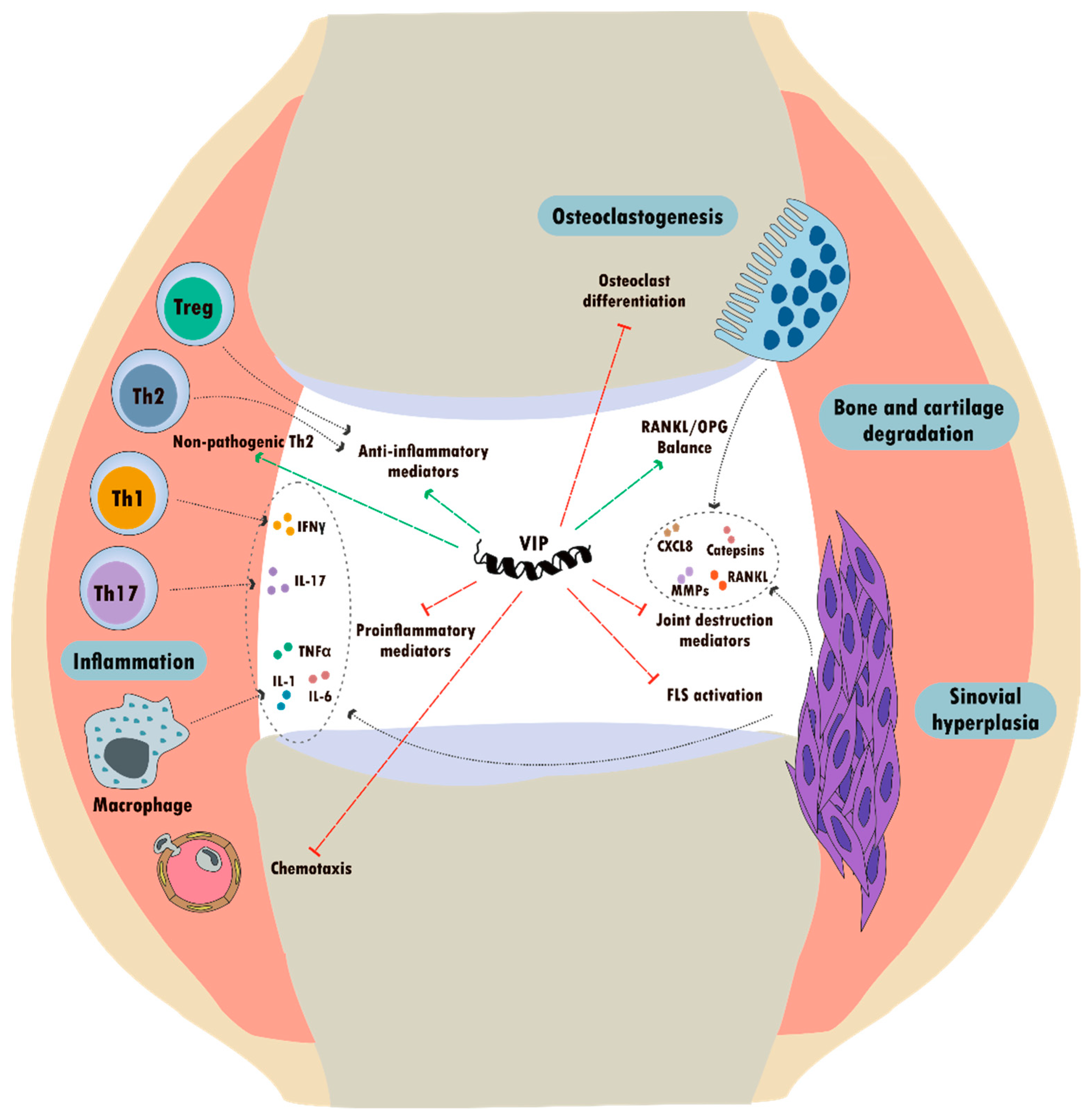
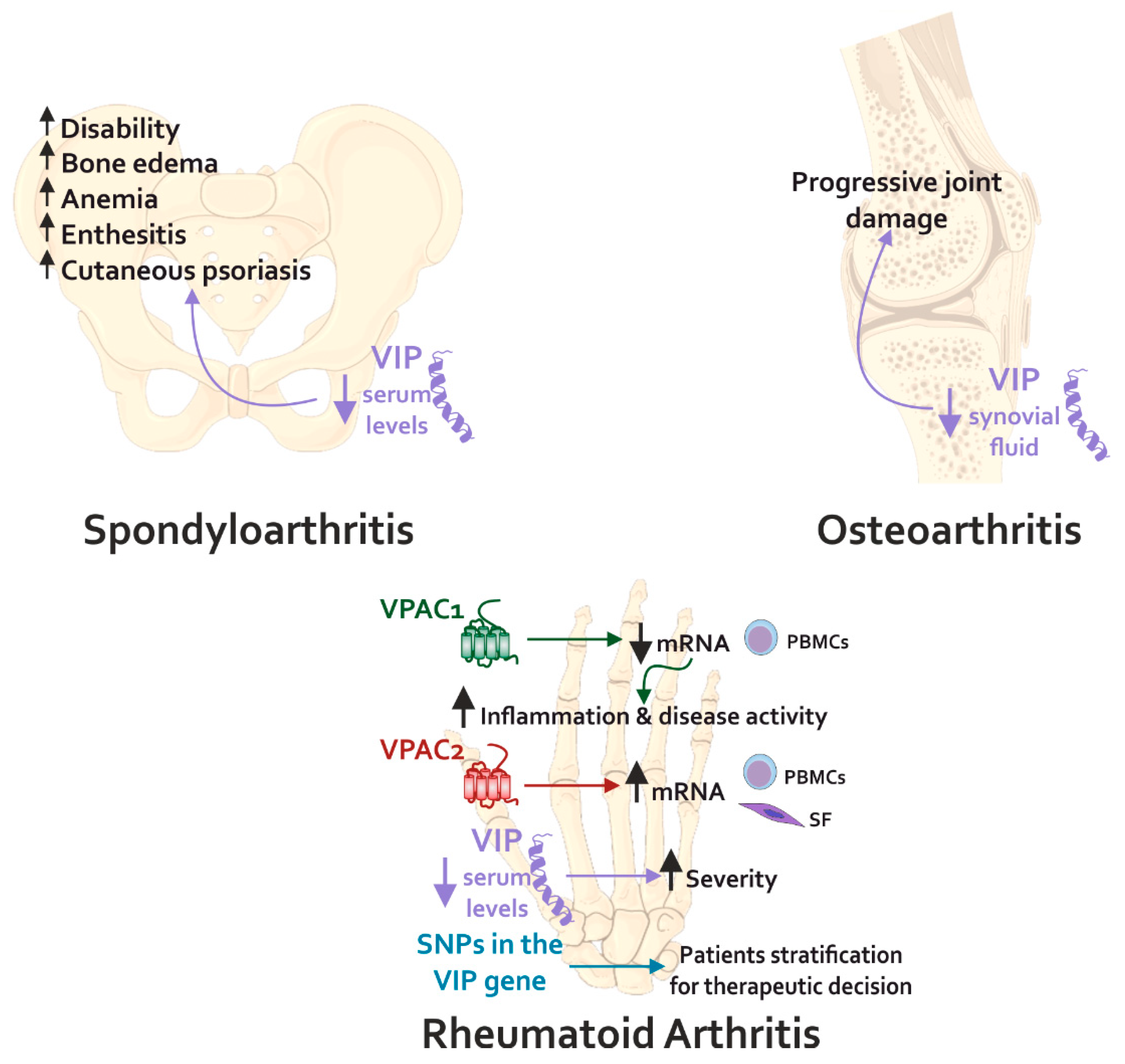
5.1. Rheumatic Diseases
5.1.1. VIP in Rheumatoid Arthritis
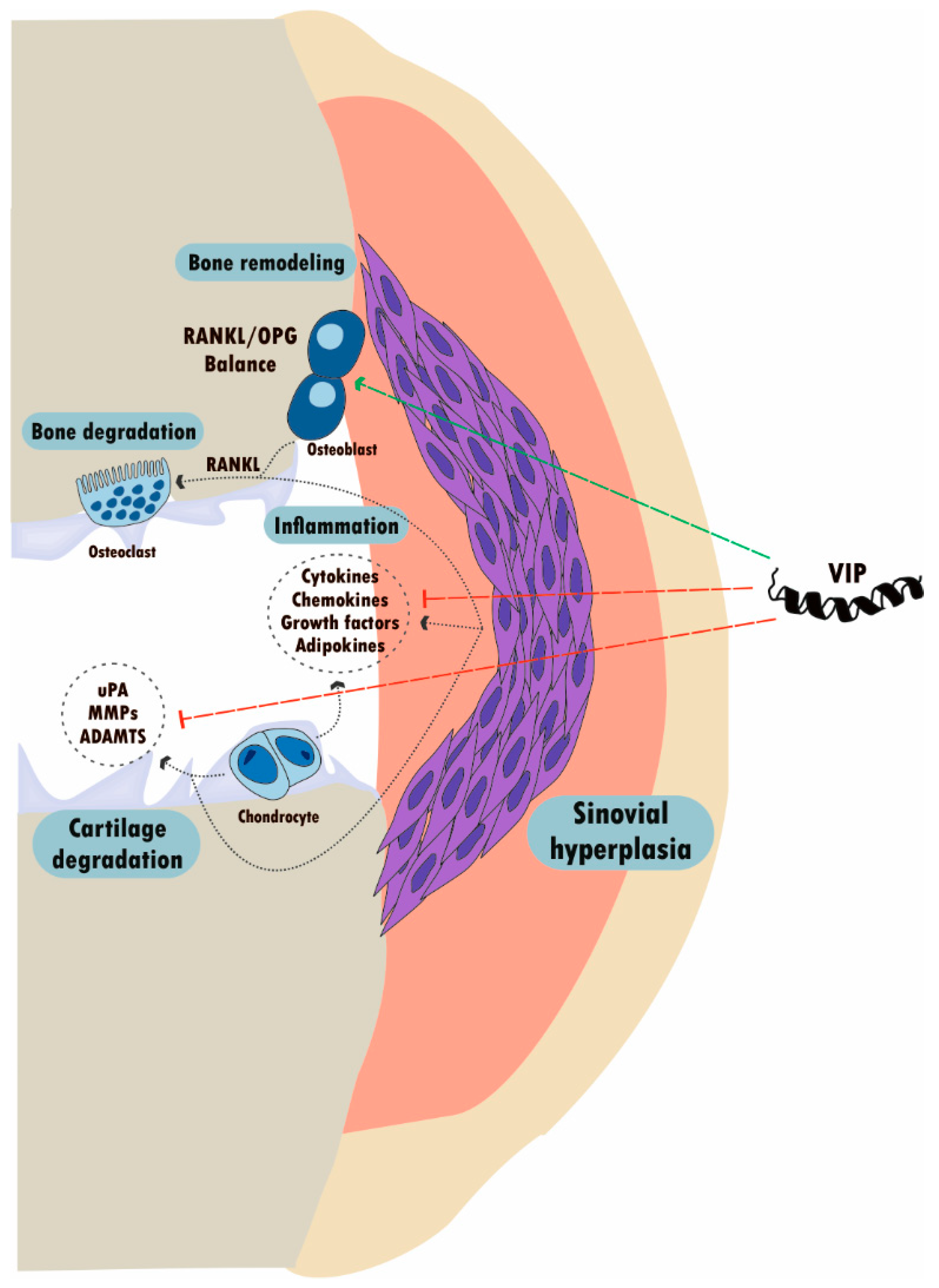
5.1.2. VIP in Osteoarthritis
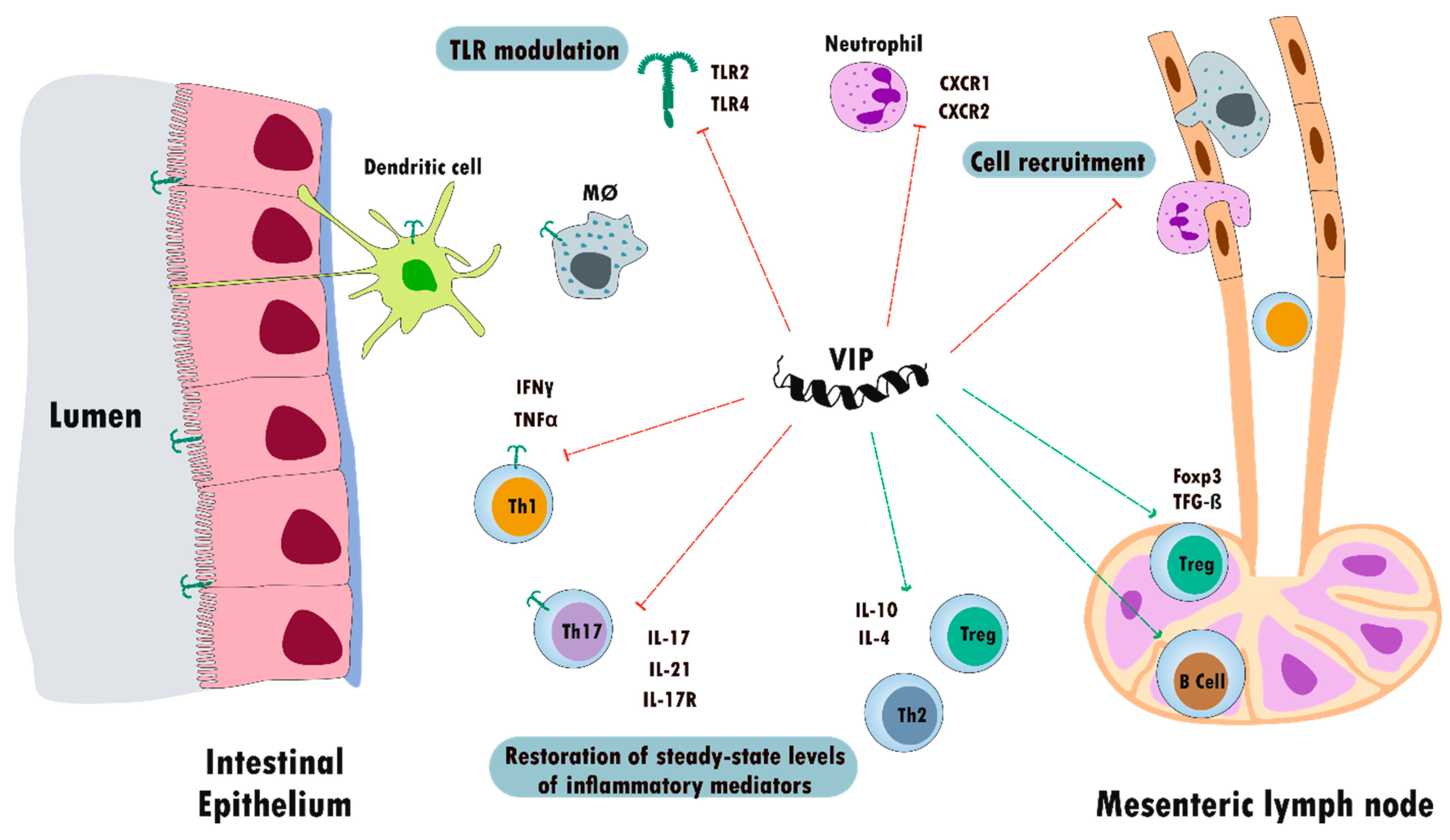
5.2. Inflammatory Bowel Disease
5.3. Central Nervous System Diseases
5.4. Other Autoimmune Disorders
6. VIP as a Therapeutic Agent: Limitations and Perspectives
7. Potential of VIP Axis as a Biomarker for Personalized Treatment in Rheumatic Diseases
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylate cyclase |
| ACPA | Anti-citrullinated protein antibodies |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motifs |
| Breg | Regulatory B cells |
| cAMP | Adenylate cyclase activity |
| CD | Crohn’s disease |
| CIA | Collagen-induced arthritis |
| CNS | Central nervous system |
| CREB | cAMP response element-binding |
| CRF | Corticotropin-releasing factor |
| DC | Dendritic cells |
| DNBS | Dinitrobenzene sulfonic acid |
| DSS | Dextran Sodium Sulfate |
| EAE | Experimental autoimmune encephalomyelitis |
| ECM | Extracellular matrix |
| ERK | Extracellular regulated kinases |
| Fn-fs | Fibronectin-fragments |
| GHRF | Growth hormone–releasing hormone |
| GPCRs | G-protein-coupled receptors |
| GRK | GPCR kinase |
| IBD | Inflammatory bowel disease |
| IFN | Interferon |
| IL- | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| IRF | Interferon regulatory factor |
| KO | Knockout |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| MS | Multiple Sclerosis |
| NFκB | Nuclear factor κB |
| NO | Nitric oxide |
| NOD | Non-obese diabetic |
| OA | Osteoarthritis |
| PACAP | Pituitary adenylate cyclase-activating polypeptide |
| PBMC | Peripheral blood mononuclear cell |
| PG | Prostaglandin |
| PKA | Protein kinase A |
| RA | Rheumatoid arthritis |
| RAMP | Receptor activity modifying protein |
| SF | Synovial fibroblasts |
| SNP | Single-nucleotide polymorphism |
| SpA | Spondyloarthritis |
| SS | Sjögren’s syndrome |
| Th | T helper |
| TLR | Toll-like receptors |
| TNBS | Trinitrobenzenesulfonic acid |
| TNF | Tumor necrosis factor |
| Treg | Regulatory T cells |
| UC | Ulcerative Colitis |
| UCN | Urocortin |
| uPA | Urokinase-type plasminogen activator |
| uPAR | Urokinase-type plasminogen activator receptor |
| VIP | Vasoactive intestinal peptide |
References
- Weigent, D.A.; Carr, D.J.; Blalock, J.E. Bidirectional communication between the neuroendocrine and immune systems. Common hormones and hormone receptors. Ann. N. Y. Acad. Sci. 1990, 579, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Sreedharan, S.P. Mediators of communication and adaptation in the neuroendocrine and immune systems. FASEB J. 1992, 6, 2646–2652. [Google Scholar] [CrossRef] [PubMed]
- Souza-Moreira, L.; Campos-Salinas, J.; Caro, M.; Gonzalez-Rey, E. Neuropeptides as pleiotropic modulators of the immune response. Neuroendocrinology 2011, 94, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Gomariz, R.P.; Martinez, C.; Abad, C.; Leceta, J.; Delgado, M. Immunology of VIP: A review and therapeutical perspectives. Curr. Pharm. Des. 2001, 7, 89–111. [Google Scholar] [CrossRef]
- Gomariz, R.P.; Juarranz, Y.; Abad, C.; Arranz, A.; Leceta, J.; Martinez, C. VIP-PACAP system in immunity: New insights for multitarget therapy. Ann. N. Y. Acad. Sci. 2006, 1070, 51–74. [Google Scholar] [CrossRef]
- Delgado, M.; Ganea, D. Vasoactive intestinal peptide: A neuropeptide with pleiotropic immune functions. Amino Acids 2013, 45, 25–39. [Google Scholar] [CrossRef]
- Abad, C.; Martinez, C.; Juarranz, M.G.; Arranz, A.; Leceta, J.; Delgado, M.; Gomariz, R.P. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn’s disease. Gastroenterology 2003, 124, 961–971. [Google Scholar] [CrossRef]
- Li, H.; Mei, Y.; Wang, Y.; Xu, L. Vasoactive intestinal polypeptide suppressed experimental autoimmune encephalomyelitis by inhibiting T helper 1 responses. J. Clin. Immunol. 2006, 26, 430–437. [Google Scholar] [CrossRef]
- Lodde, B.M.; Mineshiba, F.; Wang, J.; Cotrim, A.P.; Afione, S.; Tak, P.P.; Baum, B.J. Effect of human vasoactive intestinal peptide gene transfer in a murine model of Sjogren’s syndrome. Ann. Rheum. Dis. 2006, 65, 195–200. [Google Scholar] [CrossRef]
- Jimeno, R.; Gomariz, R.P.; Gutierrez-Canas, I.; Martinez, C.; Juarranz, Y.; Leceta, J. New insights into the role of VIP on the ratio of T-cell subsets during the development of autoimmune diabetes. Immunol. Cell Biol. 2010, 88, 734–745. [Google Scholar] [CrossRef]
- Delgado, M.; Abad, C.; Martinez, C.; Leceta, J.; Gomariz, R.P. Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat. Med. 2001, 7, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Gomariz, R.P.; Juarranz, Y.; Carrión, M.; Pérez-García, S.; Villanueva-Romero, R.; González-Álvaro, I.; Gutiérrez-Cañas, I.; Lamana, A.; Martínez, C. An Overview of VPAC Receptors in Rheumatoid Arthritis: Biological Role and Clinical Significance. Front. Endocrinol. 2019, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Elewaut, D.; McInnes, I.B.; Dayer, J.M.; Neurath, M.F. How cytokine networks fuel inflammation: Toward a cytokine-based disease taxonomy. Nat. Med. 2013, 19, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Rahman, P.; Inman, R.D.; El-Gabalawy, H.; Krause, D.O. Pathophysiology and pathogenesis of immune-mediated inflammatory diseases: Commonalities and differences. J. Rheumatol. Suppl. 2010, 85, 11–26. [Google Scholar] [CrossRef] [PubMed]
- David, T.; Ling, S.F.; Barton, A. Genetics of immune-mediated inflammatory diseases. Clin. Exp. Immunol. 2018, 193, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Perricone, C.; Versini, M.; Ben-Ami, D.; Gertel, S.; Watad, A.; Segel, M.J.; Ceccarelli, F.; Conti, F.; Cantarini, L.; Bogdanos, D.P.; et al. Smoke and autoimmunity: The fire behind the disease. Autoimmun. Rev. 2016, 15, 354–374. [Google Scholar] [CrossRef]
- Surace, A.E.A.; Hedrich, C.M. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front. Immunol. 2019, 10, 1525. [Google Scholar] [CrossRef]
- El-Gabalawy, H.; Guenther, L.C.; Bernstein, C.N. Epidemiology of immune-mediated inflammatory diseases: Incidence, prevalence, natural history, and comorbidities. J. Rheumatol. Suppl. 2010, 85, 2–10. [Google Scholar] [CrossRef]
- Cutolo, M.; Kitas, G.D.; van Riel, P.L. Burden of disease in treated rheumatoid arthritis patients: Going beyond the joint. Semin. Arthritis Rheum. 2014, 43, 479–488. [Google Scholar] [CrossRef]
- Burisch, J.; Jess, T.; Martinato, M.; Lakatos, P.L.; EpiCom, E. The burden of inflammatory bowel disease in Europe. J. Crohns Colitis 2013, 7, 322–337. [Google Scholar] [CrossRef]
- Marrie, R.A. Comorbidity in multiple sclerosis: Implications for patient care. Nat. Rev. Neurol. 2017, 13, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Said, S.I.; Mutt, V. Polypeptide with broad biological activity: Isolation from small intestine. Science 1970, 169, 1217–1218. [Google Scholar] [CrossRef] [PubMed]
- Said, S.I.; Rosenberg, R.N. Vasoactive intestinal polypeptide: Abundant immunoreactivity in neural cell lines and normal nervous tissue. Science 1976, 192, 907–908. [Google Scholar] [CrossRef] [PubMed]
- Johansson, O.; Lundberg, J.M. Ultrastructural localization of VIP-like immunoreactivity in large dense-core vesicles of ‘cholinergic-type’ nerve terminals in cat exocrine glands. Neuroscience 1981, 6, 847–862. [Google Scholar] [CrossRef]
- Staun-Olsen, P.; Ottesen, B.; Bartels, P.D.; Nielsen, M.H.; Gammeltoft, S.; Fahrenkrug, J. Receptors for vasoactive intestinal polypeptide on isolated synaptosomes from rat cerebral cortex. Heterogeneity of binding and desensitization of receptors. J. Neurochem. 1982, 39, 1242–1251. [Google Scholar] [CrossRef]
- Felten, D.L.; Felten, S.Y.; Carlson, S.L.; Olschowka, J.A.; Livnat, S. Noradrenergic and peptidergic innervation of lymphoid tissue. J. Immunol. 1985, 135, 755s–765s. [Google Scholar]
- Bellinger, D.L.; Lorton, D.; Brouxhon, S.; Felten, S.; Felten, D.L. The significance of vasoactive intestinal polypeptide (VIP) in immunomodulation. Adv. Neuroimmunol. 1996, 6, 5–27. [Google Scholar] [CrossRef]
- Cutz, E.; Chan, W.; Track, N.S.; Goth, A.; Said, S.I. Release of vasoactive intestinal polypeptide in mast cells by histamine liberators. Nature 1978, 275, 661–662. [Google Scholar] [CrossRef]
- O’Dorisio, M.S.; O’Dorisio, T.M.; Cataland, S.; Balcerzak, S.P. Vasoactive intestinal polypeptide as a biochemical marker for polymorphonuclear leukocytes. J. Lab. Clin. Med. 1980, 96, 666–672. [Google Scholar] [CrossRef]
- Aliakbari, J.; Sreedharan, S.P.; Turck, C.W.; Goetzl, E.J. Selective localization of vasoactive intestinal peptide and substance P in human eosinophils. Biochem. Biophys. Res. Commun. 1987, 148, 1440–1445. [Google Scholar] [CrossRef]
- Weinstock, J.V.; Blum, A.M. Detection of vasoactive intestinal peptide and localization of its mRNA within granulomas of murine schistosomiasis. Cell Immunol. 1990, 125, 291–300. [Google Scholar] [CrossRef]
- Carrión, M.; Pérez-García, S.; Martínez, C.; Juarranz, Y.; Estrada-Capetillo, L.; Puig-Kroger, A.; Gomariz, R.P.; Gutiérrez-Cañas, I. VIP impairs acquisition of the macrophage proinflammatory polarization profile. J. Leukoc. Biol. 2016, 100, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Gomariz, R.P.; Lorenzo, M.J.; Cacicedo, L.; Vicente, A.; Zapata, A.G. Demonstration of immunoreactive vasoactive intestinal peptide (IR-VIP) and somatostatin (IR-SOM) in rat thymus. Brain Behav. Immun. 1990, 4, 151–161. [Google Scholar] [CrossRef]
- Gomariz, R.P.; Delgado, M.; Naranjo, J.R.; Mellstrom, B.; Tormo, A.; Mata, F.; Leceta, J. VIP gene expression in rat thymus and spleen. Brain Behav. Immun. 1993, 7, 271–278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gomariz, R.P.; Garrido, E.; Leceta, J.; Martinez, C.; Abalo, R.; Delgado, M. Gene expression of VIP receptor in rat lymphocytes. Biochem. Biophys. Res. Commun. 1994, 203, 1599–1604. [Google Scholar] [CrossRef]
- Straub, R.H.; Bijlsma, J.W.; Masi, A.; Cutolo, M. Role of neuroendocrine and neuroimmune mechanisms in chronic inflammatory rheumatic diseases—The 10-year update. Semin. Arthritis Rheum. 2013, 43, 392–404. [Google Scholar] [CrossRef]
- Grassel, S.; Muschter, D. Peripheral Nerve Fibers and Their Neurotransmitters in Osteoarthritis Pathology. Int. J. Mol. Sci. 2017, 18, 931. [Google Scholar] [CrossRef]
- Juarranz, Y.; Gutiérrez-Cañas, I.; Santiago, B.; Carrión, M.; Pablos, J.L.; Gomariz, R.P. Differential expression of vasoactive intestinal peptide and its functional receptors in human osteoarthritic and rheumatoid synovial fibroblasts. Arthritis Rheum. 2008, 58, 1086–1095. [Google Scholar] [CrossRef]
- Couvineau, A.; Laburthe, M. VPAC receptors: Structure, molecular pharmacology and interaction with accessory proteins. Br. J. Pharmacol. 2012, 166, 42–50. [Google Scholar] [CrossRef]
- Dickson, L.; Finlayson, K. VPAC and PAC receptors: From ligands to function. Pharmacol. Ther. 2009, 121, 294–316. [Google Scholar] [CrossRef]
- Neumann, J.M.; Couvineau, A.; Murail, S.; Lacapere, J.J.; Jamin, N.; Laburthe, M. Class-B GPCR activation: Is ligand helix-capping the key? Trends Biochem. Sci. 2008, 33, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, N.M.; Krueckl, S.L.; McRory, J.E. The origin and function of the pituitary adenylate cyclase-activating polypeptide (PACAP)/glucagon superfamily. Endocr. Rev. 2000, 21, 619–670. [Google Scholar] [PubMed]
- Nicole, P.; Rouyer-Fessard, C.; Couvineau, A.; Drouot, C.; Fulcrand, P.; Martinez, J.; Laburthe, M. Alanine scanning of VIP. Structure-function relationship for binding to human recombinant VPAC1 receptor. Ann. N. Y. Acad. Sci. 2000, 921, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Wray, V.; Kakoschke, C.; Nokihara, K.; Naruse, S. Solution structure of pituitary adenylate cyclase activating polypeptide by nuclear magnetic resonance spectroscopy. Biochemistry 1993, 32, 5832–5841. [Google Scholar] [CrossRef]
- Ganea, D.; Hooper, K.M.; Kong, W. The neuropeptide vasoactive intestinal peptide: Direct effects on immune cells and involvement in inflammatory and autoimmune diseases. Acta Physiol. 2015, 213, 442–452. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Liao, C.; de Molliens, M.P.; Schneebeli, S.T.; Brewer, M.; Song, G.; Chatenet, D.; Braas, K.M.; May, V.; Li, J. Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr. Top. Med. Chem. 2019, 19, 1399–1417. [Google Scholar] [CrossRef]
- Ishihara, T.; Shigemoto, R.; Mori, K.; Takahashi, K.; Nagata, S. Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron 1992, 8, 811–819. [Google Scholar] [CrossRef]
- Sreedharan, S.P.; Patel, D.R.; Huang, J.X.; Goetzl, E.J. Cloning and functional expression of a human neuroendocrine vasoactive intestinal peptide receptor. Biochem. Biophys. Res. Commun. 1993, 193, 546–553. [Google Scholar] [CrossRef]
- Bokaei, P.B.; Ma, X.Z.; Byczynski, B.; Keller, J.; Sakac, D.; Fahim, S.; Branch, D.R. Identification and characterization of five-transmembrane isoforms of human vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide receptors. Genomics 2006, 88, 791–800. [Google Scholar] [CrossRef]
- Lutz, E.M.; Sheward, W.J.; West, K.M.; Morrow, J.A.; Fink, G.; Harmar, A.J. The VIP2 receptor: Molecular characterisation of a cDNA encoding a novel receptor for vasoactive intestinal peptide. FEBS Lett. 1993, 334, 3–8. [Google Scholar] [CrossRef]
- Svoboda, M.; Tastenoy, M.; Van Rampelbergh, J.; Goossens, J.F.; De Neef, P.; Waelbroeck, M.; Robberecht, P. Molecular cloning and functional characterization of a human VIP receptor from SUP-T1 lymphoblasts. Biochem. Biophys. Res. Commun. 1994, 205, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Yoshida, H.; Mizuta, M.; Mizuno, N.; Fujii, Y.; Gonoi, T.; Miyazaki, J.; Seino, S. Cloning and functional characterization of a third pituitary adenylate cyclase-activating polypeptide receptor subtype expressed in insulin-secreting cells. Proc. Natl. Acad. Sci. USA 1994, 91, 2679–2683. [Google Scholar] [CrossRef] [PubMed]
- Grinninger, C.; Wang, W.; Oskoui, K.B.; Voice, J.K.; Goetzl, E.J. A natural variant type II G protein-coupled receptor for vasoactive intestinal peptide with altered function. J. Biol. Chem. 2004, 279, 40259–40262. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Verma, D.; Grinninger, C.; Huang, M.C.; Goetzl, E.J. Functional splice variants of the type II G protein-coupled receptor (VPAC2) for vasoactive intestinal peptide in mouse and human lymphocytes. Ann. N. Y. Acad. Sci. 2006, 1070, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, P.; Coy, D.H.; De Neef, P.; Camus, J.C.; Cauvin, A.; Waelbroeck, M.; Christophe, J. [D-Phe4] peptide histidine-isoleucinamide ([D-Phe4]PHI), a highly selective vasoactive-intestinal-peptide (VIP) agonist, discriminates VIP-preferring from secretin-preferring receptors in rat pancreatic membranes. Eur. J. Biochem. 1987, 165, 243–249. [Google Scholar] [CrossRef]
- Rouyer-Fessard, C.; Couvineau, A.; Voisin, T.; Laburthe, M. Ac-Tyr1hGRF discriminates between VIP receptors from rat liver and intestinal epithelium. Life Sci. 1989, 45, 829–833. [Google Scholar] [CrossRef]
- Fishbein, V.A.; Coy, D.H.; Hocart, S.J.; Jiang, N.Y.; Mrozinski, J.E., Jr.; Mantey, S.A.; Jensen, R.T. A chimeric VIP-PACAP analogue but not VIP pseudopeptides function as VIP receptor antagonists. Peptides 1994, 15, 95–100. [Google Scholar] [CrossRef]
- Gourlet, P.; Vandermeers, A.; Vertongen, P.; Rathe, J.; De Neef, P.; Cnudde, J.; Waelbroeck, M.; Robberecht, P. Development of high affinity selective VIP1 receptor agonists. Peptides 1997, 18, 1539–1545. [Google Scholar] [CrossRef]
- Nicole, P.; Lins, L.; Rouyer-Fessard, C.; Drouot, C.; Fulcrand, P.; Thomas, A.; Couvineau, A.; Martinez, J.; Brasseur, R.; Laburthe, M. Identification of key residues for interaction of vasoactive intestinal peptide with human VPAC1 and VPAC2 receptors and development of a highly selective VPAC1 receptor agonist. Alanine scanning and molecular modeling of the peptide. J. Biol. Chem. 2000, 275, 24003–24012. [Google Scholar] [CrossRef]
- Gourlet, P.; Vandermeers-Piret, M.C.; Rathe, J.; De Neef, P.; Cnudde, J.; Robberecht, P.; Waelbroeck, M. Vasoactive intestinal peptide modification at position 22 allows discrimination between receptor subtypes. Eur. J. Pharmacol. 1998, 348, 95–99. [Google Scholar] [CrossRef]
- Van Rampelbergh, J.; Juarranz, M.G.; Perret, J.; Bondue, A.; Solano, R.M.; Delporte, C.; De Neef, P.; Robberecht, P.; Waelbroeck, M. Characterization of a novel VPAC(1) selective agonist and identification of the receptor domains implicated in the carboxyl-terminal peptide recognition. Br. J. Pharmacol. 2000, 130, 819–826. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olson, K.E.; Kosloski-Bilek, L.M.; Anderson, K.M.; Diggs, B.J.; Clark, B.E.; Gledhill, J.M., Jr.; Shandler, S.J.; Mosley, R.L.; Gendelman, H.E. Selective VIP Receptor Agonists Facilitate Immune Transformation for Dopaminergic Neuroprotection in MPTP-Intoxicated Mice. J. Neurosci. 2015, 35, 16463–16478. [Google Scholar] [CrossRef] [PubMed]
- Gourlet, P.; De Neef, P.; Cnudde, J.; Waelbroeck, M.; Robberecht, P. In vitro properties of a high affinity selective antagonist of the VIP1 receptor. Peptides 1997, 18, 1555–1560. [Google Scholar] [CrossRef]
- O’Donnell, M.; Garippa, R.J.; Rinaldi, N.; Selig, W.M.; Simko, B.; Renzetti, L.; Tannu, S.A.; Wasserman, M.A.; Welton, A.; Bolin, D.R. Ro 25-1553: A novel, long-acting vasoactive intestinal peptide agonist. Part I: In vitro and in vivo bronchodilator studies. J. Pharmacol. Exp. Ther. 1994, 270, 1282–1288. [Google Scholar]
- Xia, M.; Sreedharan, S.P.; Bolin, D.R.; Gaufo, G.O.; Goetzl, E.J. Novel cyclic peptide agonist of high potency and selectivity for the type II vasoactive intestinal peptide receptor. J. Pharmacol. Exp. Ther. 1997, 281, 629–633. [Google Scholar]
- Tsutsumi, M.; Claus, T.H.; Liang, Y.; Li, Y.; Yang, L.; Zhu, J.; Dela Cruz, F.; Peng, X.; Chen, H.; Yung, S.L.; et al. A potent and highly selective VPAC2 agonist enhances glucose-induced insulin release and glucose disposal: A potential therapy for type 2 diabetes. Diabetes 2002, 51, 1453–1460. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, M.; Dai, Y.; Hong, A. Expression, identification and biological effects of a novel VPAC2-specific agonist with high stability and bioactivity. Acta Biochim. Biophys. Sin. 2010, 42, 21–29. [Google Scholar] [CrossRef]
- Moreno, D.; Gourlet, P.; De Neef, P.; Cnudde, J.; Waelbroeck, M.; Robberecht, P. Development of selective agonists and antagonists for the human vasoactive intestinal polypeptide VPAC(2) receptor. Peptides 2000, 21, 1543–1549. [Google Scholar] [CrossRef]
- Sakamoto, K.; Koyama, R.; Kamada, Y.; Miwa, M.; Tani, A. Discovery of artificial VIPR2-antagonist peptides possessing receptor- and ligand-selectivity. Biochem. Biophys. Res. Commun. 2018, 503, 1973–1979. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Liu, H.; Li, H. In silico classification and prediction of VIP derivatives as VPAC1/ VPAC2 receptor agonists/antagonists. Comb. Chem. High Throughput Screen. 2015, 18, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.J.; Wang, D.H.; Li, Y.W.; Han, L.; Xiao, X.; Ma, M.; Wan, D.C.; Hong, A.; Ma, Y. A novel selective VPAC2 agonist peptide-conjugated chitosan modified selenium nanoparticles with enhanced anti-type 2 diabetes synergy effects. Int. J. Nanomed. 2017, 12, 2143–2160. [Google Scholar] [CrossRef] [PubMed]
- Peyrassol, X.; Laeremans, T.; Lahura, V.; Debulpaep, M.; El Hassan, H.; Steyaert, J.; Parmentier, M.; Langer, I. Development by Genetic Immunization of Monovalent Antibodies Against Human Vasoactive Intestinal Peptide Receptor 1 (VPAC1), New Innovative, and Versatile Tools to Study VPAC1 Receptor Function. Front. Endocrinol. 2018, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cañas, I.; Juarranz, M.G.; Collado, B.; Rodríguez-Henche, N.; Chiloeches, A.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide induces neuroendocrine differentiation in the LNCaP prostate cancer cell line through PKA, ERK, and PI3K. Prostate 2005, 63, 44–55. [Google Scholar] [CrossRef]
- Crespo, P.; Xu, N.; Simonds, W.F.; Gutkind, J.S. Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunits. Nature 1994, 369, 418–420. [Google Scholar] [CrossRef]
- Maier, U.; Babich, A.; Nurnberg, B. Roles of non-catalytic subunits in gbetagamma-induced activation of class I phosphoinositide 3-kinase isoforms beta and gamma. J. Biol. Chem. 1999, 274, 29311–29317. [Google Scholar] [CrossRef]
- Dickson, L.; Aramori, I.; McCulloch, J.; Sharkey, J.; Finlayson, K. A systematic comparison of intracellular cyclic AMP and calcium signalling highlights complexities in human VPAC/PAC receptor pharmacology. Neuropharmacology 2006, 51, 1086–1098. [Google Scholar] [CrossRef]
- MacKenzie, C.J.; Lutz, E.M.; Johnson, M.S.; Robertson, D.N.; Holland, P.J.; Mitchell, R. Mechanisms of phospholipase C activation by the vasoactive intestinal polypeptide/pituitary adenylate cyclase-activating polypeptide type 2 receptor. Endocrinology 2001, 142, 1209–1217. [Google Scholar] [CrossRef]
- McCulloch, D.A.; Lutz, E.M.; Johnson, M.S.; MacKenzie, C.J.; Mitchell, R. Differential activation of phospholipase D by VPAC and PAC1 receptors. Ann. N. Y. Acad. Sci. 2000, 921, 175–185. [Google Scholar] [CrossRef]
- Christopoulos, A.; Christopoulos, G.; Morfis, M.; Udawela, M.; Laburthe, M.; Couvineau, A.; Kuwasako, K.; Tilakaratne, N.; Sexton, P.M. Novel receptor partners and function of receptor activity-modifying proteins. J. Biol. Chem. 2003, 278, 3293–3297. [Google Scholar] [CrossRef]
- Wootten, D.; Lindmark, H.; Kadmiel, M.; Willcockson, H.; Caron, K.M.; Barwell, J.; Drmota, T.; Poyner, D.R. Receptor activity modifying proteins (RAMPs) interact with the VPAC2 receptor and CRF1 receptors and modulate their function. Br. J. Pharmacol. 2013, 168, 822–834. [Google Scholar] [CrossRef]
- Carrión, M.; Juarranz, Y.; Pérez-García, S.; Jimeno, R.; Pablos, J.L.; Gomariz, R.P.; Gutiérrez-Cañas, I. RNA sensors in human osteoarthritis and rheumatoid arthritis synovial fibroblasts: Immune regulation by vasoactive intestinal peptide. Arthritis Rheum. 2011, 63, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.; Gutiérrez-Cañas, I.; Carrión, M.; Juarranz, Y.; Pablos, J.L.; Martínez, C.; Gomariz, R.P. VIP reverses the expression profiling of TLR4-stimulated signaling pathway in rheumatoid arthritis synovial fibroblasts. Mol. Immunol. 2008, 45, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Juarranz, Y.; Abad, C.; Martínez, C.; Arranz, A.; Gutiérrez-Cañas, I.; Rosignoli, F.; Gomariz, R.P.; Leceta, J. Protective effect of vasoactive intestinal peptide on bone destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1034–R1045. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Munoz-Elias, E.J.; Kan, Y.; Gozes, I.; Fridkin, M.; Brenneman, D.E.; Gomariz, R.P.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun. J. Biol. Chem. 1998, 273, 31427–31436. [Google Scholar] [CrossRef]
- Shetzline, M.A.; Walker, J.K.; Valenzano, K.J.; Premont, R.T. Vasoactive intestinal polypeptide type-1 receptor regulation. Desensitization, phosphorylation, and sequestration. J. Biol. Chem. 2002, 277, 25519–25526. [Google Scholar] [CrossRef]
- Jong, Y.I.; Harmon, S.K.; O’Malley, K.L. GPCR signalling from within the cell. Br. J. Pharmacol. 2018, 175, 4026–4035. [Google Scholar] [CrossRef]
- Valdehita, A.; Bajo, A.M.; Fernandez-Martinez, A.B.; Arenas, M.I.; Vacas, E.; Valenzuela, P.; Ruiz-Villaespesa, A.; Prieto, J.C.; Carmena, M.J. Nuclear localization of vasoactive intestinal peptide (VIP) receptors in human breast cancer. Peptides 2010, 31, 2035–2045. [Google Scholar] [CrossRef]
- Vacas, E.; Arenas, M.I.; Munoz-Moreno, L.; Bajo, A.M.; Sanchez-Chapado, M.; Prieto, J.C.; Carmena, M.J. Antitumoral effects of vasoactive intestinal peptide in human renal cell carcinoma xenografts in athymic nude mice. Cancer Lett. 2013, 336, 196–203. [Google Scholar] [CrossRef]
- Barbarin, A.; Seite, P.; Godet, J.; Bensalma, S.; Muller, J.M.; Chadeneau, C. Atypical nuclear localization of VIP receptors in glioma cell lines and patients. Biochem. Biophys. Res. Commun. 2014, 454, 524–530. [Google Scholar] [CrossRef]
- Villanueva-Romero, R.; Gutierrez-Canas, I.; Carrion, M.; Gonzalez-Alvaro, I.; Rodriguez-Frade, J.M.; Mellado, M.; Martinez, C.; Gomariz, R.P.; Juarranz, Y. Activation of Th lymphocytes alters pattern expression and cellular location of VIP receptors in healthy donors and early arthritis patients. Sci. Rep. 2019, 9, 7383. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Liu, H.; Peng, X.; Cui, Y.; Song, S.; Wang, L.; Zhang, H.; Hong, A.; Zhou, T. The palmitoylation of the N-terminal extracellular Cys37 mediates the nuclear translocation of VPAC1 contributing to its anti-apoptotic activity. Oncotarget 2017, 8, 42728. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, P.; Ruscitti, P.; Vadasz, Z.; Toubi, E.; Giacomelli, R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun. Rev. 2019, 18, 102369. [Google Scholar] [CrossRef] [PubMed]
- Van Amersfoort, E.S.; Van Berkel, T.J.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U. Clinical review: A paradigm shift: The bidirectional effect of inflammation on bacterial growth. Clinical implications for patients with acute respiratory distress syndrome. Crit. Care 2002, 6, 24–29. [Google Scholar] [CrossRef]
- Navegantes, K.C.; de Souza Gomes, R.; Pereira, P.A.T.; Czaikoski, P.G.; Azevedo, C.H.M.; Monteiro, M.C. Immune modulation of some autoimmune diseases: The critical role of macrophages and neutrophils in the innate and adaptive immunity. J. Transl. Med. 2017, 15, 36. [Google Scholar] [CrossRef]
- Leceta, J.; Gomariz, R.P.; Martinez, C.; Abad, C.; Ganea, D.; Delgado, M. Receptors and transcriptional factors involved in the anti-inflammatory activity of VIP and PACAP. Ann. N. Y. Acad. Sci. 2000, 921, 92–102. [Google Scholar] [CrossRef]
- Juarranz, M.G.; Santiago, B.; Torroba, M.; Gutiérrez-Cañas, I.; Palao, G.; Galindo, M.; Abad, C.; Martínez, C.; Leceta, J.; Pablos, J.L.; et al. Vasoactive intestinal peptide modulates proinflammatory mediator synthesis in osteoarthritic and rheumatoid synovial cells. Rheumatology 2004, 43, 416–422. [Google Scholar] [CrossRef]
- Tuncel, N.; Tore, F.; Sahinturk, V.; Ak, D.; Tuncel, M. Vasoactive intestinal peptide inhibits degranulation and changes granular content of mast cells: A potential therapeutic strategy in controlling septic shock. Peptides 2000, 21, 81–89. [Google Scholar] [CrossRef]
- Delgado, M.; Ganea, D. Anti-inflammatory neuropeptides: A new class of endogenous immunoregulatory agents. Brain Behav. Immun. 2008, 22, 1146–1151. [Google Scholar] [CrossRef]
- Delgado, M.; Jonakait, G.M.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit chemokine production in activated microglia. Glia 2002, 39, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Szema, A.M.; Hamidi, S.A.; Lyubsky, S.; Dickman, K.G.; Mathew, S.; Abdel-Razek, T.; Chen, J.J.; Waschek, J.A.; Said, S.I. Mice lacking the VIP gene show airway hyperresponsiveness and airway inflammation, partially reversible by VIP. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L880–L886. [Google Scholar] [CrossRef] [PubMed]
- Said, S.I.; Hamidi, S.A.; Dickman, K.G.; Szema, A.M.; Lyubsky, S.; Lin, R.Z.; Jiang, Y.P.; Chen, J.J.; Waschek, J.A.; Kort, S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 2007, 115, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.; Tan, Y.V.; Cheung-Lau, G.; Nobuta, H.; Waschek, J.A. VIP deficient mice exhibit resistance to lipopolysaccharide induced endotoxemia with an intrinsic defect in proinflammatory cellular responses. PLoS ONE 2012, 7, e36922. [Google Scholar] [CrossRef]
- Hamidi, S.A.; Szema, A.M.; Lyubsky, S.; Dickman, K.G.; Degene, A.; Mathew, S.M.; Waschek, J.A.; Said, S.I. Clues to VIP function from knockout mice. Ann. N. Y. Acad. Sci. 2006, 1070, 5–9. [Google Scholar] [CrossRef]
- Said, S.I. Vasoactive intestinal peptide and nitric oxide: Divergent roles in relation to tissue injury. Ann. N. Y. Acad. Sci. 1996, 805, 379–387; discussion 387-8. [Google Scholar] [CrossRef]
- Said, S.I. Molecules that protect: The defense of neurons and other cells. J. Clin. Investig. 1996, 97, 2163–2164. [Google Scholar] [CrossRef]
- Waschek, J.A. VIP and PACAP: Neuropeptide modulators of CNS inflammation, injury, and repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef]
- Delgado, M.; Martinez, C.; Pozo, D.; Calvo, J.R.; Leceta, J.; Ganea, D.; Gomariz, R.P. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activation polypeptide (PACAP) protect mice from lethal endotoxemia through the inhibition of TNF-alpha and IL-6. J. Immunol. 1999, 162, 1200–1205. [Google Scholar]
- Said, S.I.; Dickman, K.; Dey, R.D.; Bandyopadhyay, A.; De Stefanis, P.; Raza, S.; Pakbaz, H.; Berisha, H.I. Glutamate toxicity in the lung and neuronal cells: Prevention or attenuation by VIP and PACAP. Ann. N. Y. Acad. Sci. 1998, 865, 226–237. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2015, 109, 14.12.1–14.12.10. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Ospelt, C.; Gay, S. TLRs and chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 495–505. [Google Scholar] [CrossRef]
- Lebre, M.C.; van der Aar, A.M.; van Baarsen, L.; van Capel, T.M.; Schuitemaker, J.H.; Kapsenberg, M.L.; de Jong, E.C. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef]
- Platz, J.; Beisswenger, C.; Dalpke, A.; Koczulla, R.; Pinkenburg, O.; Vogelmeier, C.; Bals, R. Microbial DNA induces a host defense reaction of human respiratory epithelial cells. J. Immunol. 2004, 173, 1219–1223. [Google Scholar] [CrossRef]
- Gewirtz, A.T. Intestinal epithelial toll-like receptors: To protect. And serve? Curr. Pharm. Des. 2003, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef]
- Brasier, A.R. The NF-κB Signaling Network: Insights from Systems Approaches; American Society for Microbiology: Washington, DC, USA, 2008; pp. 119–135. [Google Scholar]
- Goh, F.G.; Midwood, K.S. Intrinsic danger: Activation of Toll-like receptors in rheumatoid arthritis. Rheumatology 2012, 51, 7–23. [Google Scholar] [CrossRef]
- Fischer, M.; Ehlers, M. Toll-like receptors in autoimmunity. Ann. N. Y. Acad. Sci. 2008, 1143, 21–34. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T.; Akira, S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol. Rev. 2011, 243, 61–73. [Google Scholar] [CrossRef]
- Farrugia, M.; Baron, B. The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int. J. Inflam. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Gomariz, R.P.; Arranz, A.; Juarranz, Y.; Gutiérrez-Cañas, I.; García-Gómez, M.; Leceta, J.; Martínez, C. Regulation of TLR expression, a new perspective for the role of VIP in immunity. Peptides 2007, 28, 1825–1832. [Google Scholar] [CrossRef]
- Gomariz, R.P.; Gutierrez-Canas, I.; Arranz, A.; Carrion, M.; Juarranz, Y.; Leceta, J.; Martinez, C. Peptides targeting Toll-like receptor signalling pathways for novel immune therapeutics. Curr. Pharm. Des. 2010, 16, 1063–1080. [Google Scholar] [CrossRef]
- Smalley, S.G.; Barrow, P.A.; Foster, N. Immunomodulation of innate immune responses by vasoactive intestinal peptide (VIP): Its therapeutic potential in inflammatory disease. Clin. Exp. Immunol. 2009, 157, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.; Barrow, P.; Foster, N. Transcriptional modulation by VIP: A rational target against inflammatory disease. Clin. Epigenetics 2011, 2, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Gomariz, R.P.; Arranz, A.; Abad, C.; Torroba, M.; Martínez, C.; Rosignoli, F.; García-Gómez, M.; Leceta, J.; Juarranz, Y. Time-course expression of Toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP. J. Leukoc. Biol. 2005, 78, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.; Abad, C.; Juarranz, Y.; Torroba, M.; Rosignoli, F.; Leceta, J.; Gomariz, R.P.; Martínez, C. Effect of VIP on TLR2 and TLR4 expression in lymph node immune cells during TNBS-induced colitis. Ann. N. Y. Acad. Sci. 2006, 1070, 129–134. [Google Scholar] [CrossRef]
- Musikacharoen, T.; Matsuguchi, T.; Kikuchi, T.; Yoshikai, Y. NF-kappa B and STAT5 play important roles in the regulation of mouse Toll-like receptor 2 gene expression. J. Immunol. 2001, 166, 4516–4524. [Google Scholar] [CrossRef]
- Arranz, A.; Androulidaki, A.; Zacharioudaki, V.; Martínez, C.; Margioris, A.N.; Gomariz, R.P.; Tsatsanis, C. Vasoactive intestinal peptide suppresses toll-like receptor 4 expression in macrophages via Akt1 reducing their responsiveness to lipopolysaccharide. Mol. Immunol. 2008, 45, 2970–2980. [Google Scholar] [CrossRef]
- Foster, N.; Lea, S.R.; Preshaw, P.M.; Taylor, J.J. Pivotal advance: Vasoactive intestinal peptide inhibits up-regulation of human monocyte TLR2 and TLR4 by LPS and differentiation of monocytes to macrophages. J. Leukoc. Biol. 2007, 81, 893–903. [Google Scholar] [CrossRef]
- Haehnel, V.; Schwarzfischer, L.; Fenton, M.J.; Rehli, M. Transcriptional regulation of the human toll-like receptor 2 gene in monocytes and macrophages. J. Immunol. 2002, 168, 5629–5637. [Google Scholar] [CrossRef]
- Rehli, M.; Poltorak, A.; Schwarzfischer, L.; Krause, S.W.; Andreesen, R.; Beutler, B. PU. 1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human Toll-like receptor 4 gene. J. Biol. Chem. 2000, 275, 9773–9781. [Google Scholar] [CrossRef]
- Jiang, X.; McClellan, S.A.; Barrett, R.P.; Zhang, Y.; Hazlett, L.D. Vasoactive intestinal peptide downregulates proinflammatory TLRs while upregulating anti-inflammatory TLRs in the infected cornea. J. Immunol. 2012, 189, 269–278. [Google Scholar] [CrossRef]
- Gutiérrez-Cañas, I.; Juarranz, Y.; Santiago, B.; Arranz, A.; Martínez, C.; Galindo, M.; Paya, M.; Gomariz, R.P.; Pablos, J.L. VIP down-regulates TLR4 expression and TLR4-mediated chemokine production in human rheumatoid synovial fibroblasts. Rheumatology 2006, 45, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Neidhart, M.; Gay, R.E.; Gay, S. Synovial activation in rheumatoid arthritis. Front. Biosci. 2004, 9, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Juarranz, Y.; Gutiérrez-Cañas, I.; Arranz, A.; Martínez, C.; Abad, C.; Leceta, J.; Pablos, J.L.; Gomariz, R.P. VIP decreases TLR4 expression induced by LPS and TNF-alpha treatment in human synovial fibroblasts. Ann. N. Y. Acad. Sci. 2006, 1070, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb. Perspect. Biol. 2018, 10, a030338. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Fahmy, T.M.; Piccirillo, C.A.; La Cava, A. Rebalancing Immune Homeostasis to Treat Autoimmune Diseases. Trends Immunol. 2019, 40, 888–908. [Google Scholar] [CrossRef]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef]
- Stadhouders, R.; Lubberts, E.; Hendriks, R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15. [Google Scholar] [CrossRef]
- Van Hamburg, J.P.; Tas, S.W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 2018, 87, 69–81. [Google Scholar] [CrossRef]
- Sallusto, F. Heterogeneity of Human CD4+ T Cells Against Microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Bending, D.; De la Pena, H.; Veldhoen, M.; Phillips, J.M.; Uyttenhove, C.; Stockinger, B.; Cooke, A. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J. Clin. Investig. 2009, 119, 565–572. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Hirahara, K.; O’Shea, J.J. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011, 32, 395–401. [Google Scholar] [CrossRef]
- Jimeno, R.; Gomariz, R.P.; Garin, M.; Gutierrez-Canas, I.; Gonzalez-Alvaro, I.; Carrion, M.; Galindo, M.; Leceta, J.; Juarranz, Y. The pathogenic Th profile of human activated memory Th cells in early rheumatoid arthritis can be modulated by VIP. J. Mol. Med. 2015, 93, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Yago, T.; Kobashigawa, T.; Nanke, Y. The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M. VIP: A very important peptide in T helper differentiation. Trends Immunol. 2003, 24, 221–224. [Google Scholar] [CrossRef]
- Arranz, A.; Abad, C.; Juarranz, Y.; Leceta, J.; Martinez, C.; Gomariz, R.P. Vasoactive intestinal peptide as a healing mediator in Crohn’s disease. Neuroimmunomodulation 2008, 15, 46–53. [Google Scholar] [CrossRef]
- Jimeno, R.; Leceta, J.; Martinez, C.; Gutierrez-Canas, I.; Perez-Garcia, S.; Carrion, M.; Gomariz, R.P.; Juarranz, Y. Effect of VIP on the balance between cytokines and master regulators of activated helper T cells. Immunol. Cell Biol. 2012, 90, 178–186. [Google Scholar] [CrossRef]
- Villanueva-Romero, R.; Gutierrez-Canas, I.; Carrion, M.; Perez-Garcia, S.; Seoane, I.V.; Martinez, C.; Gomariz, R.P.; Juarranz, Y. The Anti-Inflammatory Mediator, Vasoactive Intestinal Peptide, Modulates the Differentiation and Function of Th Subsets in Rheumatoid Arthritis. J. Immunol. Res. 2018, 2018, 6043710. [Google Scholar] [CrossRef]
- Leceta, J.; Gomariz, R.P.; Martinez, C.; Carrion, M.; Arranz, A.; Juarranz, Y. Vasoactive intestinal peptide regulates Th17 function in autoimmune inflammation. Neuroimmunomodulation 2007, 14, 134–138. [Google Scholar] [CrossRef]
- Rosignoli, F.; Torroba, M.; Juarranz, Y.; Garcia-Gomez, M.; Martinez, C.; Gomariz, R.P.; Perez-Leiros, C.; Leceta, J. VIP and tolerance induction in autoimmunity. Ann. N. Y. Acad. Sci. 2006, 1070, 525–530. [Google Scholar] [CrossRef]
- Roca, V.; Calafat, M.; Larocca, L.; Ramhorst, R.; Farina, M.; Franchi, A.M.; Leiros, C.P. Potential immunomodulatory role of VIP in the implantation sites of prediabetic nonobese diabetic mice. Reproduction 2009, 138, 733–742. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Fernandez-Martin, A.; Chorny, A.; Martin, J.; Pozo, D.; Ganea, D.; Delgado, M. Therapeutic effect of vasoactive intestinal peptide on experimental autoimmune encephalomyelitis: Down-regulation of inflammatory and autoimmune responses. Am. J. Pathol. 2006, 168, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.; Waschek, J.A. Immunomodulatory roles of VIP and PACAP in models of multiple sclerosis. Curr. Pharm. Des. 2011, 17, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Benitez, R.; Delgado-Maroto, V.; Caro, M.; Forte-Lago, I.; Duran-Prado, M.; O’Valle, F.; Lichtman, A.H.; Gonzalez-Rey, E.; Delgado, M. Vasoactive Intestinal Peptide Ameliorates Acute Myocarditis and Atherosclerosis by Regulating Inflammatory and Autoimmune Responses. J. Immunol. 2018, 200, 3697–3710. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Senouthai, S.; Wang, J.; You, Y. Vasoactive intestinal peptide ameliorates renal injury in a pristane-induced lupus mouse model by modulating Th17/Treg balance. BMC Nephrol. 2019, 20, 350. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Leceta, J.; Gomariz, R.P.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide stimulate the induction of Th2 responses by up-regulating B7.2 expression. J. Immunol. 1999, 163, 3629–3635. [Google Scholar]
- Gutiérrez-Cañas, I.; Juarranz, Y.; Santiago, B.; Martínez, C.; Gomariz, R.P.; Pablos, J.L.; Leceta, J. Immunoregulatory properties of vasoactive intestinal peptide in human T cell subsets: Implications for rheumatoid arthritis. Brain Behav. Immun. 2008, 22, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Xi, Y.; Wang, H.; Hao, J.; Niu, X.; Li, W.; Tao, Y.; Chen, G. Regulatory effect of vasoactive intestinal peptide on the balance of Treg and Th17 in collagen-induced arthritis. Cell Immunol. 2010, 265, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, R.; Leceta, J.; Martinez, C.; Gutierrez-Canas, I.; Carrion, M.; Perez-Garcia, S.; Garin, M.; Mellado, M.; Gomariz, R.P.; Juarranz, Y. Vasoactive intestinal peptide maintains the nonpathogenic profile of human th17-polarized cells. J. Mol. Neurosci. 2014, 54, 512–525. [Google Scholar] [CrossRef]
- Jimeno, R.; Leceta, J.; Garin, M.; Ortiz, A.M.; Mellado, M.; Rodriguez-Frade, J.M.; Martínez, C.; Pérez-García, S.; Gomariz, R.P.; Juarranz, Y. Th17 polarization of memory Th cells in early arthritis: The vasoactive intestinal peptide effect. J. Leukoc. Biol. 2015, 98, 257–269. [Google Scholar] [CrossRef]
- Hubo, M.; Trinschek, B.; Kryczanowsky, F.; Tuettenberg, A.; Steinbrink, K.; Jonuleit, H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front. Immunol. 2013, 4, 82. [Google Scholar] [CrossRef]
- Ritprajak, P.; Kaewraemruaen, C.; Hirankarn, N. Current Paradigms of Tolerogenic Dendritic Cells and Clinical Implications for Systemic Lupus Erythematosus. Cells 2019, 8, 1291. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Danese, S.; Leone, G. Tolerogenic dendritic cells: Cytokine modulation comes of age. Blood 2006, 108, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Berkun, Y.; Verbovetski, I.; Ben-Ami, A.; Paran, D.; Caspi, D.; Krispin, A.; Trahtemberg, U.; Gill, O.; Naparstek, Y.; Mevorach, D. Altered dendritic cells with tolerizing phenotype in patients with systemic lupus erythematosus. Eur. J. Immunol. 2008, 38, 2896–2904. [Google Scholar] [CrossRef] [PubMed]
- Hackstein, H.; Thomson, A.W. Dendritic cells: Emerging pharmacological targets of immunosuppressive drugs. Nat. Rev. Immunol. 2004, 4, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Svajger, U.; Rozman, P. Induction of Tolerogenic Dendritic Cells by Endogenous Biomolecules: An Update. Front. Immunol. 2018, 9, 2482. [Google Scholar] [CrossRef] [PubMed]
- Chorny, A.; Gonzalez-Rey, E.; Fernandez-Martin, A.; Pozo, D.; Ganea, D.; Delgado, M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 13562–13567. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Chorny, A.; Fernandez-Martin, A.; Ganea, D.; Delgado, M. Vasoactive intestinal peptide generates human tolerogenic dendritic cells that induce CD4 and CD8 regulatory T cells. Blood 2006, 107, 3632–3638. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.G.; Delgado, M.; Kong, W.; Martin, F.; Skarica, M.; Ganea, D. Dendritic cells transduced with lentiviral vectors expressing VIP differentiate into VIP-secreting tolerogenic-like DCs. Mol. Ther. 2010, 18, 1035–1045. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Delgado, M. Therapeutic treatment of experimental colitis with regulatory dendritic cells generated with vasoactive intestinal peptide. Gastroenterology 2006, 131, 1799–1811. [Google Scholar] [CrossRef]
- Wu, H.; Shen, J.; Liu, L.; Lu, X.; Xue, J. Vasoactive intestinal peptide-induced tolerogenic dendritic cells attenuated arthritis in experimental collagen-induced arthritic mice. Int. J. Rheum. Dis. 2019, 22, 1255–1262. [Google Scholar] [CrossRef]
- Yalvac, M.E.; Arnold, W.D.; Hussain, S.A.; Braganza, C.; Shontz, K.M.; Clark, K.R.; Walker, C.M.; Ubogu, E.E.; Mendell, J.R.; Sahenk, Z. VIP-expressing dendritic cells protect against spontaneous autoimmune peripheral polyneuropathy. Mol. Ther. 2014, 22, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, M.H.; Yan, J.; Chen, Y.P.; Pan, J.P. Effects of vasoactive intestinal peptide on phenotypic and functional maturation of dendritic cells. Int. Immunopharmacol. 2008, 8, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Sun, J.; Wu, Q.; Pan, J. Regulatory effects of vasoactive intestinal peptide on the migration of mature dendritic cells. J. Neuroimmunol. 2007, 182, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Reduta, A.; Sharma, V.; Ganea, D. VIP/PACAP oppositely affects immature and mature dendritic cell expression of CD80/CD86 and the stimulatory activity for CD4+ T cells. J. Leukoc. Biol. 2004, 75, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 9–17. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Orr, C.; Vieira-Sousa, E.; Boyle, D.L.; Buch, M.H.; Buckley, C.D.; Canete, J.D.; Catrina, A.I.; Choy, E.H.S.; Emery, P.; Fearon, U.; et al. Synovial tissue research: A state-of-the-art review. Nat. Rev. Rheumatol. 2017, 13, 630. [Google Scholar] [CrossRef]
- Dakin, S.G.; Coles, M.; Sherlock, J.P.; Powrie, F.; Carr, A.J.; Buckley, C.D. Pathogenic stromal cells as therapeutic targets in joint inflammation. Nat. Rev. Rheumatol. 2018, 14, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Lefevre, S.; Zimmermann, B.; Gay, S.; Muller-Ladner, U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol. Med. 2010, 16, 458–468. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, H.M.; Butler, L.M.; Buckley, C.D.; Rainger, G.E.; Nash, G.B. Tissue stroma as a regulator of leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012, 91, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef]
- Mulherin, D.; Fitzgerald, O.; Bresnihan, B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 115–124. [Google Scholar] [CrossRef]
- Firestein, G.S. VIP: A very important protein in arthritis. Nat. Med. 2001, 7, 537–538. [Google Scholar] [CrossRef]
- El Karim, I.A.; Linden, G.J.; Orr, D.F.; Lundy, F.T. Antimicrobial activity of neuropeptides against a range of micro-organisms from skin, oral, respiratory and gastrointestinal tract sites. J. Neuroimmunol. 2008, 200, 11–16. [Google Scholar] [CrossRef]
- Campos-Salinas, J.; Cavazzuti, A.; O’Valle, F.; Forte-Lago, I.; Caro, M.; Beverley, S.M.; Delgado, M.; Gonzalez-Rey, E. Therapeutic efficacy of stable analogues of vasoactive intestinal peptide against pathogens. J. Biol. Chem. 2014, 289, 14583–14599. [Google Scholar] [CrossRef]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Hao, J.; Xi, Y.; Wang, W.; Wang, Z.; Li, N.; Li, W. The therapeutic effect of vasoactive intestinal peptide on experimental arthritis is associated with CD4+CD25+ T regulatory cells. Scand. J. Immunol. 2008, 68, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Muschter, D.; Schafer, N.; Stangl, H.; Straub, R.H.; Grassel, S. Sympathetic Neurotransmitters Modulate Osteoclastogenesis and Osteoclast Activity in the Context of Collagen-Induced Arthritis. PLoS ONE 2015, 10, e0139726. [Google Scholar] [CrossRef]
- Carrión, M.; Juarranz, Y.; Seoane, I.V.; Martínez, C.; González-Álvaro, I.; Pablos, J.L.; Gutiérrez-Cañas, I.; Gomariz, R.P. VIP modulates IL-22R1 expression and prevents the contribution of rheumatoid synovial fibroblasts to IL-22-mediated joint destruction. J. Mol. Neurosci. 2014, 52, 10–17. [Google Scholar] [CrossRef]
- Carrión, M.; Pérez-García, S.; Jimeno, R.; Juarranz, Y.; González-Álvaro, I.; Pablos, J.L.; Gutiérrez-Cañas, I.; Gomariz, R.P. Inflammatory mediators alter interleukin-17 receptor, interleukin-12 and -23 expression in human osteoarthritic and rheumatoid arthritis synovial fibroblasts: Immunomodulation by vasoactive intestinal Peptide. Neuroimmunomodulation 2013, 20, 274–284. [Google Scholar] [CrossRef]
- Pérez-García, S.; Juarranz, Y.; Carrión, M.; Gutiérrez-Cañas, I.; Margioris, A.; Pablos, J.L.; Tsatsanis, C.; Gomariz, R.P. Mapping the CRF-urocortins system in human osteoarthritic and rheumatoid synovial fibroblasts: Effect of vasoactive intestinal peptide. J. Cell. Physiol. 2011, 226, 3261–3269. [Google Scholar] [CrossRef]
- Monfort, J. Artrosis: Fisiopatología, Diagnóstico y Tratamiento; Médica Panamericana: Madrid, Spain, 2010. [Google Scholar]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- Mimpen, J.Y.; Snelling, S.J.B. Chondroprotective Factors in Osteoarthritis: A Joint Affair. Curr. Rheumatol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef]
- Brooks, P. Inflammation as an important feature of osteoarthritis. Bull. World Health Organ. 2003, 81, 689–690. [Google Scholar]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell. Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, J.W.; Berenbaum, F.; Lafeber, F.P. Osteoarthritis: An update with relevance for clinical practice. Lancet 2011, 377, 2115–2126. [Google Scholar] [CrossRef]
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Karsdal, M.A.; Lohmander, L.S. Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthr. Cartil. 2015, 23, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; FitzGerald, U.; Murphy, J.M. Interplay of Inflammatory Mediators with Epigenetics and Cartilage Modifications in Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Benito, M.J.; Veale, D.J.; FitzGerald, O.; van den Berg, W.B.; Bresnihan, B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis. 2005, 64, 1263–1267. [Google Scholar] [CrossRef]
- Batlle-Gualda, E.; Benito-Ruiz, P.; Blanco, F.J.; Martín, E. Manual SER de la Artrosis; IM&C: Madrid, Spain, 2002. [Google Scholar]
- Pérez-García, S.; Carrión, M.; Jimeno, R.; Ortiz, A.M.; González-Álvaro, I.; Fernández, J.; Gomariz, R.P.; Juarranz, Y. Urokinase plasminogen activator system in synovial fibroblasts from osteoarthritis patients: Modulation by inflammatory mediators and neuropeptides. J. Mol. Neurosci. 2014, 52, 18–27. [Google Scholar] [CrossRef]
- Pérez-García, S.; Gutiérrez-Cañas, I.; Seoane, I.V.; Fernández, J.; Mellado, M.; Leceta, J.; Tío, L.; Villanueva-Romero, R.; Juarranz, Y.; Gomariz, R.P. Healthy and Osteoarthritic Synovial Fibroblasts Produce a Disintegrin and Metalloproteinase with Thrombospondin Motifs 4, 5, 7, and 12: Induction by IL-1beta and Fibronectin and Contribution to Cartilage Damage. Am. J. Pathol. 2016, 186, 2449–2461. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Pelletier, J.P. Is osteoarthritis a disease involving only cartilage or other articular tissues? Eklem Hastalik Cerrahisi 2010, 21, 2–14. [Google Scholar]
- Sharma, L. Osteoarthritis year in review 2015: Clinical. Osteoarthr. Cartil. 2016, 24, 36–48. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; McDougall, J.J.; Keefe, F.J. The symptoms of osteoarthritis and the genesis of pain. Rheum. Dis. Clin. N. Am. 2008, 34, 623–643. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.B.; Attur, M. Developments in the scientific understanding of osteoarthritis. Arthritis Res. Ther. 2009, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Wildi, L.M.; Pelletier, J.P. Future therapeutics for osteoarthritis. Bone 2012, 51, 297–311. [Google Scholar] [CrossRef]
- Ghouri, A.; Conaghan, P.G. Update on novel pharmacological therapies for osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720X19864492. [Google Scholar] [CrossRef]
- Sutton, S.; Clutterbuck, A.; Harris, P.; Gent, T.; Freeman, S.; Foster, N.; Barrett-Jolley, R.; Mobasheri, A. The contribution of the synovium, synovial derived inflammatory cytokines and neuropeptides to the pathogenesis of osteoarthritis. Vet. J. 2009, 179, 10–24. [Google Scholar] [CrossRef]
- Carrión, M.; Juarranz, Y.; Martínez, C.; González-Álvaro, I.; Pablos, J.L.; Gutiérrez-Cañas, I.; Gomariz, R.P. IL-22/IL-22R1 axis and S100A8/A9 alarmins in human osteoarthritic and rheumatoid arthritis synovial fibroblasts. Rheumatology 2013, 52, 2177–2186. [Google Scholar] [CrossRef]
- Jiang, W.; Gao, S.G.; Chen, X.G.; Xu, X.C.; Xu, M.; Luo, W.; Tu, M.; Zhang, F.J.; Zeng, C.; Lei, G.H. Expression of synovial fluid and articular cartilage VIP in human osteoarthritic knee: A new indicator of disease severity? Clin. Biochem. 2012, 45, 1607–1612. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, H.; Li, Y.S.; Luo, W. Role of vasoactive intestinal peptide in osteoarthritis. J. Biomed. Sci. 2016, 23, 63. [Google Scholar] [CrossRef]
- Liang, Y.; Chen, S.; Yang, Y.; Lan, C.; Zhang, G.; Ji, Z.; Lin, H. Vasoactive intestinal peptide alleviates osteoarthritis effectively via inhibiting NF-kappaB signaling pathway. J. Biomed. Sci. 2018, 25, 25. [Google Scholar] [CrossRef]
- Delgado, M.; Munoz-Elias, E.J.; Gomariz, R.P.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: In vitro and in vivo studies. J. Immunol. 1999, 162, 1707–1716. [Google Scholar] [PubMed]
- Kambayashi, T.; Jacob, C.O.; Zhou, D.; Mazurek, N.; Fong, M.; Strassmann, G. Cyclic nucleotide phosphodiesterase type IV participates in the regulation of IL-10 and in the subsequent inhibition of TNF-alpha and IL-6 release by endotoxin-stimulated macrophages. J. Immunol. 1995, 155, 4909–4916. [Google Scholar] [PubMed]
- Strassmann, G.; Patil-Koota, V.; Finkelman, F.; Fong, M.; Kambayashi, T. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J. Exp. Med. 1994, 180, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, A.; Medina, S.; de Miguel, E.; Martin-Mola, E. Effect of calcitonin gene-related peptide, neuropeptide Y, substance P, and vasoactive intestinal peptide on interleukin-1beta, interleukin-6 and tumor necrosis factor-alpha production by peripheral whole blood cells from rheumatoid arthritis and osteoarthritis patients. Regul. Pept. 2003, 115, 19–24. [Google Scholar] [PubMed]
- Schuelert, N.; McDougall, J.J. Electrophysiological evidence that the vasoactive intestinal peptide receptor antagonist VIP6-28 reduces nociception in an animal model of osteoarthritis. Osteoarthr. Cartil. 2006, 14, 1155–1162. [Google Scholar] [CrossRef]
- Rahman, S.; Dobson, P.R.; Bunning, R.A.; Russell, R.G.; Brown, B.L. The regulation of connective tissue metabolism by vasoactive intestinal polypeptide. Regul. Pept. 1992, 37, 111–121. [Google Scholar] [CrossRef]
- Tío, L.; Orellana, C.; Pérez-García, S.; Piqueras, L.; Escudero, P.; Juarranz, Y.; García-Giralt, N.; Montañés, F.; Farran, A.; Benito, P.; et al. Effect of chondroitin sulphate on synovitis of knee osteoarthritic patients. Med. Clin. 2017, 149, 9–16. [Google Scholar] [CrossRef]
- Pérez-García, S.; Carrión, M.; Gutiérrez-Cañas, I.; González-Álvaro, I.; Gomariz, R.P.; Juarranz, Y. VIP and CRF reduce ADAMTS expression and function in osteoarthritis synovial fibroblasts. J. Cell. Mol. Med. 2016, 20, 678–687. [Google Scholar] [CrossRef]
- Juhász, T.; Helgadottir, S.L.; Tamás, A.; Reglődi, D.; Zákány, R. PACAP and VIP signaling in chondrogenesis and osteogenesis. Peptides 2015, 66, 51–57. [Google Scholar] [CrossRef]
- Juhasz, T.; Matta, C.; Katona, E.; Somogyi, C.; Takacs, R.; Gergely, P.; Csernoch, L.; Panyi, G.; Toth, G.; Reglodi, D.; et al. Pituitary adenylate cyclase activating polypeptide (PACAP) signalling exerts chondrogenesis promoting and protecting effects: Implication of calcineurin as a downstream target. PLoS ONE 2014, 9, e91541. [Google Scholar] [CrossRef]
- Lerner, U.H.; Persson, E. Osteotropic effects by the neuropeptides calcitonin gene-related peptide, substance P and vasoactive intestinal peptide. J. Musculoskelet Neuronal Interact 2008, 8, 154–165. [Google Scholar] [PubMed]
- Grassel, S.; Muschter, D. Do Neuroendocrine Peptides and Their Receptors Qualify as Novel Therapeutic Targets in Osteoarthritis? Int. J. Mol. Sci. 2018, 19, 367. [Google Scholar] [CrossRef]
- Persson, E.; Lerner, U.H. The neuropeptide VIP regulates the expression of osteoclastogenic factors in osteoblasts. J. Cell. Biochem. 2011, 112, 3732–3741. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Bernard, G.W. Neurogenic substance P stimulates osteogenesis in vitro. Peptides 1997, 18, 323–326. [Google Scholar] [CrossRef]
- Xiao, J.; Yu, W.; Wang, X.; Wang, B.; Chen, J.; Liu, Y.; Li, Z. Correlation between neuropeptide distribution, cancellous bone microstructure and joint pain in postmenopausal women with osteoarthritis and osteoporosis. Neuropeptides 2016, 56, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.K.; Hirano, I. The enteric nervous system. N. Engl. J. Med. 1996, 334, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Gross, K.J.; Pothoulakis, C. Role of neuropeptides in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 918–932. [Google Scholar] [CrossRef]
- Margolis, K.G.; Gershon, M.D. Neuropeptides and inflammatory bowel disease. Curr. Opin. Gastroenterol. 2009, 25, 503–511. [Google Scholar] [CrossRef]
- Perse, M.; Cerar, A. Dextran sodium sulphate colitis mouse model: Traps and tricks. J. Biomed Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef]
- Wirtz, S.; Neufert, C.; Weigmann, B.; Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2007, 2, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Morampudi, V.; Bhinder, G.; Wu, X.; Dai, C.; Sham, H.P.; Vallance, B.A.; Jacobson, K. DNBS/TNBS colitis models: Providing insights into inflammatory bowel disease and effects of dietary fat. J. Vis. Exp. 2014, 84, e51297. [Google Scholar] [CrossRef] [PubMed]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.; Juarranz, Y.; Martinez, C.; Arranz, A.; Rosignoli, F.; Garcia-Gomez, M.; Leceta, J.; Gomariz, R.P. cDNA array analysis of cytokines, chemokines, and receptors involved in the development of TNBS-induced colitis: Homeostatic role of VIP. Inflamm. Bowel Dis. 2005, 11, 674–684. [Google Scholar] [CrossRef]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Fitzpatrick, L.R. Inhibition of IL-17 as a pharmacological approach for IBD. Int. Rev. Immunol. 2013, 32, 544–555. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef]
- Arranz, A.; Juarranz, Y.; Leceta, J.; Gomariz, R.P.; Martínez, C. VIP balances innate and adaptive immune responses induced by specific stimulation of TLR2 and TLR4. Peptides 2008, 29, 948–956. [Google Scholar] [CrossRef]
- Abad, C.; Gomariz, R.; Waschek, J.; Leceta, J.; Martinez, C.; Juarranz, Y.; Arranz, A. VIP in inflammatory bowel disease: State of the art. Endocr. Metab. Immune Disords Drug Targets 2012, 12, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.; Cuan, N.; Hampartzoumian, T.; Connor, S.J.; Lloyd, A.R.; Grimm, M.C. Vasoactive intestinal peptide impairs leucocyte migration but fails to modify experimental murine colitis. Clin. Exp. Immunol. 2005, 139, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, D.; Anbazhagan, A.N.; Guzman, G.; Dudeja, P.K.; Onyuksel, H. Vasoactive Intestinal Peptide Nanomedicine for the Management of Inflammatory Bowel Disease. Mol. Pharm. 2017, 14, 3698–3708. [Google Scholar] [CrossRef] [PubMed]
- Conlin, V.S.; Wu, X.; Nguyen, C.; Dai, C.; Vallance, B.A.; Buchan, A.M.; Boyer, L.; Jacobson, K. Vasoactive intestinal peptide ameliorates intestinal barrier disruption associated with Citrobacter rodentium-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G735–G750. [Google Scholar] [CrossRef]
- Sun, X.; Guo, C.; Zhao, F.; Zhu, J.; Xu, Y.; Liu, Z.Q.; Yang, G.; Zhang, Y.Y.; Gu, X.; Xiao, L.; et al. Vasoactive intestinal peptide stabilizes intestinal immune homeostasis through maintaining interleukin-10 expression in regulatory B cells. Theranostics 2019, 9, 2800–2811. [Google Scholar] [CrossRef]
- Abad, C.; Tan, Y.V. Immunomodulatory Roles of PACAP and VIP: Lessons from Knockout Mice. J. Mol. Neurosci. 2018, 66, 102–113. [Google Scholar] [CrossRef]
- Yadav, M.; Huang, M.C.; Goetzl, E.J. VPAC1 (vasoactive intestinal peptide (VIP) receptor type 1) G protein-coupled receptor mediation of VIP enhancement of murine experimental colitis. Cell Immunol. 2011, 267, 124–132. [Google Scholar] [CrossRef]
- Wu, X.; Conlin, V.S.; Morampudi, V.; Ryz, N.R.; Nasser, Y.; Bhinder, G.; Bergstrom, K.S.; Yu, H.B.; Waterhouse, C.C.; Buchan, A.M.; et al. Vasoactive intestinal polypeptide promotes intestinal barrier homeostasis and protection against colitis in mice. PLoS ONE 2015, 10, e0125225. [Google Scholar] [CrossRef]
- Abad, C.; Cheung-Lau, G.; Coute-Monvoisin, A.C.; Waschek, J.A. Vasoactive intestinal peptide-deficient mice exhibit reduced pathology in trinitrobenzene sulfonic acid-induced colitis. Neuroimmunomodulation 2015, 22, 203–212. [Google Scholar] [CrossRef]
- Schulte-Bockholt, A.; Fink, J.G.; Meier, D.A.; Otterson, M.F.; Telford, G.L.; Hopp, K.; Koch, T.R. Expression of mRNA for vasoactive intestinal peptide in normal human colon and during inflammation. Mol. Cell Biochem. 1995, 142, 1–7. [Google Scholar] [CrossRef]
- Kubota, Y.; Petras, R.E.; Ottaway, C.A.; Tubbs, R.R.; Farmer, R.G.; Fiocchi, C. Colonic vasoactive intestinal peptide nerves in inflammatory bowel disease. Gastroenterology 1992, 102, 1242–1251. [Google Scholar] [CrossRef]
- Martinez, C.; Ortiz, A.M.; Juarranz, Y.; Lamana, A.; Seoane, I.V.; Leceta, J.; Garcia-Vicuna, R.; Gomariz, R.P.; Gonzalez-Alvaro, I. Serum levels of vasoactive intestinal peptide as a prognostic marker in early arthritis. PLoS ONE 2014, 9, e85248. [Google Scholar] [CrossRef] [PubMed]
- Seoane, I.V.; Tomero, E.; Martinez, C.; Garcia-Vicuna, R.; Juarranz, Y.; Lamana, A.; Ocon, E.; Ortiz, A.M.; Gomez-Leon, N.; Gonzalez-Alvaro, I.; et al. Vasoactive Intestinal Peptide in Early Spondyloarthritis: Low Serum Levels as a Potential Biomarker for Disease Severity. J. Mol. Neurosci. 2015, 56, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Arru, G.; Hosseini, S.; Niegowska, M.; Sechi, G.; Zarbo, I.R.; Sechi, L.A. Inflammation, Infectious Triggers, and Parkinson’s Disease. Front. Neurol. 2019, 10, 122. [Google Scholar] [CrossRef]
- Garretti, F.; Agalliu, D.; Lindestam Arlehamn, C.S.; Sette, A.; Sulzer, D. Autoimmunity in Parkinson’s Disease: The Role of alpha-Synuclein-Specific T Cells. Front. Immunol. 2019, 10, 303. [Google Scholar] [CrossRef]
- Martinez, B.; Peplow, P.V. Neuroprotection by immunomodulatory agents in animal models of Parkinson’s disease. Neural Regen. Res. 2018, 13, 1493–1506. [Google Scholar]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Deng, G.; Jin, L. The effects of vasoactive intestinal peptide in neurodegenerative disorders. Neurol Res. 2017, 39, 65–72. [Google Scholar] [CrossRef]
- Tan, Y.V.; Waschek, J.A. Targeting VIP and PACAP receptor signalling: New therapeutic strategies in multiple sclerosis. ASN Neuro 2011, 3, e00065. [Google Scholar] [CrossRef]
- Fernandez-Martin, A.; Gonzalez-Rey, E.; Chorny, A.; Martin, J.; Pozo, D.; Ganea, D.; Delgado, M. VIP prevents experimental multiple sclerosis by downregulating both inflammatory and autoimmune components of the disease. Ann. N. Y. Acad. Sci. 2006, 1070, 276–281. [Google Scholar] [CrossRef]
- Reddy, J.; Illes, Z.; Zhang, X.; Encinas, J.; Pyrdol, J.; Nicholson, L.; Sobel, R.A.; Wucherpfennig, K.W.; Kuchroo, V.K. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2004, 101, 15434–15439. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002, 169, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Gregg, R.K.; Bell, J.J.; Ellis, J.S.; Divekar, R.; Lee, H.H.; Jain, R.; Waldner, H.; Hardaway, J.C.; Collins, M.; et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J. Immunol. 2005, 174, 6772–6780. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Immunology of relapse and remission in multiple sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martin, A.; Gonzalez-Rey, E.; Chorny, A.; Ganea, D.; Delgado, M. Vasoactive intestinal peptide induces regulatory T cells during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2006, 36, 318–326. [Google Scholar] [CrossRef]
- Tan, Y.V.; Abad, C.; Wang, Y.; Lopez, R.; Waschek, J. VPAC2 (vasoactive intestinal peptide receptor type 2) receptor deficient mice develop exacerbated experimental autoimmune encephalomyelitis with increased Th1/Th17 and reduced Th2/Treg responses. Brain Behav. Immun. 2015, 44, 167–175. [Google Scholar] [CrossRef]
- Abad, C.; Tan, Y.V.; Lopez, R.; Nobuta, H.; Dong, H.; Phan, P.; Feng, J.M.; Campagnoni, A.T.; Waschek, J.A. Vasoactive intestinal peptide loss leads to impaired CNS parenchymal T-cell infiltration and resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 19555–19560. [Google Scholar] [CrossRef]
- Abad, C.; Jayaram, B.; Becquet, L.; Wang, Y.; O’Dorisio, M.S.; Waschek, J.A.; Tan, Y.V. VPAC1 receptor (Vipr1)-deficient mice exhibit ameliorated experimental autoimmune encephalomyelitis, with specific deficits in the effector stage. J. Neuroinflamm. 2016, 13, 169. [Google Scholar] [CrossRef]
- Andersen, O.; Fahrenkrug, J.; Wikkelso, C.; Johansson, B.B. VIP in cerebrospinal fluid of patients with multiple sclerosis. Peptides 1984, 5, 435–437. [Google Scholar] [CrossRef]
- Baranowska-Bik, A.; Kochanowski, J.; Uchman, D.; Wolinska-Witort, E.; Kalisz, M.; Martynska, L.; Baranowska, B.; Bik, W. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase activating polypeptide (PACAP) in humans with multiple sclerosis. J. Neuroimmunol. 2013, 263, 159–161. [Google Scholar] [CrossRef]
- Sun, W.; Hong, J.; Zang, Y.C.; Liu, X.; Zhang, J.Z. Altered expression of vasoactive intestinal peptide receptors in T lymphocytes and aberrant Th1 immunity in multiple sclerosis. Int. Immunol. 2006, 18, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.H.; Beers, D.R.; Henkel, J.S. T cell-microglial dialogue in Parkinson’s disease and amyotrophic lateral sclerosis: Are we listening? Trends Immunol. 2010, 31, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Mosley, R.L.; Lu, Y.; Olson, K.E.; Machhi, J.; Yan, W.; Namminga, K.L.; Smith, J.R.; Shandler, S.J.; Gendelman, H.E. A Synthetic Agonist to Vasoactive Intestinal Peptide Receptor-2 Induces Regulatory T Cell Neuroprotective Activities in Models of Parkinson’s Disease. Front. Cell Neurosci. 2019, 13, 421. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Mosley, R.L. A Perspective on Roles Played by Innate and Adaptive Immunity in the Pathobiology of Neurodegenerative Disorders. J. Neuroimmune Pharmacol. 2015, 10, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Shin, Y.K.; White, C.M.; Ji, S.; Kim, W.; Carlson, O.D.; Napora, J.K.; Chadwick, W.; Chapter, M.; Waschek, J.A.; et al. Vasoactive intestinal peptide-null mice demonstrate enhanced sweet taste preference, dysglycemia, and reduced taste bud leptin receptor expression. Diabetes 2010, 59, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, D.; Karacay, B.; Shutt, D.; Leverich, W.; Schafer, B.; Takle, E.; Thedens, D.; Khanna, G.; Raikwar, S.; Yang, B.; et al. Characterization of intestinal and pancreatic dysfunction in VPAC1-null mutant mouse. Pancreas 2011, 40, 861–871. [Google Scholar] [CrossRef]
- Kato, I.; Suzuki, Y.; Akabane, A.; Yonekura, H.; Tanaka, O.; Kondo, H.; Takasawa, S.; Yoshimoto, T.; Okamoto, H. Transgenic mice overexpressing human vasoactive intestinal peptide (VIP) gene in pancreatic beta cells. Evidence for improved glucose tolerance and enhanced insulin secretion by VIP and PHM-27 in vivo. J. Biol. Chem. 1994, 269, 21223–21228. [Google Scholar]
- Li, C.; Zhu, F.; Wu, B.; Wang, Y. Vasoactive Intestinal Peptide Protects Salivary Glands against Structural Injury and Secretory Dysfunction via IL-17A and AQP5 Regulation in a Model of Sjogren Syndrome. Neuroimmunomodulation 2017, 24, 300–309. [Google Scholar] [CrossRef]
- Hauk, V.; Calafat, M.; Larocca, L.; Fraccaroli, L.; Grasso, E.; Ramhorst, R.; Leiros, C.P. Vasoactive intestinal peptide/vasoactive intestinal peptide receptor relative expression in salivary glands as one endogenous modulator of acinar cell apoptosis in a murine model of Sjogren’s syndrome. Clin. Exp. Immunol. 2011, 166, 309–316. [Google Scholar] [CrossRef]
- Hauk, V.; Fraccaroli, L.; Grasso, E.; Eimon, A.; Ramhorst, R.; Hubscher, O.; Perez Leiros, C. Monocytes from Sjogren’s syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin. Exp. Immunol. 2014, 177, 662–670. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Springer, J.; Fischer, A. Vasoactive intestinal polypeptide as mediator of asthma. Pulm. Pharmacol. Ther. 2001, 14, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Petkov, V.; Mosgoeller, W.; Ziesche, R.; Raderer, M.; Stiebellehner, L.; Vonbank, K.; Funk, G.C.; Hamilton, G.; Novotny, C.; Burian, B.; et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J. Clin. Investig. 2003, 111, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Zissel, G.; Lutzen, N.; Schupp, J.; Schmiedlin, R.; Gonzalez-Rey, E.; Rensing-Ehl, A.; Bacher, G.; Cavalli, V.; Bevec, D.; et al. Inhaled vasoactive intestinal peptide exerts immunoregulatory effects in sarcoidosis. Am. J. Respir. Crit. Care Med. 2010, 182, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Bardea, A.; Reshef, A.; Zamostiano, R.; Zhukovsky, S.; Rubinraut, S.; Fridkin, M.; Brenneman, D.E. Neuroprotective strategy for Alzheimer disease: Intranasal administration of a fatty neuropeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 427–432. [Google Scholar] [CrossRef]
- Korkmaz, O.T.; Tuncel, N.; Tuncel, M.; Oncu, E.M.; Sahinturk, V.; Celik, M. Vasoactive intestinal peptide (VIP) treatment of Parkinsonian rats increases thalamic gamma-aminobutyric acid (GABA) levels and alters the release of nerve growth factor (NGF) by mast cells. J. Mol. Neurosci. 2010, 41, 278–287. [Google Scholar] [CrossRef]
- Vacas, E.; Bajo, A.M.; Schally, A.V.; Sanchez-Chapado, M.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide induces oxidative stress and suppresses metastatic potential in human clear cell renal cell carcinoma. Mol. Cell Endocrinol. 2013, 365, 212–222. [Google Scholar] [CrossRef]
- Chu, T.G.; Orlowski, M. Soluble metalloendopeptidase from rat brain: Action on enkephalin-containing peptides and other bioactive peptides. Endocrinology 1985, 116, 1418–1425. [Google Scholar] [CrossRef]
- Vessillier, S.; Adams, G.; Montero-Melendez, T.; Jones, R.; Seed, M.; Perretti, M.; Chernajovsky, Y. Molecular engineering of short half-life small peptides (VIP, alphaMSH and gamma(3)MSH) fused to latency-associated peptide results in improved anti-inflammatory therapeutics. Ann. Rheum. Dis. 2012, 71, 143–149. [Google Scholar] [CrossRef]
- Bloom, S.R.; Polak, J.M.; Pearse, A.G. Vasoactive intestinal peptide and watery-diarrhoea syndrome. Lancet 1973, 2, 14–16. [Google Scholar] [CrossRef]
- Henning, R.J.; Sawmiller, D.R. Vasoactive intestinal peptide: Cardiovascular effects. Cardiovasc. Res. 2001, 49, 27–37. [Google Scholar] [CrossRef]
- Fernandez-Montesinos, R.; Castillo, P.M.; Klippstein, R.; Gonzalez-Rey, E.; Mejias, J.A.; Zaderenko, A.P.; Pozo, D. Chemical synthesis and characterization of silver-protected vasoactive intestinal peptide nanoparticles. Nanomedicine 2009, 4, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Masaka, T.; Li, Y.; Kawatobi, S.; Koide, Y.; Takami, A.; Yano, K.; Imai, R.; Yagi, N.; Suzuki, H.; Hikawa, H.; et al. Liposome modified with VIP-lipopeptide as a new drug delivery system. Yakugaku Zasshi 2014, 134, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Onyuksel, H.; Mohanty, P.S.; Rubinstein, I. VIP-grafted sterically stabilized phospholipid nanomicellar 17-allylamino-17-demethoxy geldanamycin: A novel targeted nanomedicine for breast cancer. Int. J. Pharm. 2009, 365, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Koo, O.M.; Rubinstein, I.; Onyuksel, H. Actively targeted low-dose camptothecin as a safe, long-acting, disease-modifying nanomedicine for rheumatoid arthritis. Pharm. Res. 2011, 28, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Toscano, M.G.; Benabdellah, K.; Cobo, M.; O’Valle, F.; Gonzalez-Rey, E.; Martin, F. In vivo delivery of lentiviral vectors expressing vasoactive intestinal peptide complementary DNA as gene therapy for collagen-induced arthritis. Arthritis Rheum. 2008, 58, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, Z.A.; Mostafa, G.A.; Aly, G.S.; El-Shahed, G.S.; El-Aziz, M.M.; El-Emam, S.M. Cardiovascular autonomic function assessed by autonomic function tests and serum autonomic neuropeptides in Egyptian children and adolescents with rheumatic diseases. Rheumatology 2009, 48, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Seoane, I.V.; Ortiz, A.M.; Piris, L.; Lamana, A.; Juarranz, Y.; Garcia-Vicuna, R.; Gonzalez-Alvaro, I.; Gomariz, R.P.; Martinez, C. Clinical Relevance of VPAC1 Receptor Expression in Early Arthritis: Association with IL-6 and Disease Activity. PLoS ONE 2016, 11, e0149141. [Google Scholar] [CrossRef]
- Choy, E.; Taylor, P.; McAuliffe, S.; Roberts, K.; Sargeant, I. Variation in the use of biologics in the management of rheumatoid arthritis across the UK. Curr. Med. Res. Opin. 2012, 28, 1733–1741. [Google Scholar] [CrossRef]
- Felson, D.T.; Smolen, J.S.; Wells, G.; Zhang, B.; van Tuyl, L.H.; Funovits, J.; Aletaha, D.; Allaart, C.F.; Bathon, J.; Bombardieri, S.; et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum. 2011, 63, 573–586. [Google Scholar] [CrossRef]
- Van Leeuwen, M.A.; van Rijswijk, M.H.; van der Heijde, D.M.; Te Meerman, G.J.; van Riel, P.L.; Houtman, P.M.; van De Putte, L.B.; Limburg, P.C. The acute-phase response in relation to radiographic progression in early rheumatoid arthritis: A prospective study during the first three years of the disease. Br. J. Rheumatol. 1993, 32, 9–13. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Castrejon, I.; Ortiz, A.M.; Garcia-Vicuna, R.; Lopez-Bote, J.P.; Humbria, A.; Carmona, L.; Gonzalez-Alvaro, I. Are the C-reactive protein values and erythrocyte sedimentation rate equivalent when estimating the 28-joint disease activity score in rheumatoid arthritis? Clin. Exp. Rheumatol. 2008, 26, 769–775. [Google Scholar] [PubMed]
- Centola, M.; Cavet, G.; Shen, Y.; Ramanujan, S.; Knowlton, N.; Swan, K.A.; Turner, M.; Sutton, C.; Smith, D.R.; Haney, D.J.; et al. Development of a multi-biomarker disease activity test for rheumatoid arthritis. PLoS ONE 2013, 8, e60635. [Google Scholar] [CrossRef] [PubMed]
- Seoane, I.V.; Martinez, C.; Garcia-Vicuna, R.; Ortiz, A.M.; Juarranz, Y.; Talayero, V.C.; Gonzalez-Alvaro, I.; Gomariz, R.P.; Lamana, A. Vasoactive intestinal peptide gene polymorphisms, associated with its serum levels, predict treatment requirements in early rheumatoid arthritis. Sci. Rep. 2018, 8, 2035. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, C.; Juarranz, Y.; Gutiérrez-Cañas, I.; Carrión, M.; Pérez-García, S.; Villanueva-Romero, R.; Castro, D.; Lamana, A.; Mellado, M.; González-Álvaro, I.; et al. A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 65. https://doi.org/10.3390/ijms21010065
Martínez C, Juarranz Y, Gutiérrez-Cañas I, Carrión M, Pérez-García S, Villanueva-Romero R, Castro D, Lamana A, Mellado M, González-Álvaro I, et al. A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases. International Journal of Molecular Sciences. 2020; 21(1):65. https://doi.org/10.3390/ijms21010065
Chicago/Turabian StyleMartínez, Carmen, Yasmina Juarranz, Irene Gutiérrez-Cañas, Mar Carrión, Selene Pérez-García, Raúl Villanueva-Romero, David Castro, Amalia Lamana, Mario Mellado, Isidoro González-Álvaro, and et al. 2020. "A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases" International Journal of Molecular Sciences 21, no. 1: 65. https://doi.org/10.3390/ijms21010065
APA StyleMartínez, C., Juarranz, Y., Gutiérrez-Cañas, I., Carrión, M., Pérez-García, S., Villanueva-Romero, R., Castro, D., Lamana, A., Mellado, M., González-Álvaro, I., & Gomariz, R. P. (2020). A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases. International Journal of Molecular Sciences, 21(1), 65. https://doi.org/10.3390/ijms21010065