Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
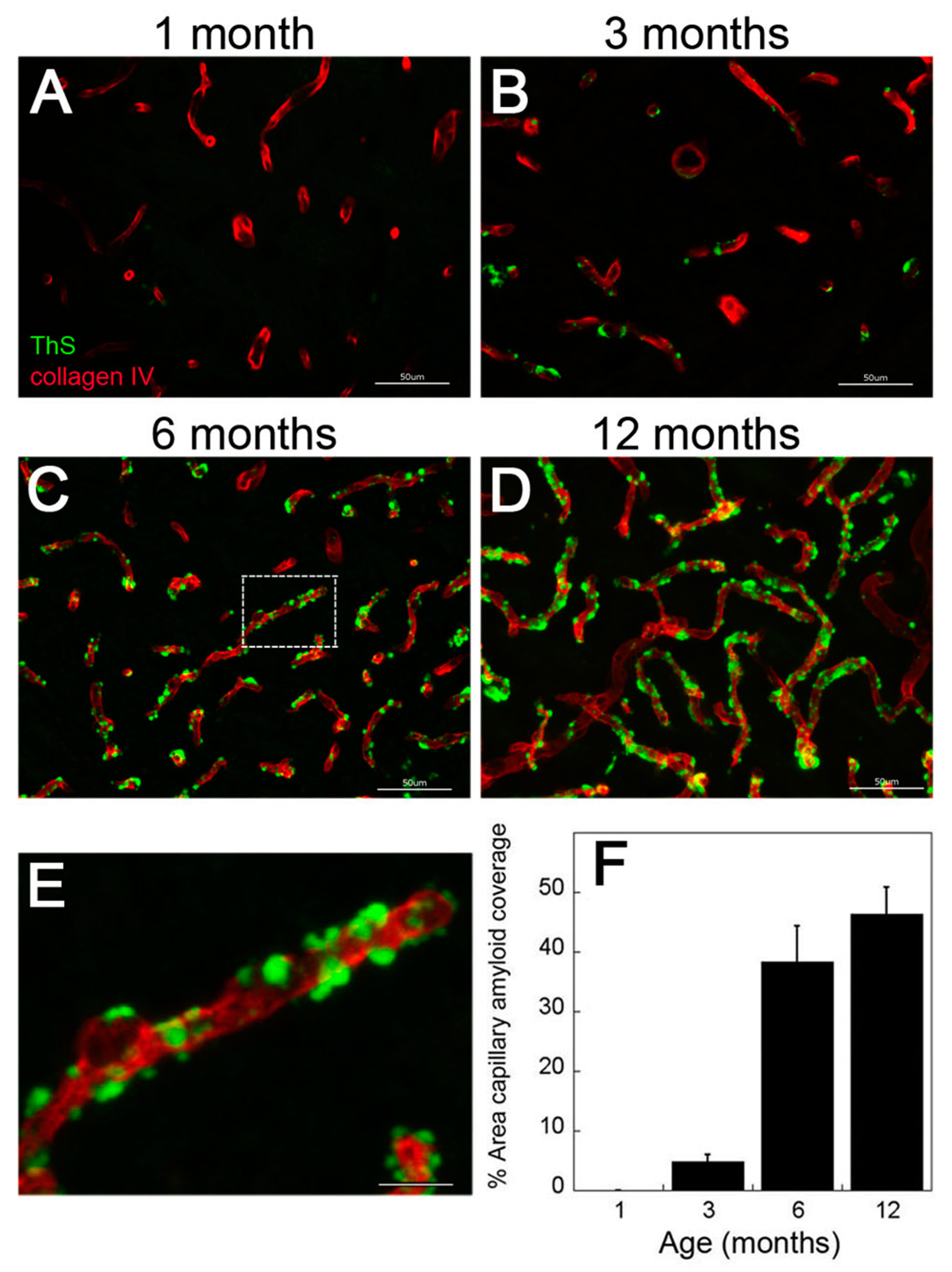
2.1. rTg-DI Rats Show Early-Onset and Progressive Accumulation of Cerebral Vascular Amyloid
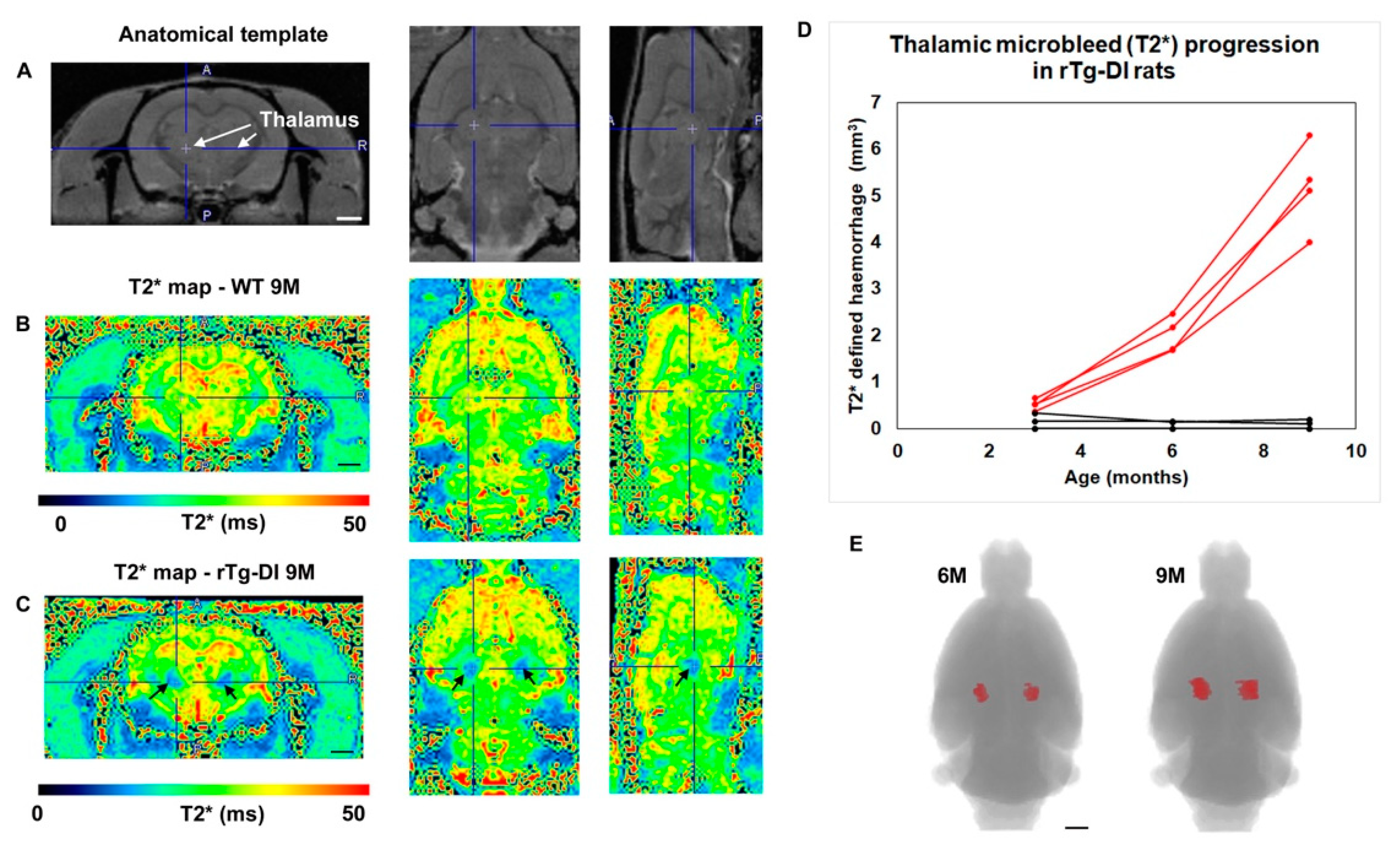
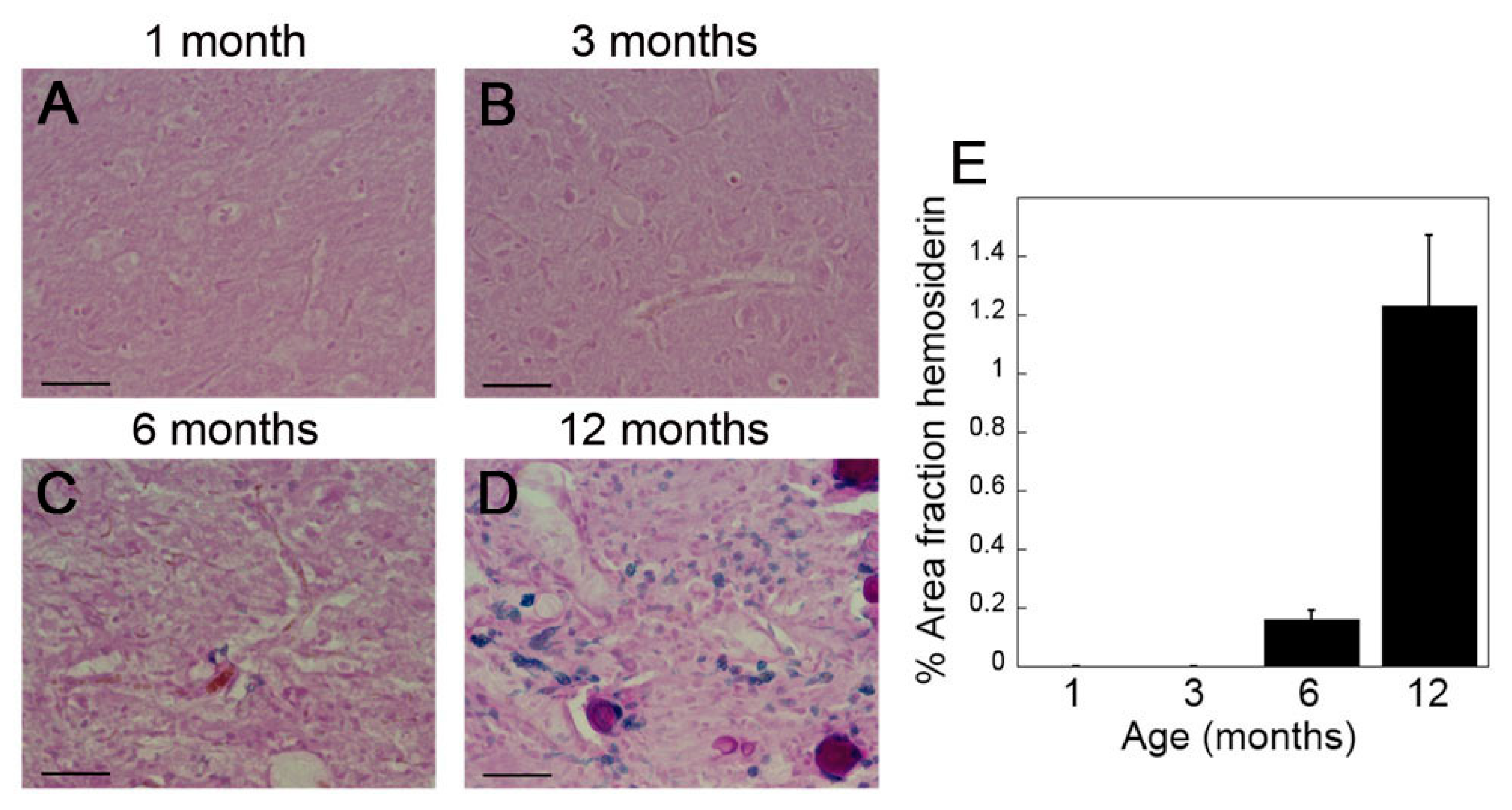
2.2. rTg-DI Rats Develop Consistent Thalamic Microbleeds Detected by MRI and Histology
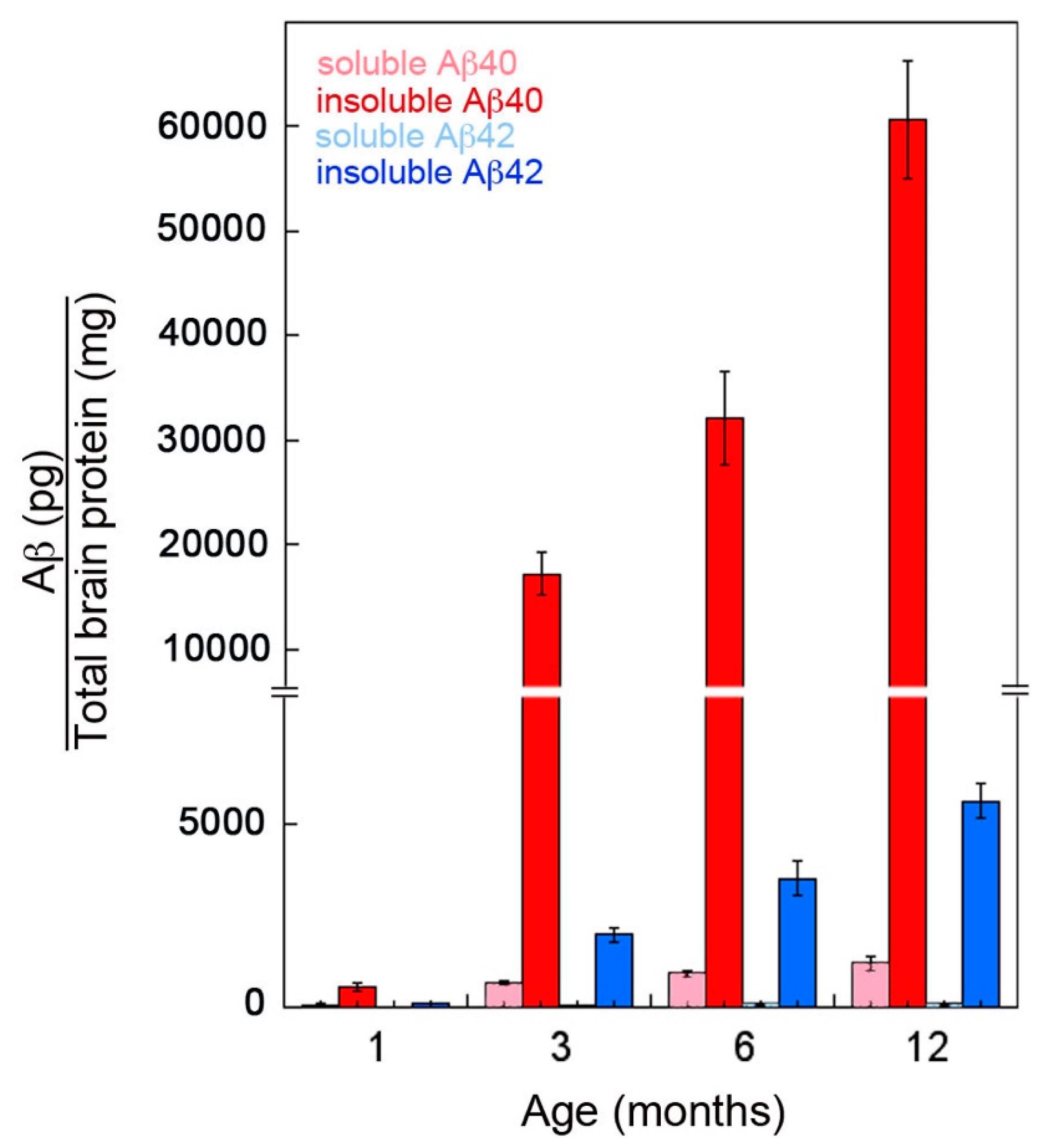
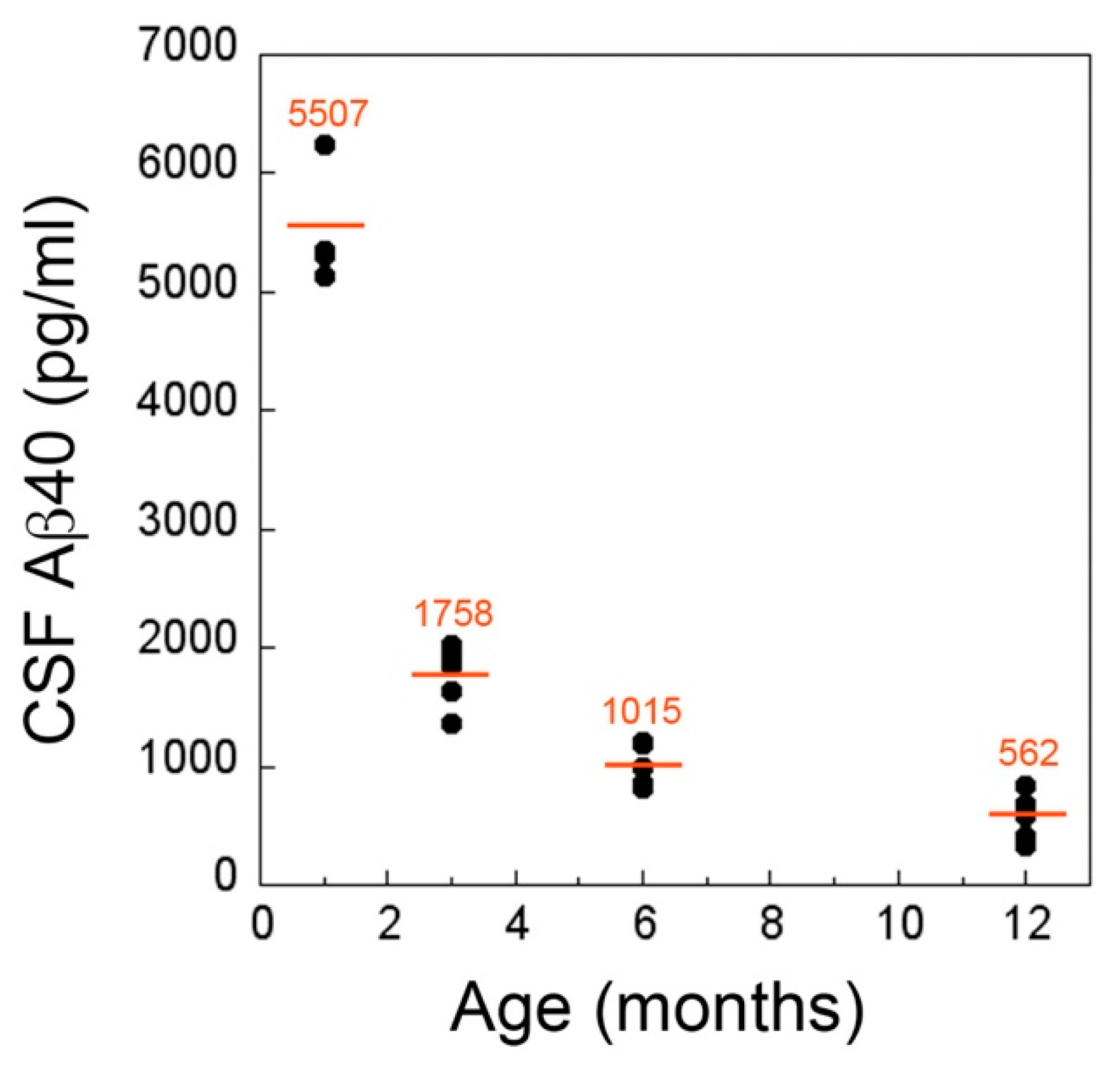
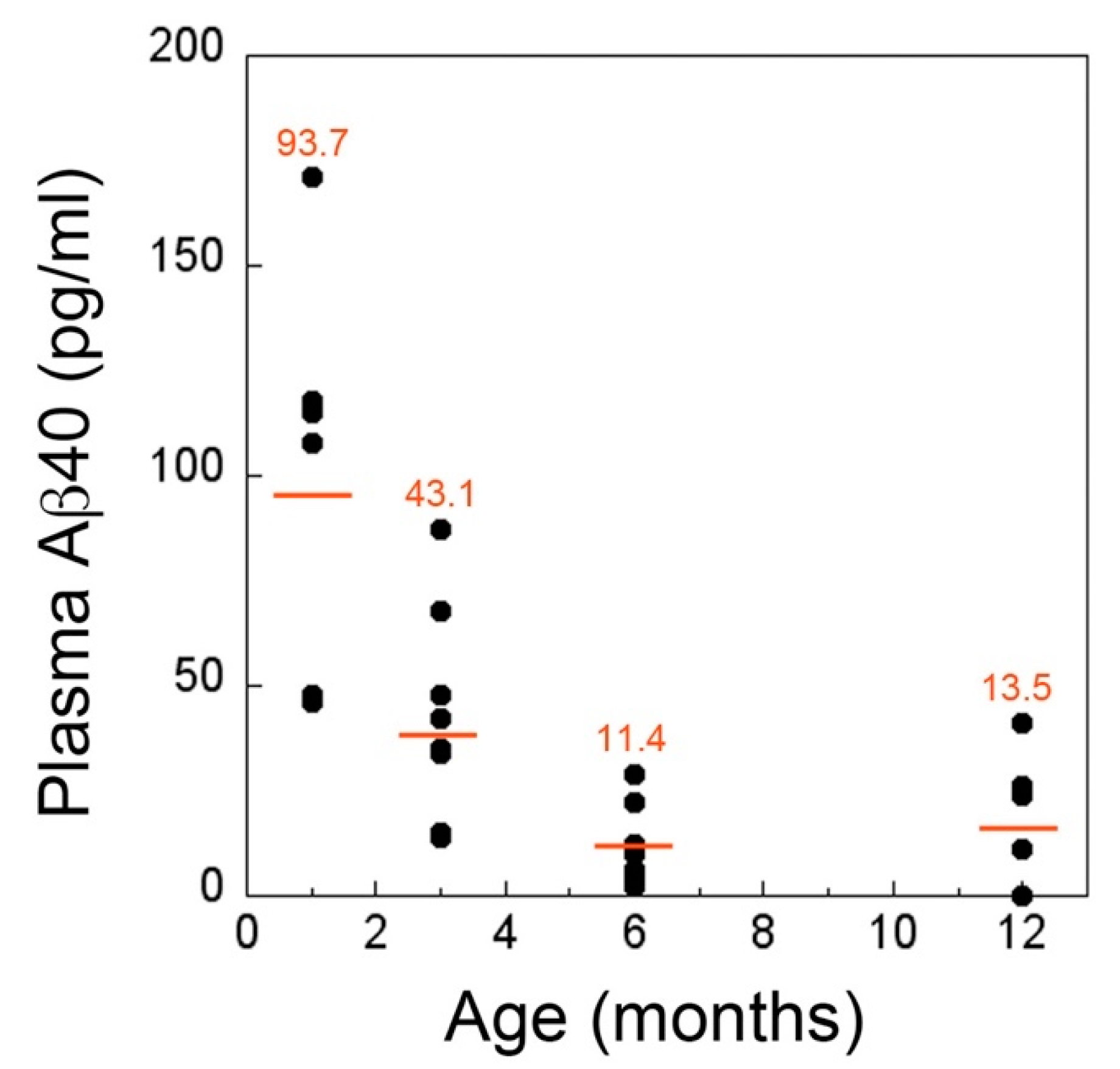
2.3. CSF and Plasma Levels of Aβ40 Markedly Drop at the Inception of Microvascular CAA in rTg-DI Rats
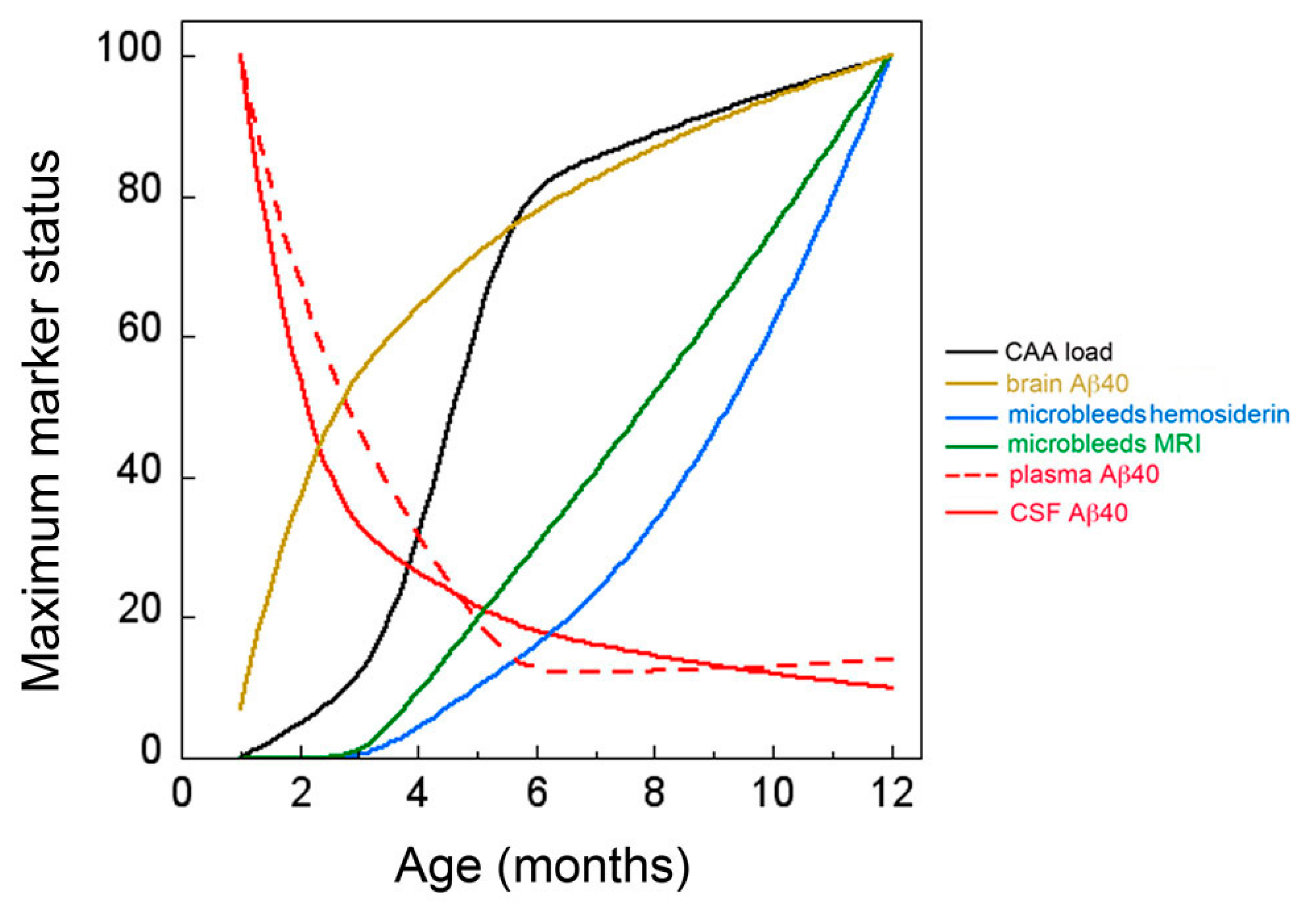
3. Discussion
4. Materials and Methods
4.1. rTg-DI Rats
4.2. CSF Collection
4.3. Plasma Isolation
4.4. Brain Tissue Collection and Preparation
4.5. ELISA Quantitation of Aβ Peptides
4.6. Immunohistochemical Analysis
4.7. Quantitative Histological Analysis of CAA Load and Microbleeds
4.8. Magnetic Resonance Imaging Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β protein |
| AβPP | Aβ precursor protein |
| AD | Alzheimer’s disease |
| CAA | Cerebral amyloid angiopathy |
| CNS | Central nervous system |
| CSF | Cerebral spinal fluid |
| ELISA | Enzyme linked immunosorbent assay |
| ISF | Interstitial fluid |
| LRP1 | low density lipoprotein receptor related protein 1 |
| MRI | Magnetic resonance imaging |
| rTg-DI | Transgenic rats expressing Dutch/Iowa mutant AβPP |
| VCID | Vascular mediated cognitive impairment and dementia |
References
- Rensink, A.A.; De Waal, R.M.; Kremer, B.; Verbeek, M.M. Pathogenesis of cerebral amyloid angiopathy. Brain Res. Brain Res. Rev. 2003, 43, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.; Thal, D.R.; Van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Auriel, E.; Greenberg, S.M. The pathophysiology and clinical presentation of cerebral amyloid angiopathy. Curr. Atheroscler. Rep. 2012, 14, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Carare, R.; Cordonnier, C.; Greenberg, S.M.; Schneider, J.A.; Smith, E.E.; Buchem, M.V.; Grond, J.V.; Verbeek, M.M.; Werring, D.J. The increasing impact of cerebral amyloid angiopathy: Essential new insights for clinical practice. J. Neurol. Neurosurg. Psychiatry 2017, 88, 982–994. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Leurgans, S.E.; Wang, Z.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 2011, 69, 320–327. [Google Scholar] [CrossRef]
- Boyle, P.A.; Yu, L.; Nag, S.; Leurgans, S.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015, 85, 1930–1936. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Gurol, M.E.; Rosand, J.; Smith, E.E. Amyloid angiopathy-related vascular cognitive impairment. Stroke 2004, 35, 2616–2619. [Google Scholar] [CrossRef]
- Martinez-Ramirez, S.; van Rooden, S.; Charidimou, A.; van Opstal, A.M.; Wermer, M.; Gurol, M.E.; Terwindt, G.; van der Grond, J.; Greenberg, S.M.; van Buchem, M.; et al. Perivascular Spaces Volume in Sporadic and Hereditary (Dutch-Type) Cerebral Amyloid Angiopathy. Stroke 2018, 49, 1913–1919. [Google Scholar] [CrossRef]
- Xiong, L.; van Veluw, S.J.; Bounemia, N.; Charidimou, A.; Pasi, M.; Boulouis, G.; Reijmer, Y.D.; Giese, A.K.; Davidsdottir, S.; Fotiadis, P.; et al. Cerebral Cortical Microinfarcts on Magnetic Resonance Imaging and Their Association with Cognition in Cerebral Amyloid Angiopathy. Stroke 2018, 49, 2330–2336. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef]
- Knudsen, K.A.; Rosand, J.; Karluk, D.; Greenberg, S.M. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the Boston criteria. Neurology 2001, 56, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Charidimou, A. Diagnosis of Cerebral Amyloid Angiopathy: Evolution of the Boston Criteria. Stroke 2018, 49, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Frosch, M.P.; Salman, R.A.; Baron, J.C.; Cordonnier, C.; Hernandez-Guillamon, M.; Linn, J.; Raposo, N.; Rodrigues, M.; Romero, J.R.; et al. Advancing diagnostic criteria for sporadic cerebral amyloid angiopathy: Study protocol for a multicenter MRI-pathology validation of Boston criteria v2.0. Int. J. Stroke 2019, 14, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Caetano, A.; Ladeira, F.; Barbosa, R.; Calado, S.; Viana-Baptista, M. Cerebral amyloid angiopathy—The modified Boston criteria in clinical practice. J. Neurol. Sci. 2018, 384, 55–57. [Google Scholar] [CrossRef]
- Johnson, K.A.; Gregas, M.; Becker, J.A.; Kinnecom, C.; Salat, D.H.; Moran, E.K.; Smith, E.E.; Rosand, J.; Rentz, D.M.; Klunk, W.E.; et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol. 2007, 62, 229–234. [Google Scholar] [CrossRef]
- Ly, J.V.; Donnan, G.A.; Villemagne, V.L.; Zavala, J.A.; Ma, H.; O’Keefe, G.; Gong, S.J.; Gunawan, R.M.; Saunder, T.; Ackerman, U.; et al. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology 2010, 74, 487–493. [Google Scholar] [CrossRef]
- Felling, R.J.; Faigle, R.; Ho, C.Y.; Llinas, R.H.; Urrutia, V.C. Cerebral Amyloid Angiopathy: A Hidden Risk for IV Thrombolysis? J. Neurol. Transl. Neurosci. 2014, 2, 1034. [Google Scholar]
- Charidimou, A.; Nicoll, J.A.; McCarron, M.O. Thrombolysis-related intracerebral hemorrhage and cerebral amyloid angiopathy: Accumulating evidence. Front. Neurol. 2015, 6, 99. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Hatfield, J.; Lee, H.; Hoos, M.D.; Popescu, D.; Crooks, E.; Kim, R.; Smith, S.O.; Robinson, J.K.; et al. A Novel Transgenic Rat Model of Robust Cerebral Microvascular Amyloid with Prominent Vasculopathy. Am. J. Pathol. 2018, 188, 2877–2889. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Kremer, B.P.; Rikkert, M.O.; Van Domburg, P.H.; Skehan, M.E.; Greenberg, S.M. Cerebrospinal fluid amyloid beta (40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 2009, 66, 245–249. [Google Scholar] [CrossRef]
- Renard, D.; Castelnovo, G.; Wacongne, A.; Le Floch, A.; Thouvenot, E.; Mas, J.; Gabelle, A.; Labauge, P.; Lehmann, S. Interest of CSF biomarker analysis in possible cerebral amyloid angiopathy cases defined by the modified Boston criteria. J. Neurol. 2012, 259, 2429–2433. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, E.S.; Verbeek, M.M.; van der Grond, J.; Zielman, R.; van Rooden, S.; van Zwet, E.W.; van Opstal, A.M.; Haan, J.; Greenberg, S.M.; van Buchem, M.A.; et al. beta-Amyloid in CSF: Biomarker for preclinical cerebral amyloid angiopathy. Neurology 2017, 88, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Roongpiboonsopit, D.; Auriel, E.; Pasi, M.; Haley, K.; van Etten, E.S.; Martinez-Ramirez, S.; Ayres, A.; Vashkevich, A.; et al. Cortical superficial siderosis multifocality in cerebral amyloid angiopathy: A prospective study. Neurology 2017, 89, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, S.; Pontes-Neto, O.M.; Dumas, A.P.; Auriel, E.; Halpin, A.; Quimby, M.; Gurol, M.E.; Greenberg, S.M.; Viswanathan, A. Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology 2013, 80, 1551–1556. [Google Scholar] [CrossRef]
- Brown, R.; Benveniste, H.; Black, S.E.; Charpak, S.; Dichgans, M.; Joutel, A.; Nedergaard, M.; Smith, K.J.; Zlokovic, B.V.; Wardlaw, J.M. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc. Res. 2018, 114, 1462–1473. [Google Scholar] [CrossRef]
- Ueno, M.; Chiba, Y.; Murakami, R.; Matsumoto, K.; Fujihara, R.; Uemura, N.; Yanase, K.; Kamada, M. Disturbance of Intracerebral Fluid Clearance and Blood-Brain Barrier in Vascular Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 2600. [Google Scholar] [CrossRef]
- Albargothy, N.J.; Johnston, D.A.; MacGregor-Sharp, M.; Weller, R.O.; Verma, A.; Hawkes, C.A.; Carare, R.O. Convective influx/glymphatic system: Tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018, 136, 139–152. [Google Scholar] [CrossRef]
- Bakker, E.N.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef]
- Palmqvist, S.; Zetterberg, H.; Blennow, K.; Vestberg, S.; Andreasson, U.; Brooks, D.J.; Owenius, R.; Hagerstrom, D.; Wollmer, P.; Minthon, L.; et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta-amyloid 42: A cross-validation study against amyloid positron emission tomography. JAMA Neurol. 2014, 71, 1282–1289. [Google Scholar] [CrossRef]
- Grimmer, T.; Riemenschneider, M.; Forstl, H.; Henriksen, G.; Klunk, W.E.; Mathis, C.A.; Shiga, T.; Wester, H.J.; Kurz, A.; Drzezga, A. Beta amyloid in Alzheimer’s disease: Increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol. Psychiatry 2009, 65, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Miebach, L.; Wolfsgruber, S.; Polcher, A.; Peters, O.; Menne, F.; Luther, K.; Incesoy, E.; Priller, J.; Spruth, E.; Altenstein, S.; et al. Which features of subjective cognitive decline are related to amyloid pathology? Findings from the DELCODE study. Alzheimers Res. Ther. 2019, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Villemagne, V.L.; Doecke, J.D.; Rembach, A.; Sarros, S.; Varghese, S.; McGlade, A.; Laughton, K.M.; Pertile, K.K.; Fowler, C.J.; et al. Alzheimer’s Disease Normative Cerebrospinal Fluid Biomarkers Validated in PET Amyloid-beta Characterized Subjects from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study. J. Alzheimers Dis. 2015, 48, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Catak, C.; Zedde, M.; Malik, R.; Janowitz, D.; Soric, V.; Seegerer, A.; Krebs, A.; During, M.; Opherk, C.; Linn, J.; et al. Decreased CSF Levels of ss-Amyloid in Patients with Cortical Superficial Siderosis. Front. Neurol. 2019, 10, 439. [Google Scholar] [CrossRef] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Burki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Sturchler-Pierrat, C.; Burki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef]
- Herzig, M.C.; Van Nostrand, W.E.; Jucker, M. Mechanism of cerebral beta-amyloid angiopathy: Murine and cellular models. Brain Pathol. 2006, 16, 40–54. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Ramanathan, A.; Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: The role, regulation and restoration of LRP1. Front. Aging Neurosci. 2015, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid beta -Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Lopez, O.L.; Kuller, L.H.; Mehta, P.D.; Becker, J.T.; Gach, H.M.; Sweet, R.A.; Chang, Y.F.; Tracy, R.; DeKosky, S.T. Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology 2008, 70, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Alzheimer disease: Biomarkers of AD risk—The end of the road for plasma amyloid-beta? Nat. Rev. Neurol. 2016, 12, 613. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Shi, D.Y.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fu, Z.; Dass, S.; Kotarba, A.E.; Davis, J.; Smith, S.O.; Van Nostrand, W.E. Cerebral vascular amyloid seeds drive amyloid beta-protein fibril assembly with a distinct anti-parallel structure. Nat. Commun. 2016, 7, 13527. [Google Scholar] [CrossRef]
- Johnson-Wood, K.; Lee, M.; Motter, R.; Hu, K.; Gordon, G.; Barbour, R.; Khan, K.; Gordon, M.; Tan, H.; Games, D.; et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 1550–1555. [Google Scholar] [CrossRef]
- DeMattos, R.B.; O’Dell, M.A.; Parsadanian, M.; Taylor, J.W.; Harmony, J.A.; Bales, K.R.; Paul, S.M.; Aronow, B.J.; Holtzman, D.M. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10843–10848. [Google Scholar] [CrossRef]
- Benveniste, H.; Lee, H.; Ding, F.; Sun, Q.; Al-Bizri, E.; Makaryus, R.; Probst, S.; Nedergaard, M.; Stein, E.A.; Lu, H. Anesthesia with Dexmedetomidine and Low-dose Isoflurane Increases Solute Transport via the Glymphatic Pathway in Rat Brain When Compared with High-dose Isoflurane. Anesthesiology 2017, 127, 976–988. [Google Scholar] [CrossRef]
- Peran, P.; Hagberg, G.; Luccichenti, G.; Cherubini, A.; Brainovich, V.; Celsis, P.; Caltagirone, C.; Sabatini, U. Voxel-based analysis of R2* maps in the healthy human brain. J. Magn. Reson. Imaging 2007, 26, 1413–1420. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Xu, F.; Hoos, M.D.; Lee, H.; Benveniste, H.; Van Nostrand, W.E. Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats. Int. J. Mol. Sci. 2020, 21, 303. https://doi.org/10.3390/ijms21010303
Zhu X, Xu F, Hoos MD, Lee H, Benveniste H, Van Nostrand WE. Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats. International Journal of Molecular Sciences. 2020; 21(1):303. https://doi.org/10.3390/ijms21010303
Chicago/Turabian StyleZhu, Xiaoyue, Feng Xu, Michael D. Hoos, Hedok Lee, Helene Benveniste, and William E. Van Nostrand. 2020. "Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats" International Journal of Molecular Sciences 21, no. 1: 303. https://doi.org/10.3390/ijms21010303
APA StyleZhu, X., Xu, F., Hoos, M. D., Lee, H., Benveniste, H., & Van Nostrand, W. E. (2020). Reduced Levels of Cerebrospinal Fluid/Plasma Aβ40 as an Early Biomarker for Cerebral Amyloid Angiopathy in RTg-DI Rats. International Journal of Molecular Sciences, 21(1), 303. https://doi.org/10.3390/ijms21010303