A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs

Abstract
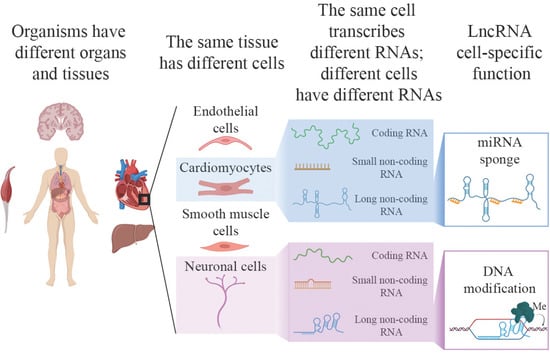
1. Introduction
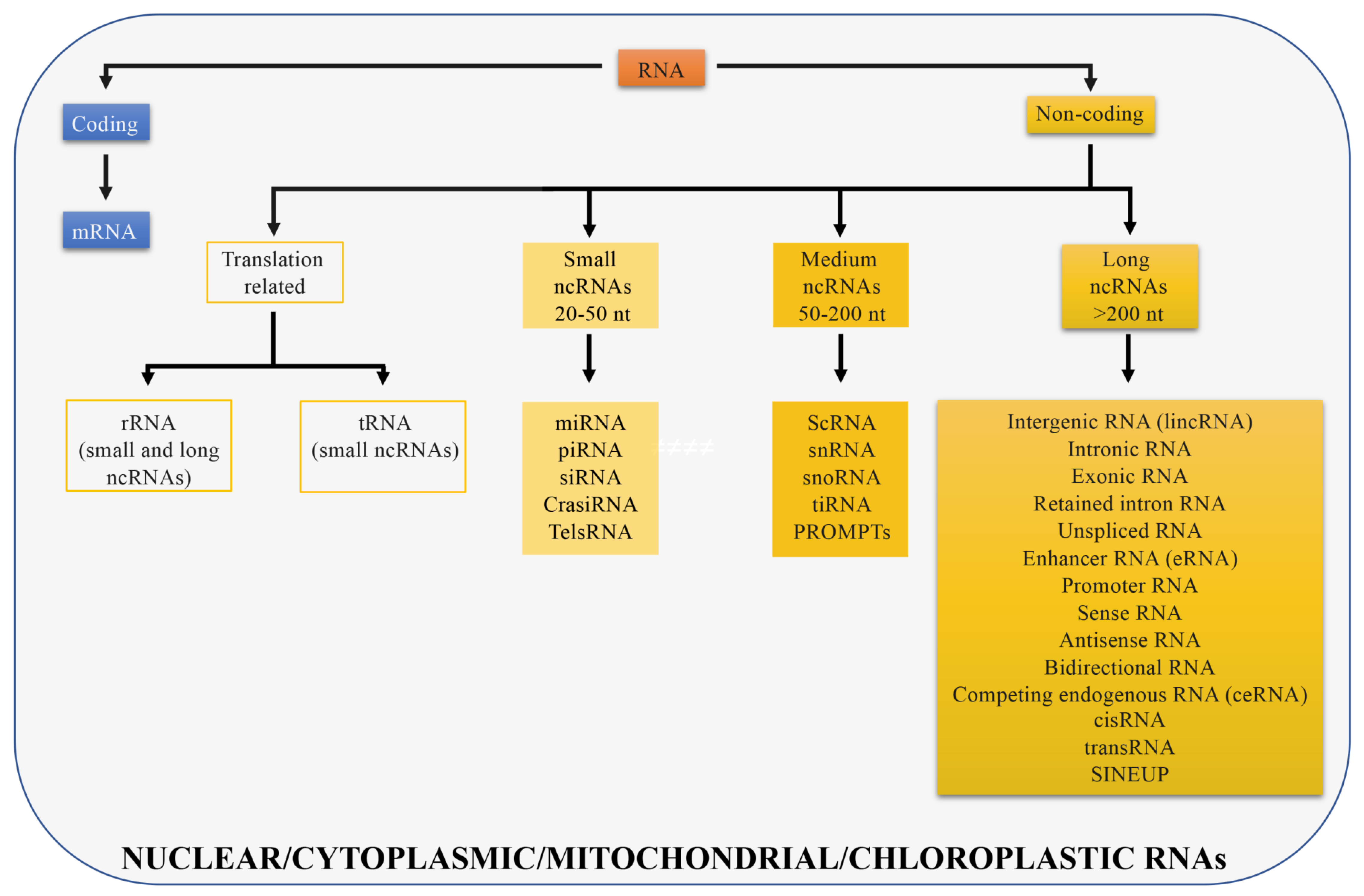
2. Classification of LncRNAs according to Genomic Position
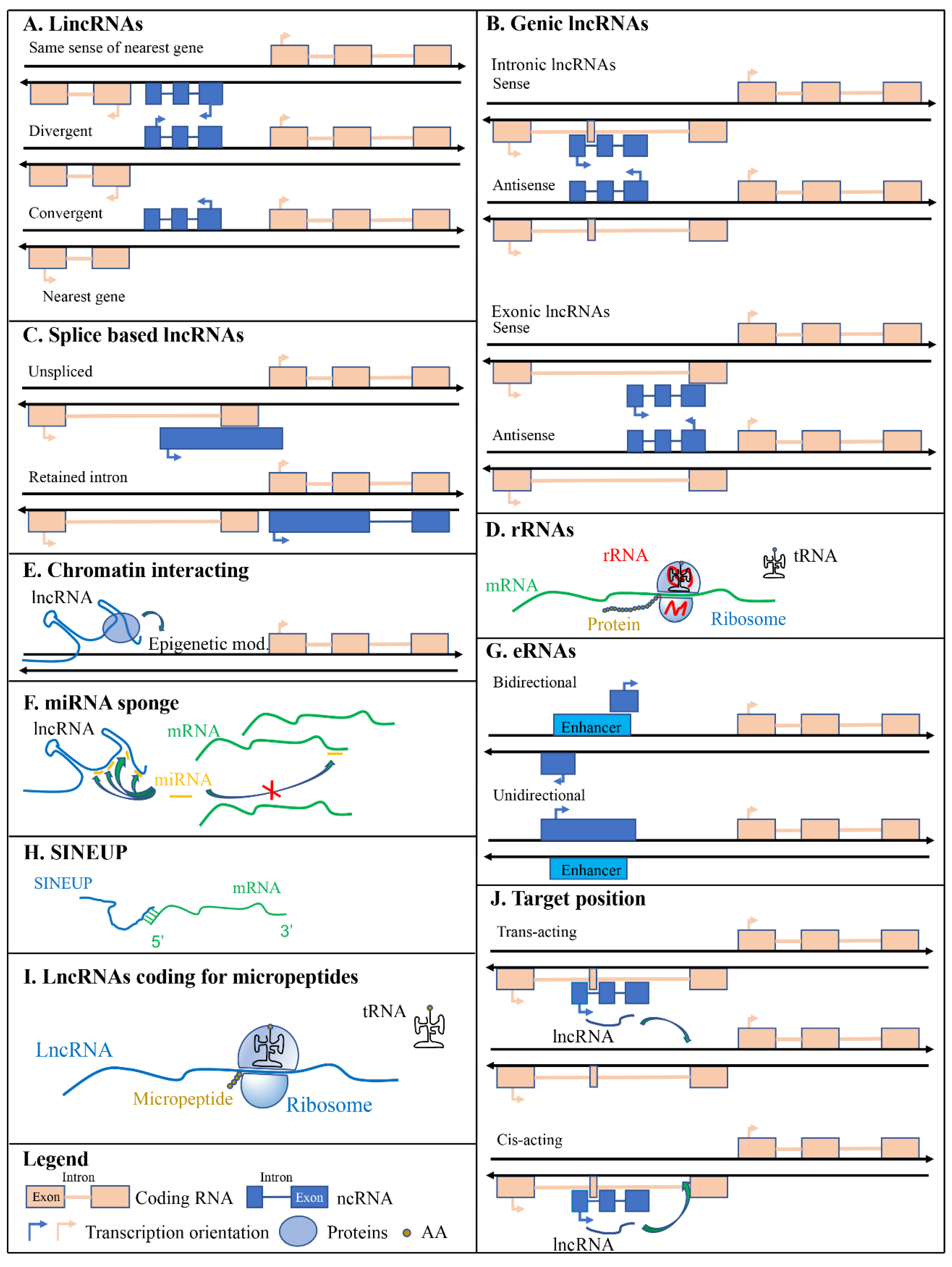
2.1. Long Intergenic Non-Coding RNAs (LincRNAs)
2.2. Genic LncRNAs: Intronic and Exonic
2.3. Splicing based Classification
3. Classification of LncRNAs as Specified by Their Function
3.1. Ribosomal RNAs
3.2. Chromatin Interacting RNAs
3.3. miRNA Sponges
3.4. Enhancer RNAs
3.5. SINEUPs
3.6. LncRNAs Coding for Micropeptides
3.7. Target Position
4. Classification of LncRNAs according to Their Subcellular Localization
4.1. Nuclear LncRNAs
4.2. Cytoplasmic LncRNAs
4.3. Mitochondrial and Chloroplastic LncRNAs
5. Methods for Transcriptional Analysis of Single Cells: Progresses and Limitations
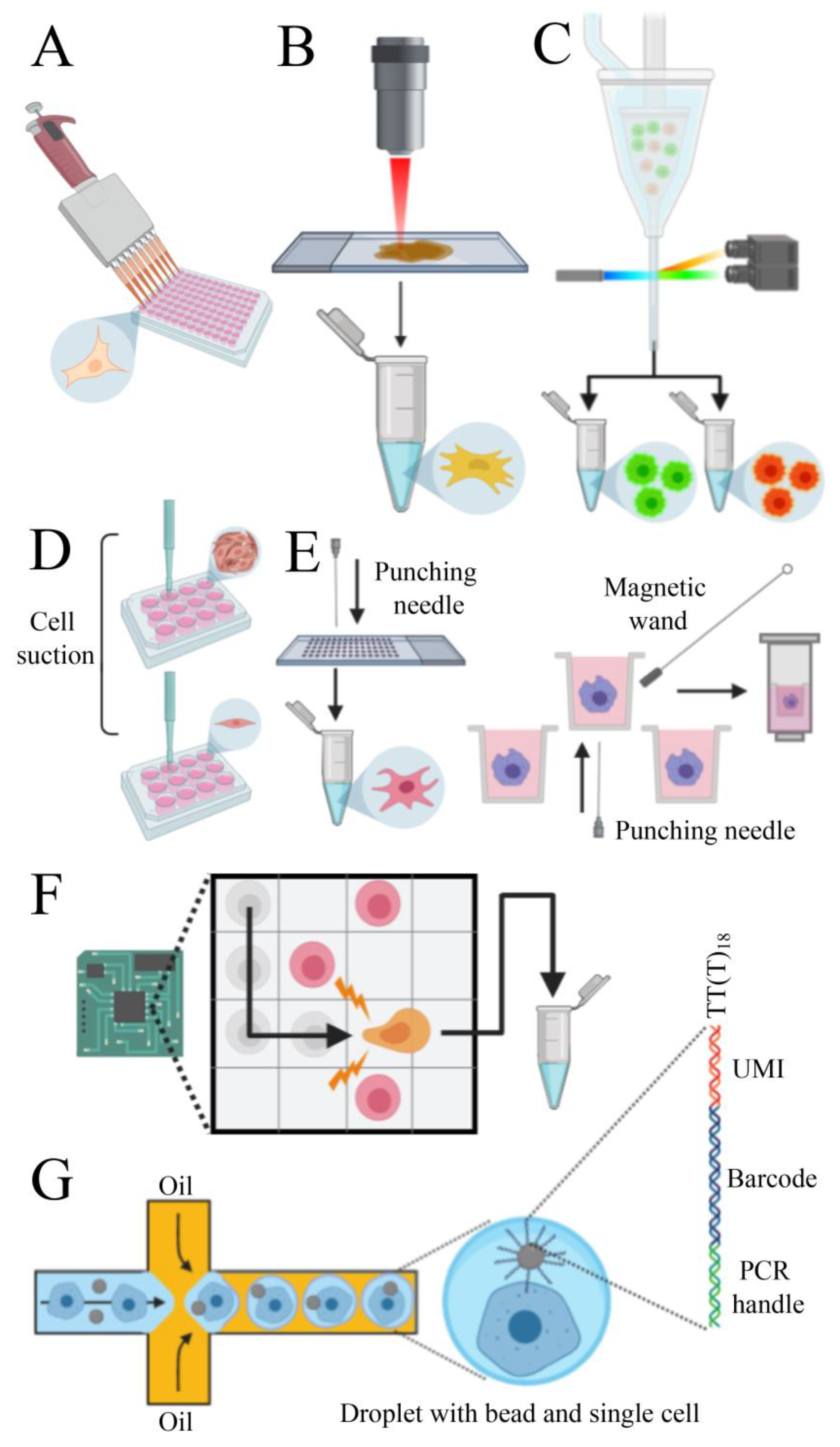
5.1. Single-Cell Isolation
5.2. Library Preparation
5.3. RNA Sequencing and Bioinformatic Analysis
6. Single-Cell Analysis of LncRNAs
6.1. LncRNAs in Embryo-Derived Cells
6.2. LncRNAs in Stem Cells
6.3. LncRNAs in Differentiated Cells
6.4. LncRNAs in Tumors
7. Databases
7.1. Collection of Single-Cell Gene Expression
7.2. Databases of LncRNAs
8. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Liu, C. Coding or noncoding, the converging concepts of RNAs. Front. Genet. 2019, 10, 496. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Vandevenne, M.; Delmarcelle, M.; Galleni, M. RNA regulatory networks as a control of stochasticity in biological systems. Front. Genet. 2019, 10, 403. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M. The human transcriptome: An unfinished story. Genes 2012, 3, 344–360. [Google Scholar] [CrossRef]
- Li, S.; Wan, C.; Zheng, R.; Fan, J.; Dong, X.; Meyer, C.A.; Liu, X.S. Cistrome-GO: A web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res. 2019, 47, W206–W211. [Google Scholar] [CrossRef]
- Ahnert, S.E.; Fink, T.M.; Zinovyev, A. How much non-coding DNA do eukaryotes require? J. Theor. Biol. 2008, 252, 587–592. [Google Scholar] [CrossRef]
- Taft, R.J.; Pheasant, M.; Mattick, J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 2007, 29, 288–299. [Google Scholar] [CrossRef]
- Kellis, M.; Wold, B.; Snyder, M.P.; Bernstein, B.E.; Kundaje, A.; Marinov, G.K.; Ward, L.D.; Birney, E.; Crawford, G.E.; Dekker, J.; et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 2014, 111, 6131–6138. [Google Scholar] [CrossRef]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.; Carone, D.M.; Brown, J.; Hall, L.; Qureshi, S.; Mitchell, S.E.; Jannetty, N.; Hannon, G.; Renfree, M.; Pask, A.; et al. Unique small RNA signatures uncovered in the tammar wallaby genome. BMC Genom. 2012, 13, 559. [Google Scholar] [CrossRef]
- Cao, F.; Li, X.; Hiew, S.; Brady, H.; Liu, Y.; Dou, Y. Dicer independent small RNAs associate with telomeric heterochromatin. RNA 2009, 15, 1274–1281. [Google Scholar] [CrossRef]
- Bird, R.C.; Sells, B.H. Small cytoplasmic RNAs and their location within the cytoplasm. Biochem. Cell Biol. 1987, 65, 582–587. [Google Scholar] [CrossRef]
- Valadkhan, S.; Gunawardane, L.S. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem. 2013, 54, 79–90. [Google Scholar] [CrossRef]
- Kiss, T. Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell 2002, 109, 145–148. [Google Scholar] [CrossRef]
- Taft, R.J.; Hawkins, P.G.; Mattick, J.S.; Morris, K.V. The relationship between transcription initiation RNAs and CCCTC-binding factor (CTCF) localization. Epigenet. Chromatin 2011, 4, 13. [Google Scholar] [CrossRef]
- Preker, P.; Nielsen, J.; Kammler, S.; Lykke-Andersen, S.; Christensen, M.S.; Mapendano, C.K.; Schierup, M.H.; Jensen, T.H. RNA exosome depletion reveals transcription upstream of active human promoters. Science 2008, 322, 1851–1854. [Google Scholar] [CrossRef]
- Pennisi, E. Chronicling embryos, cell by cell, gene by gene. Science 2018, 360, 367. [Google Scholar] [CrossRef] [PubMed]
- Harland, R.M. A new view of embryo development and regeneration. Science 2018, 360, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Wagner, D.E.; McKenna, A.; Pandey, S.; Klein, A.M.; Shendure, J.; Gagnon, J.A.; Schier, A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018, 36, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Olmos, D.; Arkenau, H.T.; Ang, J.E.; Ledaki, I.; Attard, G.; Carden, C.P.; Reid, A.H.; A’Hern, R.; Fong, P.C.; Oomen, N.B.; et al. Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): A single-centre experience. Ann. Oncol. 2009, 20, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A cancer cell program promotes t cell exclusion and resistance to checkpoint blockade. Cell 2018, 175, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Levitin, H.M.; Yuan, J.; Sims, P.A. Single-cell transcriptomic analysis of tumor heterogeneity. Trends Cancer 2018, 4, 264–268. [Google Scholar] [CrossRef]
- Ranzoni, A.M.; Strzelecka, P.M.; Cvejic, A. Application of single-cell RNA sequencing methodologies in understanding haematopoiesis and immunology. Essays Biochem. 2019, 63, 217–225. [Google Scholar] [CrossRef]
- Stephenson, W.; Donlin, L.T.; Butler, A.; Rozo, C.; Bracken, B.; Rashidfarrokhi, A.; Goodman, S.M.; Ivashkiv, L.B.; Bykerk, V.P.; Orange, D.E.; et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat. Commun. 2018, 9, 791. [Google Scholar] [CrossRef]
- Bossel Ben-Moshe, N.; Hen-Avivi, S.; Levitin, N.; Yehezkel, D.; Oosting, M.; Joosten, L.A.B.; Netea, M.G.; Avraham, R. Predicting bacterial infection outcomes using single cell RNA-sequencing analysis of human immune cells. Nat. Commun. 2019, 10, 3266. [Google Scholar] [CrossRef]
- Chiu, I.M.; Barrett, L.B.; Williams, E.K.; Strochlic, D.E.; Lee, S.; Weyer, A.D.; Lou, S.; Bryman, G.S.; Roberson, D.P.; Ghasemlou, N.; et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. Elife 2014, 3, e04660. [Google Scholar] [CrossRef]
- Shekhar, K.; Lapan, S.W.; Whitney, I.E.; Tran, N.M.; Macosko, E.Z.; Kowalczyk, M.; Adiconis, X.; Levin, J.Z.; Nemesh, J.; Goldman, M.; et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 2016, 166, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lonnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, W.; Gao, L.; Zhao, L. Genome-wide profiling of long non-coding RNAs from tomato and a comparison with mRNAs associated with the regulation of fruit ripening. BMC Plant Biol. 2018, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Bon, C.; Luffarelli, R.; Russo, R.; Fortuni, S.; Pierattini, B.; Santulli, C.; Fimiani, C.; Persichetti, F.; Cotella, D.; Mallamaci, A.; et al. SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 2019, 47, 10728–10743. [Google Scholar] [CrossRef]
- Espinoza, S.; Scarpato, M.; Damiani, D.; Manago, F.; Mereu, M.; Contestabile, A.; Peruzzo, O.; Carninci, P.; Santoro, C.; Papaleo, F.; et al. SINEUP Non-coding RNA Targeting GDNF rescues motor deficits and neurodegeneration in a mouse model of parkinson’s disease. Mol. Ther. 2019. [Google Scholar] [CrossRef]
- Podbevsek, P.; Fasolo, F.; Bon, C.; Cimatti, L.; Reisser, S.; Carninci, P.; Bussi, G.; Zucchelli, S.; Plavec, J.; Gustincich, S. Structural determinants of the SINE B2 element embedded in the long non-coding RNA activator of translation AS Uchl1. Sci. Rep. 2018, 8, 3189. [Google Scholar] [CrossRef]
- Takahashi, H.; Kozhuharova, A.; Sharma, H.; Hirose, M.; Ohyama, T.; Fasolo, F.; Yamazaki, T.; Cotella, D.; Santoro, C.; Zucchelli, S.; et al. Identification of functional features of synthetic SINEUPs, antisense lncRNAs that specifically enhance protein translation. PLoS ONE 2018, 13, e0183229. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef]
- Dieci, G.; Fiorino, G.; Castelnuovo, M.; Teichmann, M.; Pagano, A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007, 23, 614–622. [Google Scholar] [CrossRef]
- Lafontaine, D.L.; Tollervey, D. The function and synthesis of ribosomes. Nat. Rev. Mol. Cell Biol. 2001, 2, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Bhagavan, N.V.; Ha, C.-E. RNA and protein synthesis. In Essentials of Medical Biochemistry; Academic Press: Cambridge, MA, USA, 2011; pp. 301–320. [Google Scholar] [CrossRef]
- Cramer, P.; Armache, K.J.; Baumli, S.; Benkert, S.; Brueckner, F.; Buchen, C.; Damsma, G.E.; Dengl, S.; Geiger, S.R.; Jasiak, A.J.; et al. Structure of eukaryotic RNA polymerases. Annu. Rev. Biophys. 2008, 37, 337–352. [Google Scholar] [CrossRef]
- Holmes, D.S.; Mayfield, J.E.; Sander, G.; Bonner, J. Chromosomal RNA: Its properties. Science 1972, 177, 72–74. [Google Scholar] [CrossRef]
- Shao, Z.; Raible, F.; Mollaaghababa, R.; Guyon, J.R.; Wu, C.T.; Bender, W.; Kingston, R.E. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 1999, 98, 37–46. [Google Scholar] [CrossRef]
- Levine, S.S.; Weiss, A.; Erdjument-Bromage, H.; Shao, Z.; Tempst, P.; Kingston, R.E. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol. Cell. Biol. 2002, 22, 6070–6078. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. Elife 2012, 1, e00205. [Google Scholar] [CrossRef]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Chiang, A.; Bender, W.; Shimell, M.J.; O’Connor, M. Elements of the Drosophila bithorax complex that mediate repression by Polycomb group products. Dev. Biol. 1993, 158, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef]
- van Kruijsbergen, I.; Hontelez, S.; Veenstra, G.J. Recruiting polycomb to chromatin. Int. J. Biochem. Cell Biol. 2015, 67, 177–187. [Google Scholar] [CrossRef]
- Maclary, E.; Hinten, M.; Harris, C.; Kalantry, S. Long nonoding RNAs in the X-inactivation center. Chromosome Res. 2013, 21, 601–614. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Patop, I.L.; Wust, S.; Kadener, S. Past, present, and future of circRNAs. EMBO J. 2019, 38, e100836. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Kadonaga, J.T. Going the distance: A current view of enhancer action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef]
- Banerji, J.; Rusconi, S.; Schaffner, W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 1981, 27, 299–308. [Google Scholar] [CrossRef]
- Chen, H.; Du, G.; Song, X.; Li, L. Non-coding transcripts from enhancers: New insights into enhancer activity and gene expression regulation. Genom. Proteom. Bioinform. 2017, 15, 201–207. [Google Scholar] [CrossRef]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef]
- Schaukowitch, K.; Joo, J.Y.; Liu, X.; Watts, J.K.; Martinez, C.; Kim, T.K. Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 2014, 56, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Koch, F.; Fenouil, R.; Gut, M.; Cauchy, P.; Albert, T.K.; Zacarias-Cabeza, J.; Spicuglia, S.; de la Chapelle, A.L.; Heidemann, M.; Hintermair, C.; et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol. 2011, 18, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; Andrau, J.C. Noncoding transcription at enhancers: General principles and functional models. Annu. Rev. Genet. 2012, 46, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Cotella, D.; Takahashi, H.; Carrieri, C.; Cimatti, L.; Fasolo, F.; Jones, M.H.; Sblattero, D.; Sanges, R.; Santoro, C.; et al. SINEUPs: A new class of natural and synthetic antisense long non-coding RNAs that activate translation. RNA Biol. 2015, 12, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Fasolo, F.; Russo, R.; Cimatti, L.; Patrucco, L.; Takahashi, H.; Jones, M.H.; Santoro, C.; Sblattero, D.; Cotella, D.; et al. SINEUPs are modular antisense long non-coding RNAs that increase synthesis of target proteins in cells. Front. Cell. Neurosci. 2015, 9, 174. [Google Scholar] [CrossRef]
- Takahashi, H.; Sharma, H.; Carninci, P. Cell based assays of SINEUP non-coding RNAs that can specifically enhance mRNA translation. J. Vis. Exp. 2019, 144, e58627. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- Ulveling, D.; Francastel, C.; Hube, F. When one is better than two: RNA with dual functions. Biochimie 2011, 93, 633–644. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Xie, S.; Liu, Y.; Xie, Z. Global and cell-type specific properties of lincRNAs with ribosome occupancy. Nucleic Acids Res. 2017, 45, 2786–2796. [Google Scholar] [CrossRef] [PubMed]
- Bazzini, A.A.; Johnstone, T.G.; Christiano, R.; Mackowiak, S.D.; Obermayer, B.; Fleming, E.S.; Vejnar, C.E.; Lee, M.T.; Rajewsky, N.; Walther, T.C.; et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014, 33, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Pasut, A.; Matsumoto, M.; Yamashita, R.; Fung, J.; Monteleone, E.; Saghatelian, A.; Nakayama, K.I.; Clohessy, J.G.; Pandolfi, P.P. mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 2017, 541, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vashisht, A.A.; O’Rourke, J.; Corbel, S.Y.; Moran, R.; Romero, A.; Miraglia, L.; Zhang, J.; Durrant, E.; Schmedt, C.; et al. The microprotein Minion controls cell fusion and muscle formation. Nat. Commun. 2017, 8, 15664. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Z.; Chen, M.; Chen, D.; Gao, X.C.; Zhu, S.; Huang, H.; Hu, M.; Zhu, H.; Yan, G.R. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol. Cell 2017, 68, 171–184. [Google Scholar] [CrossRef]
- Hanyu-Nakamura, K.; Sonobe-Nojima, H.; Tanigawa, A.; Lasko, P.; Nakamura, A. Drosophila Pgc protein inhibits P-TEFb recruitment to chromatin in primordial germ cells. Nature 2008, 451, 730–733. [Google Scholar] [CrossRef]
- Choi, S.W.; Kim, H.W.; Nam, J.W. The small peptide world in long noncoding RNAs. Brief. Bioinform. 2018, 20, 1853–1864. [Google Scholar] [CrossRef]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2019. [Google Scholar] [CrossRef]
- Lubelsky, Y.; Ulitsky, I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature 2018, 555, 107–111. [Google Scholar] [CrossRef]
- Shukla, C.J.; McCorkindale, A.L.; Gerhardinger, C.; Korthauer, K.D.; Cabili, M.N.; Shechner, D.M.; Irizarry, R.A.; Maass, P.G.; Rinn, J.L. High-throughput identification of RNA nuclear enrichment sequences. EMBO J. 2018, 37, e98452. [Google Scholar] [CrossRef]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafreniere, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Truong, M. Single particle imaging of mRNAs crossing the nuclear pore: Surfing on the edge. Bioessays 2016, 38, 744–750. [Google Scholar] [CrossRef]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Rackham, O.; Shearwood, A.M.; Mercer, T.R.; Davies, S.M.; Mattick, J.S.; Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 2011, 17, 2085–2093. [Google Scholar] [CrossRef]
- Villegas, J.; Zarraga, A.M.; Muller, I.; Montecinos, L.; Werner, E.; Brito, M.; Meneses, A.M.; Burzio, L.O. A novel chimeric mitochondrial RNA localized in the nucleus of mouse sperm. DNA Cell Biol. 2000, 19, 579–588. [Google Scholar] [CrossRef]
- Villegas, J.; Araya, P.; Bustos-Obregon, E.; Burzio, L.O. Localization of the 16S mitochondrial rRNA in the nucleus of mammalian spermatogenic cells. Mol. Hum. Reprod. 2002, 8, 977–983. [Google Scholar] [CrossRef]
- Kobayashi, S.; Amikura, R.; Okada, M. Presence of mitochondrial large ribosomal RNA outside mitochondria in germ plasm of Drosophila melanogaster. Science 1993, 260, 1521–1524. [Google Scholar] [CrossRef]
- Kobayashi, S.; Amikura, R.; Mukai, M. Localization of mitochondrial large ribosomal RNA in germ plasm of Xenopus embryos. Curr. Biol. 1998, 8, 1117–1120. [Google Scholar] [CrossRef]
- Iida, T.; Kobayashi, S. Essential role of mitochondrially encoded large rRNA for germ-line formation in Drosophila embryos. Proc. Natl. Acad. Sci. USA 1998, 95, 11274–11278. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.; Burzio, V.; Villota, C.; Landerer, E.; Martinez, R.; Santander, M.; Martinez, R.; Pinto, R.; Vera, M.I.; Boccardo, E.; et al. Expression of a novel non-coding mitochondrial RNA in human proliferating cells. Nucleic Acids Res. 2007, 35, 7336–7347. [Google Scholar] [CrossRef] [PubMed]
- Burzio, V.A.; Villota, C.; Villegas, J.; Landerer, E.; Boccardo, E.; Villa, L.L.; Martinez, R.; Lopez, C.; Gaete, F.; Toro, V.; et al. Expression of a family of noncoding mitochondrial RNAs distinguishes normal from cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9430–9434. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Hotto, A.M.; Schmitz, R.J.; Fei, Z.; Ecker, J.R.; Stern, D.B. Unexpected Diversity of Chloroplast Noncoding RNAs as Revealed by Deep Sequencing of the Arabidopsis Transcriptome. G3 2011, 1, 559–570. [Google Scholar] [CrossRef]
- Rurek, M. Participation of non-coding RNAs in plant organelle biogenesis. Acta Biochim. Pol. 2016, 63, 653–663. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Lao, K.Q.; Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Tuch, B.; Bodeau, J.; et al. mRNA-sequencing whole transcriptome analysis of a single cell on the SOLiD system. J. Biomol. Tech. 2009, 20, 266–271. [Google Scholar]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for single-cell collection and analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef] [PubMed]
- Chemello, F.; Grespi, F.; Zulian, A.; Cancellara, P.; Hebert-Chatelain, E.; Martini, P.; Bean, C.; Alessio, E.; Buson, L.; Bazzega, M.; et al. Transcriptomic analysis of single isolated myofibers identifies miR-27a-3p and miR-142-3p as Regulators of metabolism in skeletal muscle. Cell Rep. 2019, 26, 3784–3797. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Buson, L.; Chemello, F.; Peggion, C.; Grespi, F.; Martini, P.; Massimino, M.L.; Pacchioni, B.; Millino, C.; Romualdi, C.; et al. Single cell analysis reveals the involvement of the long non-coding RNA Pvt1 in the modulation of muscle atrophy and mitochondrial network. Nucleic Acids Res. 2019, 47, 1653–1670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Gao, M.; Chong, Z.; Li, Y.; Han, X.; Chen, R.; Qin, L. Single-cell isolation by a modular single-cell pipette for RNA-sequencing. Lab Chip 2016, 16, 4742–4748. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, S.; MacLennan, G.T.; Williamson, S.R.; Davidson, D.D.; Wang, M.; Jones, T.D.; Lopez-Beltran, A.; Montironi, R. Laser-assisted microdissection in translational research: Theory, technical considerations, and future applications. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 31–47. [Google Scholar] [CrossRef]
- Nichterwitz, S.; Chen, G.; Aguila Benitez, J.; Yilmaz, M.; Storvall, H.; Cao, M.; Sandberg, R.; Deng, Q.; Hedlund, E. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 2016, 7, 12139. [Google Scholar] [CrossRef]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Stevens, M.; Oomens, L.; Broekmaat, J.; Weersink, J.; Abali, F.; Swennenhuis, J.; Tibbe, A. VyCAP’s puncher technology for single cell identification, isolation, and analysis. Cytom. A 2018, 93, 1255–1259. [Google Scholar] [CrossRef]
- Wang, Y.; Phillips, C.; Xu, W.; Pai, J.H.; Dhopeshwarkar, R.; Sims, C.E.; Allbritton, N. Micromolded arrays for separation of adherent cells. Lab Chip 2010, 10, 2917–2924. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Trapani, M.; Manaresi, N.; Medoro, G. DEPArray system: An automatic image-based sorter for isolation of pure circulating tumor cells. Cytom. A 2018, 93, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Utada, A.S.; Lorenceau, E.; Link, D.R.; Kaplan, P.D.; Stone, H.A.; Weitz, D.A. Monodisperse double emulsions generated from a microcapillary device. Science 2005, 308, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Bakken, T.E.; Hodge, R.D.; Miller, J.A.; Yao, Z.; Nguyen, T.N.; Aevermann, B.; Barkan, E.; Bertagnolli, D.; Casper, T.; Dee, N.; et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 2018, 13, e0209648. [Google Scholar] [CrossRef]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Arauzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lonnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015, 16, 148. [Google Scholar] [CrossRef]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 2017, 14, 267–270. [Google Scholar] [CrossRef]
- Hayashi, T.; Ozaki, H.; Sasagawa, Y.; Umeda, M.; Danno, H.; Nikaido, I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat. Commun. 2018, 9, 619. [Google Scholar] [CrossRef]
- Faridani, O.R.; Abdullayev, I.; Hagemann-Jensen, M.; Schell, J.P.; Lanner, F.; Sandberg, R. Single-cell sequencing of the small-RNA transcriptome. Nat. Biotechnol. 2016, 34, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Wellenreuther, R.; Schupp, I.; Poustka, A.; Wiemann, S. SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genom. 2004, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Biscontin, A.; Casara, S.; Cagnin, S.; Tombolan, L.; Rosolen, A.; Lanfranchi, G.; De Pitta, C. New miRNA labeling method for bead-based quantification. BMC Mol. Biol. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. RNA amplification for successful gene profiling analysis. J. Transl. Med. 2005, 3, 28. [Google Scholar] [CrossRef]
- Day, R.C.; McNoe, L.; Macknight, R.C. Evaluation of global RNA amplification and its use for high-throughput transcript analysis of laser-microdissected endosperm. Int. J. Plant Genom. 2007, 2007, 61028. [Google Scholar] [CrossRef][Green Version]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- White, A.K.; VanInsberghe, M.; Petriv, O.I.; Hamidi, M.; Sikorski, D.; Marra, M.A.; Piret, J.; Aparicio, S.; Hansen, C.L. High-throughput microfluidic single-cell RT-qPCR. Proc. Natl. Acad. Sci. USA 2011, 108, 13999–14004. [Google Scholar] [CrossRef]
- Hebenstreit, D. Methods, Challenges and Potentials of Single Cell RNA-seq. Biology 2012, 1, 658–667. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef] [PubMed]
- Ballarino, M.; Morlando, M.; Fatica, A.; Bozzoni, I. Non-coding RNAs in muscle differentiation and musculoskeletal disease. J. Clin. Investig. 2016, 126, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lonnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, J.; Zhang, L.; Liu, S.; Ma, Y.; Wen, X.; Hao, J.; Li, Z.; Ni, Y.; Li, X.; et al. Combined Single-cell profiling of lncRNAs and functional screening reveals that H19 is pivotal for embryonic hematopoietic stem cell development. Cell Stem Cell 2019, 24, 285–298. [Google Scholar] [CrossRef]
- Wu, Z.; Gao, S.; Zhao, X.; Chen, J.; Keyvanfar, K.; Feng, X.; Kajigaya, S.; Young, N.S. Long noncoding RNAs of single hematopoietic stem and progenitor cells in healthy and dysplastic human bone marrow. Haematologica 2019, 104, 894–906. [Google Scholar] [CrossRef]
- Kern, C.; Wang, Y.; Chitwood, J.; Korf, I.; Delany, M.; Cheng, H.; Medrano, J.F.; Van Eenennaam, A.L.; Ernst, C.; Ross, P.; et al. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genom. 2018, 19, 684. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Iyer, M.K.; Stuart, P.E.; Swindell, W.R.; Gudjonsson, J.E.; Tejasvi, T.; Sarkar, M.K.; Li, B.; Ding, J.; Voorhees, J.J.; et al. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol. 2015, 16, 24. [Google Scholar] [CrossRef]
- Schor, I.E.; Bussotti, G.; Males, M.; Forneris, M.; Viales, R.R.; Enright, A.J.; Furlong, E.E.M. Non-coding RNA Expression, Function, and Variation during Drosophila Embryogenesis. Curr. Biol. 2018, 28, 3547–3561. [Google Scholar] [CrossRef]
- Hartshorn, C.; Rice, J.E.; Wangh, L.J. Differential pattern of Xist RNA accumulation in single blastomeres isolated from 8-cell stage mouse embryos following laser zona drilling. Mol. Reprod. Dev. 2003, 64, 41–51. [Google Scholar] [CrossRef]
- Hartshorn, C.; Anshelevich, A.; Wangh, L.J. Rapid, single-tube method for quantitative preparation and analysis of RNA and DNA in samples as small as one cell. BMC Biotechnol. 2005, 5, 2. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Pierce, K.E.; Rice, J.E.; Wangh, L.J. Linear-after-the-exponential (LATE)-PCR: An advanced method of asymmetric PCR and its uses in quantitative real-time analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, C.; Eckert, J.J.; Hartung, O.; Wangh, L.J. Single-cell duplex RT-LATE-PCR reveals Oct4 and Xist RNA gradients in 8-cell embryos. BMC Biotechnol. 2007, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Hansis, C.; Tang, Y.X.; Grifo, J.A.; Krey, L.C. Analysis of Oct-4 expression and ploidy in individual human blastomeres. Mol. Hum. Reprod. 2001, 7, 155–161. [Google Scholar] [CrossRef][Green Version]
- Petropoulos, S.; Edsgard, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Plaza Reyes, A.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-cell RNA-seq reveals lineage and x chromosome dynamics in human preimplantation embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Marinov, G.K.; Pepke, S.; Singer, Z.S.; He, P.; Williams, B.; Schroth, G.P.; Elowitz, M.B.; Wold, B.J. Single-cell transcriptome analysis reveals dynamic changes in lncRNA expression during reprogramming. Cell Stem Cell 2015, 16, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Briggs, S.F.; Dominguez, A.A.; Chavez, S.L.; Reijo Pera, R.A. Single-cell XIST expression in human preimplantation embryos and newly reprogrammed female induced pluripotent stem cells. Stem Cells 2015, 33, 1771–1781. [Google Scholar] [CrossRef]
- Wainer Katsir, K.; Linial, M. Human genes escaping X-inactivation revealed by single cell expression data. BMC Genom. 2019, 20, 201. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Griffiths, J.A.; Scialdone, A.; Marioni, J.C. Using single-cell genomics to understand developmental processes and cell fate decisions. Mol. Syst. Biol. 2018, 14, e8046. [Google Scholar] [CrossRef]
- Rizvi, A.H.; Camara, P.G.; Kandror, E.K.; Roberts, T.J.; Schieren, I.; Maniatis, T.; Rabadan, R. Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat. Biotechnol. 2017, 35, 551–560. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Nordman, E.; Li, B.; Xu, N.; Bashkirov, V.I.; Lao, K.; Surani, M.A. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat. Protoc. 2010, 5, 516–535. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Nordman, E.; Bao, S.; Lee, C.; Wang, X.; Tuch, B.B.; Heard, E.; Lao, K.; Surani, M.A. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE 2011, 6, e21208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Huang, K.; Luo, Y.; Li, S. Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genom. 2014, 15, 845. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.J.; Ren, Z.R.; Yan, J.B. Identification and functional analysis of long non-coding RNAs in human and mouse early embryos based on single-cell transcriptome data. Oncotarget 2016, 7, 61215–61228. [Google Scholar] [CrossRef]
- Johnson, M.B.; Wang, P.P.; Atabay, K.D.; Murphy, E.A.; Doan, R.N.; Hecht, J.L.; Walsh, C.A. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat. Neurosci. 2015, 18, 637–646. [Google Scholar] [CrossRef]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef]
- Spaethling, J.M.; Na, Y.J.; Lee, J.; Ulyanova, A.V.; Baltuch, G.H.; Bell, T.J.; Brem, S.; Chen, H.I.; Dueck, H.; Fisher, S.A.; et al. Primary cell culture of live neurosurgically resected aged adult human brain cells and single cell transcriptomics. Cell Rep. 2017, 18, 791–803. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Cancer epigenetics: From disruption of differentiation programs to the emergence of cancer stem cells. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 251–258. [Google Scholar] [CrossRef]
- Naugler, C.T. Population genetics of cancer cell clones: Possible implications of cancer stem cells. Theor. Biol. Med. Model. 2010, 7, 42. [Google Scholar] [CrossRef]
- Wang, J.; Roy, B. Single-cell RNA-seq reveals lincRNA expression differences in Hela-S3 cells. Biotechnol. Lett. 2017, 39, 359–366. [Google Scholar] [CrossRef]
- Pang, B.; Xu, J.; Hu, J.; Guo, F.; Wan, L.; Cheng, M.; Pang, L. Single-cell RNA-seq reveals the invasive trajectory and molecular cascades underlying glioblastoma progression. Mol. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, T.; Yang, Y.; Zheng, S. Tumor heterogeneity uncovered by dynamic expression of long noncoding RNA at single-cell resolution. Cancer Genet. 2015, 208, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Konikoff, C.; Karr, T.L.; Kumar, S. Relationship between gene co-expression and sharing of transcription factor binding sites in Drosophila melanogaster. Bioinformatics 2009, 25, 2473–2477. [Google Scholar] [CrossRef] [PubMed]
- Triska, M.; Ivliev, A.; Nikolsky, Y.; Tatarinova, T.V. Analysis of cis-regulatory elements in gene co-expression networks in cancer. Methods Mol. Biol. 2017, 1613, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Sequence Read Archive. Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 6 November 2019).
- Gene Expression Omnibus. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 6 November 2019).
- Single Cell Expression Atlas. Available online: https://www.ebi.ac.uk/gxa/sc/home (accessed on 6 November 2019).
- Single-Cell Centric Database. Available online: http://single-cell.clst.riken.jp/ (accessed on 6 November 2019).
- Abugessaisa, I.; Noguchi, S.; Bottcher, M.; Hasegawa, A.; Kouno, T.; Kato, S.; Tada, Y.; Ura, H.; Abe, K.; Shin, J.W.; et al. SCPortalen: Human and mouse single-cell centric database. Nucleic Acids Res. 2018, 46, D781–D787. [Google Scholar] [CrossRef]
- Single Cell Portal. Available online: https://singlecell.broadinstitute.org/single_cell (accessed on 6 November 2019).
- PanglaoDB. Available online: https://panglaoda.se/ (accessed on 6 November 2019).
- Single Cell Database. Available online: http://betsholtzlab.org/VascularSingleCells/database.html (accessed on 6 November 2019).
- JingleBells. Available online: http://jinglebells.bgu.ac.il/nonimmune/ (accessed on 6 November 2019).
- Allen Brain Map. Available online: https://portal.brain-map.org/atlases-and-data/rnaseq (accessed on 6 November 2019).
- Human Single Cell RNA Seq. Available online: https://bioinfo.uth.edu/scrnaseqdb/ (accessed on 6 November 2019).
- Datasets Obtained with Nadia Protocol. Available online: https://www.dolomite-bio.com/support/single-cell-drop-seq-data/ (accessed on 6 November 2019).
- Sanger Institute DB. Available online: https://hemberg-lab.github.io/scRNA.seq.datasets/ (accessed on 6 November 2019).
- BioTuring. Available online: https://bioturing.com/bbrowser (accessed on 6 November 2019).
- Cancer Single Cell ATLAS. Available online: http://biocc.hrbmu.edu.cn/CancerCEA/ (accessed on 6 November 2019).
- Ensembl. Available online: http://www.ensembl.org/index.html (accessed on 6 November 2019).
- RefSeq NCBI Reference Sequence Database. Available online: https://www.ncbi.nlm.nih.gov/refseq/ (accessed on 6 November 2019).
- GENCODE. Available online: https://www.gencodegenes.org/ (accessed on 6 November 2019).
- NONCODE. Available online: http://www.noncode.org/introduce.php (accessed on 6 November 2019).
- LINCIPEDIA. Available online: https://lncipedia.org/ (accessed on 6 November 2019).
- Experimentally Validated LncRNAs. Available online: http://biophy.dzu.edu.cn/EVLncRNAs/ (accessed on 6 November 2019).
- The MOuse NonCode Lung Database. Available online: https://www.monocldb.org/ (accessed on 6 November 2019).
- Martini, P.; Paracchini, L.; Caratti, G.; Mello-Grand, M.; Fruscio, R.; Beltrame, L.; Calura, E.; Sales, G.; Ravaggi, A.; Bignotti, E.; et al. lncRNAs as novel indicators of patients’ prognosis in stage I epithelial ovarian cancer: A retrospective and multicentric study. Clin. Cancer Res. 2017, 23, 2356–2366. [Google Scholar] [CrossRef]
- Genomics of Long Noncoding RNA and Disease. Available online: https://www.gold-lab.org/ (accessed on 6 November 2019).
- The LncRNA and Disease Database. Available online: http://www.cuilab.cn/lncrnadisease (accessed on 6 November 2019).
- Mammalian NcRNA-Disease Repository. Available online: http://www.rna-society.org/mndr/ (accessed on 6 November 2019).
- Zebrafish LncRNA Database. Available online: http://www.zflnc.org/ (accessed on 6 November 2019).
- Pig LncRNA net. Available online: http://lnc.rnanet.org/ (accessed on 6 November 2019).
- LncRNA in Plants. Available online: http://cantata.amu.edu.pl/ (accessed on 6 November 2019).
- LncATLAS. Available online: http://lncatlas.crg.eu/ (accessed on 6 November 2019).
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Fernandes, J.C.R.; Acuna, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long non-coding rnas in the regulation of gene expression: Physiology and disease. Noncoding RNA 2019, 5, 17. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Wu, F.; Liu, Y.; Wu, Q.; Li, D.; Zhang, L.; Wu, X.; Wang, R.; Zhang, D.; Gao, S.; Li, W. Long non-coding RNAs potentially function synergistically in the cellular reprogramming of SCNT embryos. BMC Genom. 2018, 19, 631. [Google Scholar] [CrossRef]
- Martinez, N.J.; Gregory, R.I. MicroRNA gene regulatory pathways in the establishment and maintenance of ESC identity. Cell Stem Cell 2010, 7, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Luginbuhl, J.; Sivaraman, D.M.; Shin, J.W. The essentiality of non-coding RNAs in cell reprogramming. Noncoding RNA Res. 2017, 2, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Gawronski, K.A.B.; Kim, J. Single cell transcriptomics of noncoding RNAs and their cell-specificity. Wiley Interdiscip. Rev. RNA 2017, 8, e1433. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355, eaah7111. [Google Scholar] [CrossRef]
- Kim, J.; Eberwine, J. RNA: State memory and mediator of cellular phenotype. Trends Cell Biol. 2010, 20, 311–318. [Google Scholar] [CrossRef][Green Version]
- Kim, K.T.; Lee, H.W.; Lee, H.O.; Kim, S.C.; Seo, Y.J.; Chung, W.; Eum, H.H.; Nam, D.H.; Kim, J.; Joo, K.M.; et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015, 16, 127. [Google Scholar] [CrossRef]
- Marx, V. A dream of single-cell proteomics. Nat. Methods 2019, 16, 809–812. [Google Scholar] [CrossRef]
- Xiao, Z.; Cheng, G.; Jiao, Y.; Pan, C.; Li, R.; Jia, D.; Zhu, J.; Wu, C.; Zheng, M.; Jia, J. Holo-Seq: Single-cell sequencing of holo-transcriptome. Genome Biol. 2018, 19, 163. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Zhang, Y.; Liu, J. Advancing single-cell proteomics and metabolomics with microfluidic technologies. Analyst 2019, 144, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, V.; Corrado, B.; Sbrescia, S.; Sibilio, S.; Urciuolo, F.; Netti, P.A.; Imparato, G. Intestine-on-chip device increases ECM remodeling inducing faster epithelial cell differentiation. Biotechnol. Bioeng. 2019. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Cho, S.; Discher, D.E. Stem cell differentiation is regulated by extracellular matrix mechanics. Physiology 2018, 33, 16–25. [Google Scholar] [CrossRef]
- Hu, B.C. The human body at cellular resolution: The NIH human biomolecular atlas program. Nature 2019, 574, 187–192. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Micropipette Isolation | LCM | FACS | Capillary Based | Punching Technology | DEP | High-Throughput Droplet-Based | Low-Throughput Droplet-Based |
|---|---|---|---|---|---|---|---|---|
| Main Platforms | N/A | Several Platforms | Several Platforms | AVISO CellCelector | CellRaft AIR | DEPArray NxT | Chromium System | C1 System |
| Nadia | ||||||||
| Puncher Platform | InDrop System | |||||||
| ddSEQ Single-Cell Isolator | ||||||||
| Throughput | Low | Low | High | Low (<100 cells) | Low (<100 cells) | Low (<100 cells) | High (between 6 k and 10 k cells) | Low (<800 cells) |
| Visual Control | Yes | Yes | No | Yes | Yes | Yes | No | Yes |
| Cell selection | Yes (morphologically) | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Input of cells | Low | Low | High | Medium | Medium | Medium | High | Medium |
| Advantages | Low cost | Spatial information, storage of tissue | Capture rare cells, fast analysis | Low price of consumables | Active cell selection, high transfer efficiency | Active cell selection, cell–cell interaction analysis | Suitable for processing a high number of cells | Suitable for RNA-Seq, DNA-Seq, miRNA-Seq, epigenomics, qRT-PCR analysis |
| Disadvantages | Laborious, low efficiency | Fixatives can damage RNA and introduce bias | Require antibodies/molecular markers | Require skills to operate, bioinformatics not provided | Bioinformatic analysis not provided | High price of consumables | High cost, profiles of only polyadenylated RNAs (need specific developed protocols for example miRNAs that are not polyadenylated) | Size-based cell selection |
| Database Name | Organisms | Reference |
|---|---|---|
| PanglaoDB | Mouse, human | [182] |
| Single-cell database | Mouse | [183] |
| JingleBells | Different organisms (Mouse, human, zebrafish, brown rat) | [184] |
| Brain atlas | Mouse, human | [185] |
| Single-cell RNA sequencing | Human | [186] |
| Single-cell data with Nadia (DolomiteBio) | [187] | |
| Sanger institute experiments | Mouse, human | [188] |
| BioTuring | [189] | |
| Cancer single-cell atlas | Human | [190] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessio, E.; Bonadio, R.S.; Buson, L.; Chemello, F.; Cagnin, S. A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 302. https://doi.org/10.3390/ijms21010302
Alessio E, Bonadio RS, Buson L, Chemello F, Cagnin S. A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs. International Journal of Molecular Sciences. 2020; 21(1):302. https://doi.org/10.3390/ijms21010302
Chicago/Turabian StyleAlessio, Enrico, Raphael Severino Bonadio, Lisa Buson, Francesco Chemello, and Stefano Cagnin. 2020. "A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs" International Journal of Molecular Sciences 21, no. 1: 302. https://doi.org/10.3390/ijms21010302
APA StyleAlessio, E., Bonadio, R. S., Buson, L., Chemello, F., & Cagnin, S. (2020). A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs. International Journal of Molecular Sciences, 21(1), 302. https://doi.org/10.3390/ijms21010302