Alpha-Tocotrienol Prevents Oxidative Stress-Mediated Post-Translational Cleavage of Bcl-xL in Primary Hippocampal Neurons


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
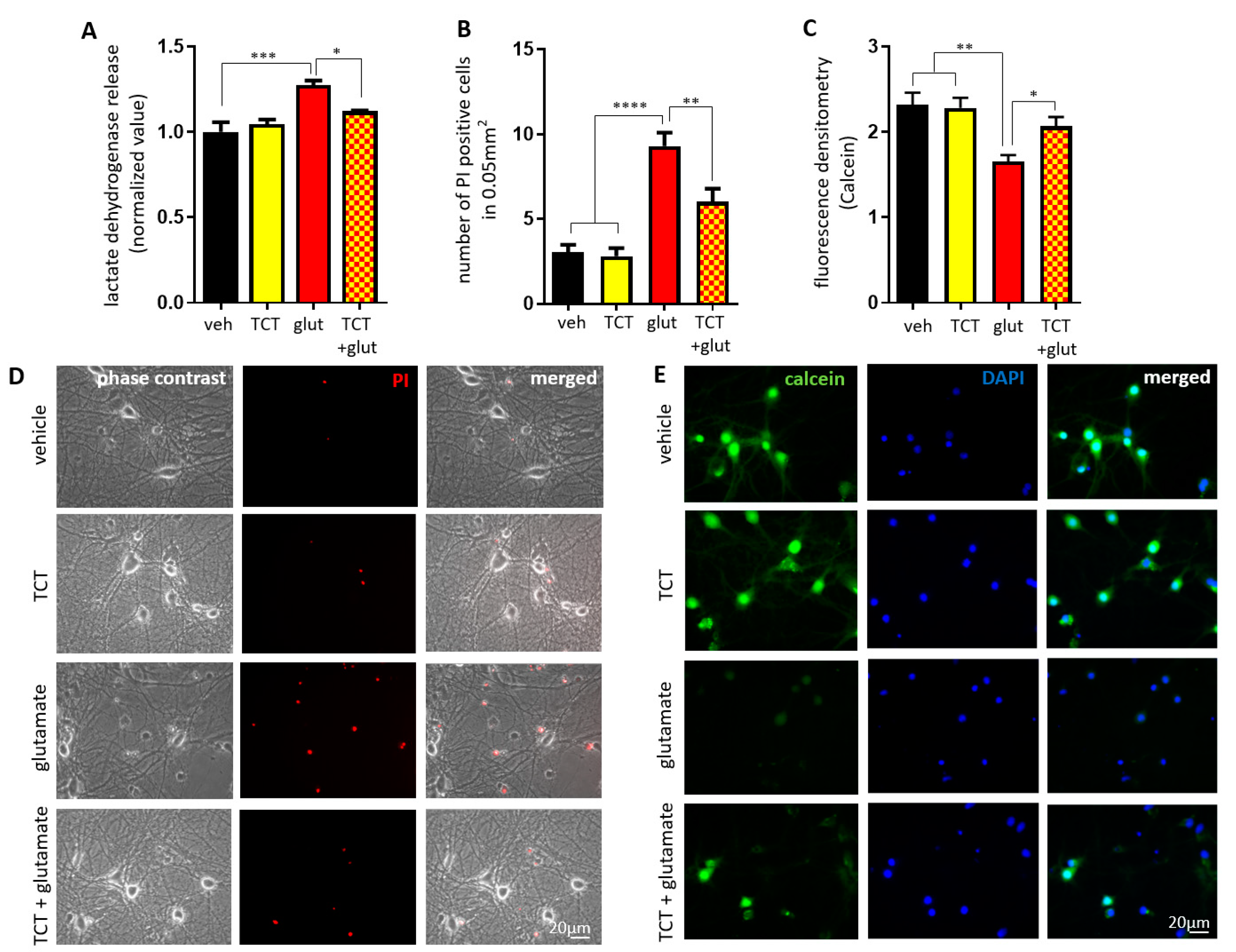
2.1. α-TCT Protects Primary Hippocampal Neurons against Excitotoxicity
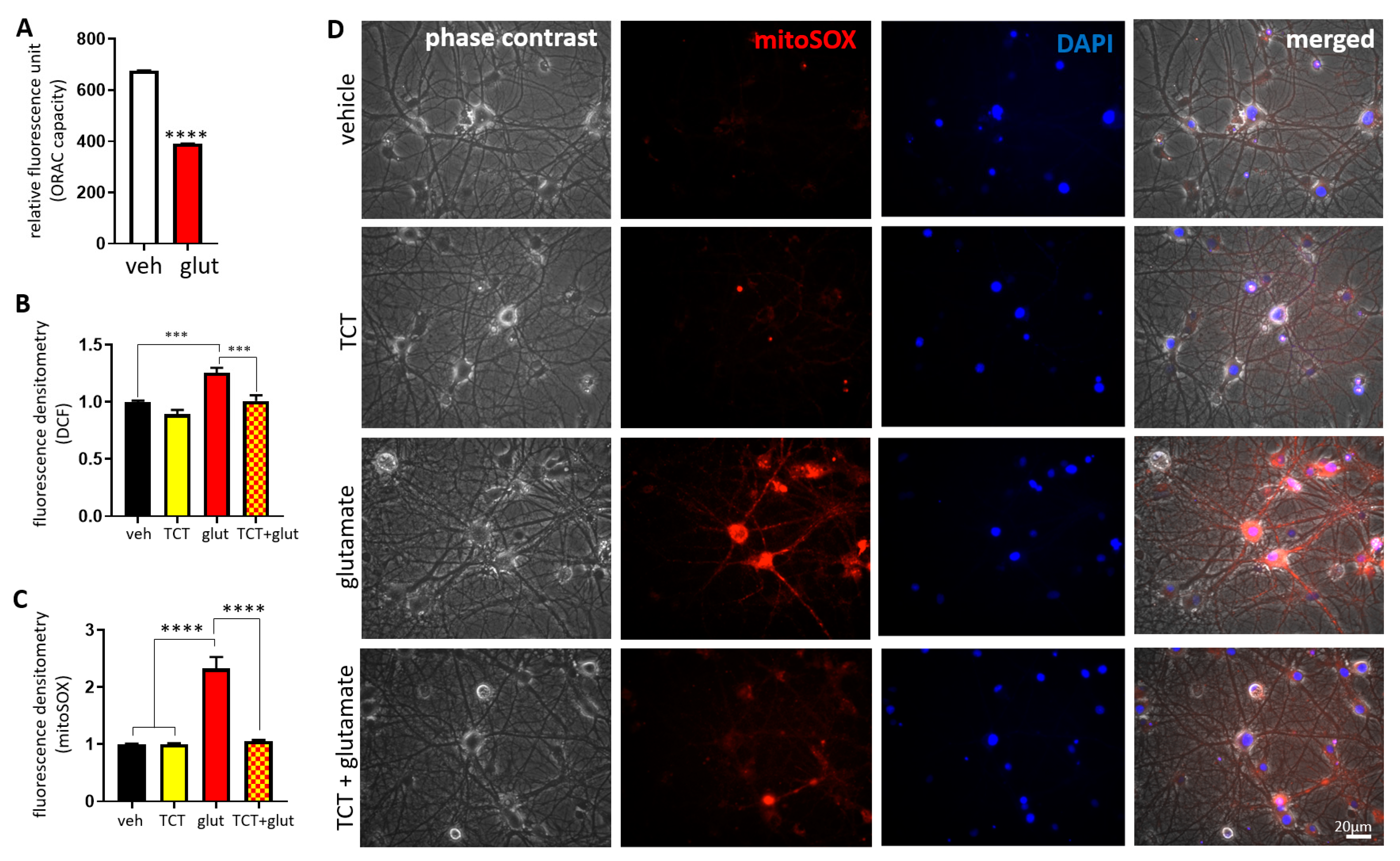
2.2. α-TCT Attenuates Glutamate-Induced Oxidative Stress in the Mitochondria.
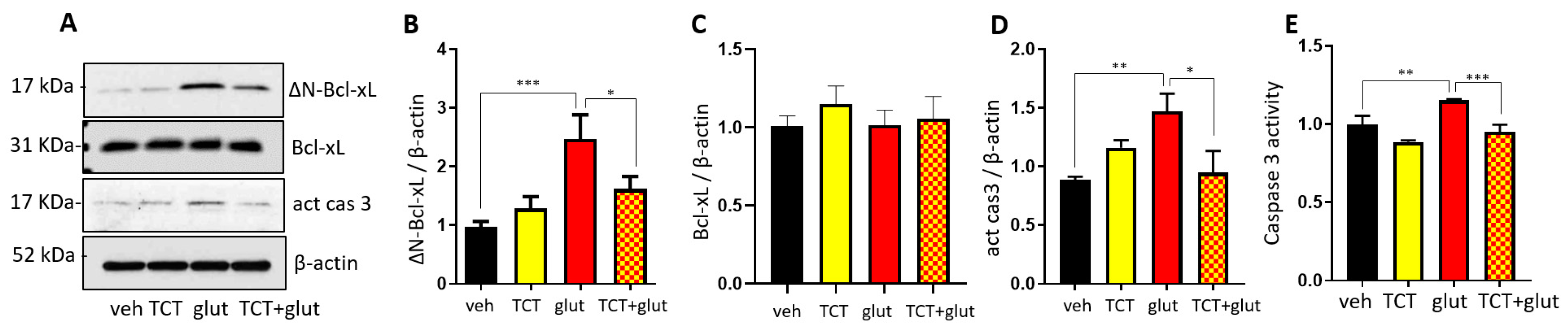
2.3. α-TCT Decreases Mitochondrial Formation of ∆N-Bcl-xL in Primary Hippocampal Neurons
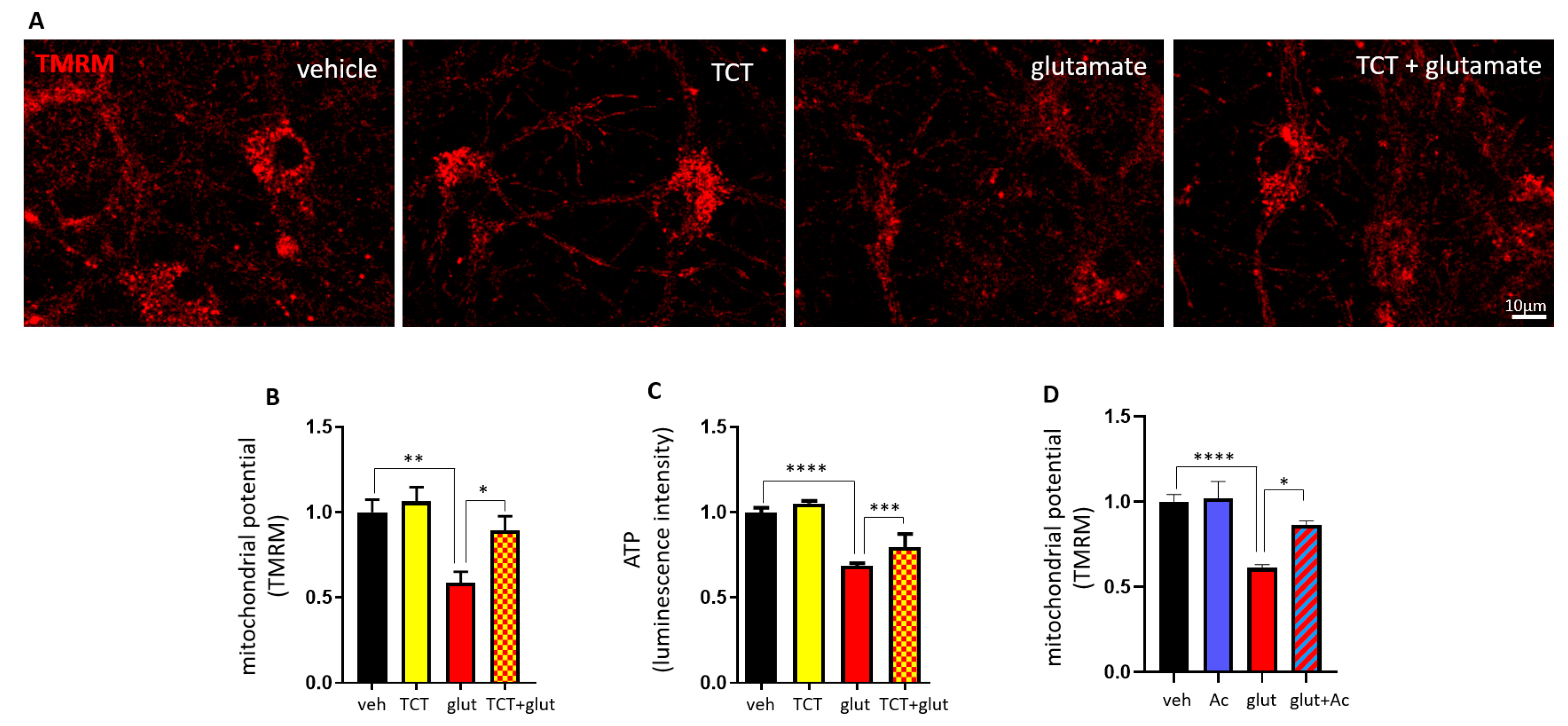
2.4. α-TCT Prevents ∆N-Bcl-xL-Induced Mitochondrial Dysfunction.
3. Discussion
4. Materials and Methods
4.1. Culture of Primary Hippocampal Neurons
4.1.1. α-tocotrienol (TCT) Treatment
4.1.2. Glutamate Treatment
4.1.3. Ac-DEVD-CHO Treatment
4.2. Viability Assay
4.2.1. Lactate Dehydrogenase (LDH) Assay
4.2.2. Calcein-AM and Propidium Iodide (PI)
4.3. Measurement of Mitochondrial Potential (Δψ)
4.4. Measurement of ATP Production
4.5. ROS Measurement
4.5.1. 2′,7′-dichlorodihydrofluorescein Diacetate (H2DCFDA) Staining
4.5.2. mitoSOX Staining
4.5.3. Antioxidant Capacity
4.6. Immunoblots
4.7. Caspase 3 Activity
4.8. Homology Modeling and Protein–Ligand Docking
4.9. Cloning and Purification of ∆N-Bcl-xL Recombinant Proteins
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Bcl-xL | B-cell lymphoma—extra large |
| TCT | α-tocotrienol |
| ROS | reactive oxygen species |
| LDH | lactate dehydrogenase |
| PI | propidium iodide |
| DCF | 2′,7′-dichlorofluorescein |
| TMRM | tetramethylrhodamine |
References
- Nakamura, A.; Swahari, V.; Plestant, C.; Smith, I.; McCoy, E.; Smith, S.; Moy, S.S.; Anton, E.S.; Deshmukh, M. Bcl-xl is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J. Neurosci. 2016, 36, 5448–5461. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of bcl-xl-bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.; Liang, C.; Jung, J.U.; Oh, B.H. Evidence that inhibition of bax activation by bcl-2 involves its tight and preferential interaction with the bh3 domain of bax. Cell Res. 2011, 21, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Soane, L.; Siegel, Z.T.; Schuh, R.A.; Fiskum, G. Postnatal developmental regulation of bcl-2 family proteins in brain mitochondria. J. Neurosci. Res. 2008, 86, 1267–1276. [Google Scholar] [CrossRef]
- Ivanovska, I.; Galonek, H.L.; Hildeman, D.A.; Hardwick, J.M. Regulation of cell death in the lymphoid system by bcl-2 family proteins. Acta Haematol. 2004, 111, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Levine, B.; Boise, L.H.; Thompson, C.B.; Hardwick, J.M. Bax-independent inhibition of apoptosis by bcl-xl. Nature 1996, 379, 554–556. [Google Scholar] [CrossRef]
- Liu, X.; Dai, S.; Zhu, Y.; Marrack, P.; Kappler, J.W. The structure of a bcl-xl/bim fragment complex: Implications for bim function. Immunity 2003, 19, 341–352. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for bcl-xl and bcl-2, displaces bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xl regulates metabolic efficiency of neurons through interaction with the mitochondrial f1fo atp synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xl regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Chen, Y.B.; Jonas, E.A. Multipolar functions of bcl-2 proteins link energetics to apoptosis. Trends Cell Biol. 2012, 22, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Tornero, D.; Posadas, I.; Cena, V. Bcl-x(l) blocks a mitochondrial inner membrane channel and prevents ca2+ overload-mediated cell death. PLoS ONE 2011, 6, e20423. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Licznerski, P.; Alavian, K.N.; Shanabrough, M.; Jonas, E.A. Bcl-xl is necessary for neurite outgrowth in hippocampal neurons. Antioxid Redox Signal 2015, 22, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.; Kugler, S.; Happold, C.; Bahr, M.; Isenmann, S. Excess bcl-xl increases the intrinsic growth potential of adult cns neurons in vitro. Mol. Cell Neurosci. 2004, 26, 63–74. [Google Scholar] [CrossRef]
- Li, H.; Alavian, K.N.; Lazrove, E.; Mehta, N.; Jones, A.; Zhang, P.; Licznerski, P.; Graham, M.; Uo, T.; Guo, J.; et al. A bcl-xl-drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat. Cell Biol. 2013, 15, 773–785. [Google Scholar] [CrossRef]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-xl induces drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef]
- Jonas, E. Bcl-xl regulates synaptic plasticity. Mol. Interv. 2006, 6, 208–222. [Google Scholar] [CrossRef]
- Jonas, E.A.; Porter, G.A.; Alavian, K.N. Bcl-xl in neuroprotection and plasticity. Front Physiol. 2014, 5, 355. [Google Scholar] [CrossRef]
- Park, H.A.; Jonas, E. Mitochondrial regulators of synaptic plasticity in the ischemic brain. In Synaptic Plasticity; Heinbockel, T., Ed.; INTECH: London, UK, 2017; pp. 39–67. [Google Scholar]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In vivo delivery of a bcl-xl fusion protein containing the tat protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [CrossRef]
- Liu, X.H.; Collier, R.J.; Youle, R.J. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a bcl-xl fusion protein. J. Biol. Chem. 2001, 276, 46326–46332. [Google Scholar] [CrossRef]
- Ofengeim, D.; Chen, Y.B.; Miyawaki, T.; Li, H.; Sacchetti, S.; Flannery, R.J.; Alavian, K.N.; Pontarelli, F.; Roelofs, B.A.; Hickman, J.A.; et al. N-terminally cleaved bcl-xl mediates ischemia-induced neuronal death. Nat. Neurosci. 2012, 15, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, T.; Mashiko, T.; Ofengeim, D.; Flannery, R.J.; Noh, K.M.; Fujisawa, S.; Bonanni, L.; Bennett, M.V.; Zukin, R.S.; Jonas, E.A. Ischemic preconditioning blocks bad translocation, bcl-xl cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4892–4897. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J.; Cheng, E.H.; Karp, C.L.; Kirsch, D.G.; Ueno, K.; Takahashi, A.; Kastan, M.B.; Griffin, D.E.; Earnshaw, W.C.; Veliuona, M.A.; et al. Modulation of cell death by bcl-xl through caspase interaction. Proc. Natl. Acad. Sci. USA 1998, 95, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Nagahashi, A.; Nagashima, K.; Rokudai, S.; Tsuruo, T. Acceleration of apoptotic cell death after the cleavage of bcl-xl protein by caspase-3-like proteases. Oncogene 1998, 17, 1295–1304. [Google Scholar] [CrossRef]
- Park, H.A.; Licznerski, P.; Mnatsakanyan, N.; Niu, Y.; Sacchetti, S.; Wu, J.; Polster, B.M.; Alavian, K.N.; Jonas, E.A. Inhibition of bcl-xl prevents pro-death actions of deltan-bcl-xl at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ. 2017, 24, 1963–1974. [Google Scholar] [CrossRef]
- Park, H.A.; Jonas, E.A. Deltan-bcl-xl, a therapeutic target for neuroprotection. Neural. Regen Res. 2017, 12, 1791–1794. [Google Scholar] [CrossRef]
- Jonas, E.A.; Hickman, J.A.; Chachar, M.; Polster, B.M.; Brandt, T.A.; Fannjiang, Y.; Ivanovska, I.; Basanez, G.; Kinnally, K.W.; Zimmerberg, J.; et al. Proapoptotic n-truncated bcl-xl protein activates endogenous mitochondrial channels in living synaptic terminals. Proc. Natl. Acad. Sci. USA 2004, 101, 13590–13595. [Google Scholar] [CrossRef]
- Hickman, J.A.; Hardwick, J.M.; Kaczmarek, L.K.; Jonas, E.A. Bcl-xl inhibitor abt-737 reveals a dual role for bcl-xl in synaptic transmission. J. Neurophysiol. 2008, 99, 1515–1522. [Google Scholar] [CrossRef]
- Bonanni, L.; Chachar, M.; Jover-Mengual, T.; Li, H.; Jones, A.; Yokota, H.; Ofengeim, D.; Flannery, R.J.; Miyawaki, T.; Cho, C.H.; et al. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J. Neurosci. 2006, 26, 6851–6862. [Google Scholar] [CrossRef]
- Serbinova, E.; Kagan, V.; Han, D.; Packer, L. Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radic Biol. Med. 1991, 10, 263–275. [Google Scholar] [CrossRef]
- Serbinova, E.A.; Packer, L. Antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Methods Enzymol. 1994, 234, 354–366. [Google Scholar] [PubMed]
- Osakada, F.; Hashino, A.; Kume, T.; Katsuki, H.; Kaneko, S.; Akaike, A. Alpha-tocotrienol provides the most potent neuroprotection among vitamin e analogs on cultured striatal neurons. Neuropharmacology 2004, 47, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Kamat, J.P.; Devasagayam, T.P. Tocotrienols from palm oil as potent inhibitors of lipid peroxidation and protein oxidation in rat brain mitochondria. Neurosci. Lett. 1995, 195, 179–182. [Google Scholar] [CrossRef]
- Shrader, W.D.; Amagata, A.; Barnes, A.; Enns, G.M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; et al. Alpha-tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg. Med. Chem. Lett. 2011, 21, 3693–3698. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Tsuchiya, M.; Wassall, S.R.; Choo, Y.M.; Govil, G.; Kagan, V.E.; Packer, L. Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: Implication to the molecular mechanism of their antioxidant potency. Biochemistry 1993, 32, 10692–10699. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Kubicki, N.; Gnyawali, S.; Chan, Y.C.; Roy, S.; Khanna, S.; Sen, C.K. Natural vitamin e alpha-tocotrienol protects against ischemic stroke by induction of multidrug resistance-associated protein 1. Stroke 2011, 42, 2308–2314. [Google Scholar] [CrossRef]
- Rink, C.; Christoforidis, G.; Khanna, S.; Peterson, L.; Patel, Y.; Abduljalil, A.; Irfanoglu, O.; Machiraju, R.; Bergdall, V.K.; Sen, C.K. Tocotrienol vitamin e protects against preclinical canine ischemic stroke by inducing arteriogenesis. J. Cereb. Blood Flow Metab. 2011, 31, 2218–2230. [Google Scholar] [CrossRef]
- Khanna, S.; Roy, S.; Slivka, A.; Craft, T.K.; Chaki, S.; Rink, C.; Notestine, M.A.; DeVries, A.C.; Parinandi, N.L.; Sen, C.K. Neuroprotective properties of the natural vitamin e alpha-tocotrienol. Stroke 2005, 36, 2258–2264. [Google Scholar] [CrossRef]
- Mishima, K.; Tanaka, T.; Pu, F.; Egashira, N.; Iwasaki, K.; Hidaka, R.; Matsunaga, K.; Takata, J.; Karube, Y.; Fujiwara, M. Vitamin e isoforms alpha-tocotrienol and gamma-tocopherol prevent cerebral infarction in mice. Neurosci. Lett. 2003, 337, 56–60. [Google Scholar] [CrossRef]
- Gopalan, Y.; Shuaib, I.L.; Magosso, E.; Ansari, M.A.; Abu Bakar, M.R.; Wong, J.W.; Khan, N.A.; Liong, W.C.; Sundram, K.; Ng, B.H.; et al. Clinical investigation of the protective effects of palm vitamin e tocotrienols on brain white matter. Stroke 2014, 45, 1422–1428. [Google Scholar] [CrossRef]
- Mangialasche, F.; Westman, E.; Kivipelto, M.; Muehlboeck, J.S.; Cecchetti, R.; Baglioni, M.; Tarducci, R.; Gobbi, G.; Floridi, P.; Soininen, H.; et al. Classification and prediction of clinical diagnosis of alzheimer’s disease based on mri and plasma measures of alpha-/gamma-tocotrienols and gamma-tocopherol. J. Intern. Med. 2013, 273, 602–621. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.F.; Yanagisawa, D.; Durani, L.W.; Hamezah, H.S.; Damanhuri, H.A.; Wan Ngah, W.Z.; Tsuji, M.; Kiuchi, Y.; Ono, K.; Tooyama, I. Tocotrienol-rich fraction modulates amyloid pathology and improves cognitive function in abetapp/ps1 mice. J. Alzheimers Dis. 2017, 55, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Wan Nasri, W.N.; Makpol, S.; Mazlan, M.; Tooyama, I.; Wan Zurinah Wan Ngah, W.Z.; Damanhuri, H.A. Tocotrienol rich fraction supplementation modulate brain hippocampal gene expression in appswe/ps1de9 alzheimer’s disease mouse model. J. Alzheimers Dis. 2018, 70, S239–S254. [Google Scholar] [CrossRef] [PubMed]
- Durani, L.W.; Hamezah, H.S.; Ibrahim, N.F.; Yanagisawa, D.; Nasaruddin, M.L.; Mori, M.; Azizan, K.A.; Damanhuri, H.A.; Makpol, S.; Wan Ngah, W.Z.; et al. Tocotrienol-rich fraction of palm oil improves behavioral impairments and regulates metabolic pathways in abetapp/ps1 mice. J. Alzheimers Dis. 2018, 64, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Taridi, N.M.; Abd Rani, N.; Abd Latiff, A.; Ngah, W.Z.; Mazlan, M. Tocotrienol rich fraction reverses age-related deficits in spatial learning and memory in aged rats. Lipids 2014, 49, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, H.; Drejer, J.; Schousboe, A.; Diemer, N.H. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 1984, 43, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Lehmann, A.; Sandberg, M.; Nystrom, B.; Jacobson, I.; Hamberger, A. Ischemia-induced shift of inhibitory and excitatory amino acids from intra- to extracellular compartments. J. Cereb. Blood Flow Metab. 1985, 5, 413–419. [Google Scholar] [CrossRef]
- Seng, N.S.; Megyesi, J.; Tarcsafalvi, A.; Price, P.M. Mimicking cdk2 phosphorylation of bcl-xl at ser73 results in caspase activation and bcl-xl cleavage. Cell Death Discov. 2016, 2, 1–6. [Google Scholar] [CrossRef]
- Basanez, G.; Zhang, J.; Chau, B.N.; Maksaev, G.I.; Frolov, V.A.; Brandt, T.A.; Burch, J.; Hardwick, J.M.; Zimmerberg, J. Pro-apoptotic cleavage products of bcl-xl form cytochrome c-conducting pores in pure lipid membranes. J. Biol. Chem. 2001, 276, 31083–31091. [Google Scholar] [CrossRef]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. Pink1 protects against cell death induced by mitochondrial depolarization, by phosphorylating bcl-xl and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef]
- Gama, V.; Deshmukh, M. Life after momp. Mol. Cell. 2015, 58, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Cassina, A.; Danial, N.N. Regulation of mitochondrial nutrient and energy metabolism by bcl-2 family proteins. Trends Endocrinol. Metab. 2015, 26, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic mcl-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef]
- Veas-Perez de Tudela, M.; Delgado-Esteban, M.; Maestre, C.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Vecino, R.; Bolanos, J.P.; Almeida, A. Regulation of bcl-xl-atp synthase interaction by mitochondrial cyclin b1-cyclin-dependent kinase-1 determines neuronal survival. J. Neurosci. 2015, 35, 9287–9301. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Samali, A.; Gahm, A.; Orrenius, S. Caspases: Their intracellular localization and translocation during apoptosis. Cell Death Differ. 1999, 6, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Tang, D.G. Mitochondrially localized active caspase-9 and caspase-3 result mostly from translocation from the cytosol and partly from caspase-mediated activation in the organelle. Lack of evidence for apaf-1-mediated procaspase-9 activation in the mitochondria. J. Biol. Chem. 2003, 278, 17408–17420. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Tyas, L.; Brophy, V.A.; Pope, A.; Rivett, A.J.; Tavare, J.M. Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Rep. 2000, 1, 266–270. [Google Scholar] [CrossRef]
- Ricci, J.E.; Gottlieb, R.A.; Green, D.R. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef]
- Polster, B.M.; Fiskum, G. Mitochondrial mechanisms of neural cell apoptosis. J. Neurochem. 2004, 90, 1281–1289. [Google Scholar] [CrossRef]
- Sridharan, V.; Tripathi, P.; Aykin-Burns, N.; Krager, K.J.; Sharma, S.K.; Moros, E.G.; Melnyk, S.B.; Pavliv, O.; Hauer-Jensen, M.; Boerma, M. A tocotrienol-enriched formulation protects against radiation-induced changes in cardiac mitochondria without modifying late cardiac function or structure. Radiat Res. 2015, 183, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Hagl, S.; Kocher, A.; Schiborr, C.; Eckert, S.H.; Ciobanu, I.; Birringer, M.; El-Askary, H.; Helal, A.; Khayyal, M.T.; Frank, J.; et al. Rice bran extract protects from mitochondrial dysfunction in guinea pig brains. Pharmacol. Res. 2013, 76, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Hagl, S.; Grewal, R.; Ciobanu, I.; Helal, A.; Khayyal, M.T.; Muller, W.E.; Eckert, G.P. Rice bran extract compensates mitochondrial dysfunction in a cellular model of early alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Page, G.P.; Kim, K.; Allison, D.B.; Meydani, M.; Weindruch, R.; Prolla, T.A. Alpha- and gamma-tocopherol prevent age-related transcriptional alterations in the heart and brain of mice. J. Nutr. 2008, 138, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Martin, E.; Dejaegere, A.; Stote, R.H. Molecular basis for bcl-2 homology 3 domain recognition in the bcl-2 protein family: Identification of conserved hot spot interactions. J. Biol. Chem. 2009, 284, 17499–17511. [Google Scholar] [CrossRef] [PubMed]
- Aritomi, M.; Kunishima, N.; Inohara, N.; Ishibashi, Y.; Ohta, S.; Morikawa, K. Crystal structure of rat bcl-xl. Implications for the function of the bcl-2 protein family. J. Biol. Chem. 1997, 272, 27886–27892. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Huang, S.; Wu, H.; Zhang, M. Molecular basis of bcl-xl’s target recognition versatility revealed by the structure of bcl-xl in complex with the bh3 domain of beclin-1. J. Mol. Biol. 2007, 372, 223–235. [Google Scholar] [CrossRef]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the bcl-xl-beclin 1 peptide complex: Beclin 1 is a novel bh3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef]
- Wakui, N.; Yoshino, R.; Yasuo, N.; Ohue, M.; Sekijima, M. Exploring the selectivity of inhibitor complexes with bcl-2 and bcl-xl: A molecular dynamics simulation approach. J. Mol. Graph Model. 2018, 79, 166–174. [Google Scholar] [CrossRef]
- Beaudoin, G.M., 3rd; Lee, S.H.; Singh, D.; Yuan, Y.; Ng, Y.G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Khanna, S.; Rink, C.; Gnyawali, S.; Roy, S.; Sen, C.K. Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ. 2009, 16, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.B.; Kurten, R.C.; McCullough, S.S.; Brock, R.W.; Hinson, J.A. Mechanisms of acetaminophen-induced hepatotoxicity: Role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J. Pharmacol. Exp. Ther. 2005, 312, 509–516. [Google Scholar] [CrossRef]
- Duan, Y.; Gross, R.A.; Sheu, S.S. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(adp-ribose) polymerase-1 activation during glutamate excitotoxicity. J. Physiol. 2007, 585, 741–758. [Google Scholar] [CrossRef]
- Crowe, K.M. Optimizing protein precipitation efficiency for assessing the contribution of low molecular weight compounds to serum antioxidant capacity. Clin. Biochem. 2014, 47, 116–118. [Google Scholar] [CrossRef]
- Prior, R.L.; Hoang, H.; Gu, L.; Wu, X.; Bacchiocca, M.; Howard, L.; Hampsch-Woodill, M.; Huang, D.; Ou, B.; Jacob, R. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (orac(fl))) of plasma and other biological and food samples. J. Agric Food Chem. 2003, 51, 3273–3279. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. Swiss-model and the swiss-pdbviewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-A.; Mnatsakanyan, N.; Broman, K.; Davis, A.U.; May, J.; Licznerski, P.; Crowe-White, K.M.; Lackey, K.H.; Jonas, E.A. Alpha-Tocotrienol Prevents Oxidative Stress-Mediated Post-Translational Cleavage of Bcl-xL in Primary Hippocampal Neurons. Int. J. Mol. Sci. 2020, 21, 220. https://doi.org/10.3390/ijms21010220
Park H-A, Mnatsakanyan N, Broman K, Davis AU, May J, Licznerski P, Crowe-White KM, Lackey KH, Jonas EA. Alpha-Tocotrienol Prevents Oxidative Stress-Mediated Post-Translational Cleavage of Bcl-xL in Primary Hippocampal Neurons. International Journal of Molecular Sciences. 2020; 21(1):220. https://doi.org/10.3390/ijms21010220
Chicago/Turabian StylePark, Han-A, Nelli Mnatsakanyan, Katheryn Broman, Abigail U. Davis, Jordan May, Pawel Licznerski, Kristi M. Crowe-White, Kimberly H. Lackey, and Elizabeth A. Jonas. 2020. "Alpha-Tocotrienol Prevents Oxidative Stress-Mediated Post-Translational Cleavage of Bcl-xL in Primary Hippocampal Neurons" International Journal of Molecular Sciences 21, no. 1: 220. https://doi.org/10.3390/ijms21010220
APA StylePark, H.-A., Mnatsakanyan, N., Broman, K., Davis, A. U., May, J., Licznerski, P., Crowe-White, K. M., Lackey, K. H., & Jonas, E. A. (2020). Alpha-Tocotrienol Prevents Oxidative Stress-Mediated Post-Translational Cleavage of Bcl-xL in Primary Hippocampal Neurons. International Journal of Molecular Sciences, 21(1), 220. https://doi.org/10.3390/ijms21010220