Transcriptome Analysis Identifies Candidate Genes and Functional Pathways Controlling the Response of Two Contrasting Barley Varieties to Powdery Mildew Infection

 ,
,
Abstract
1. Introduction
2. Results
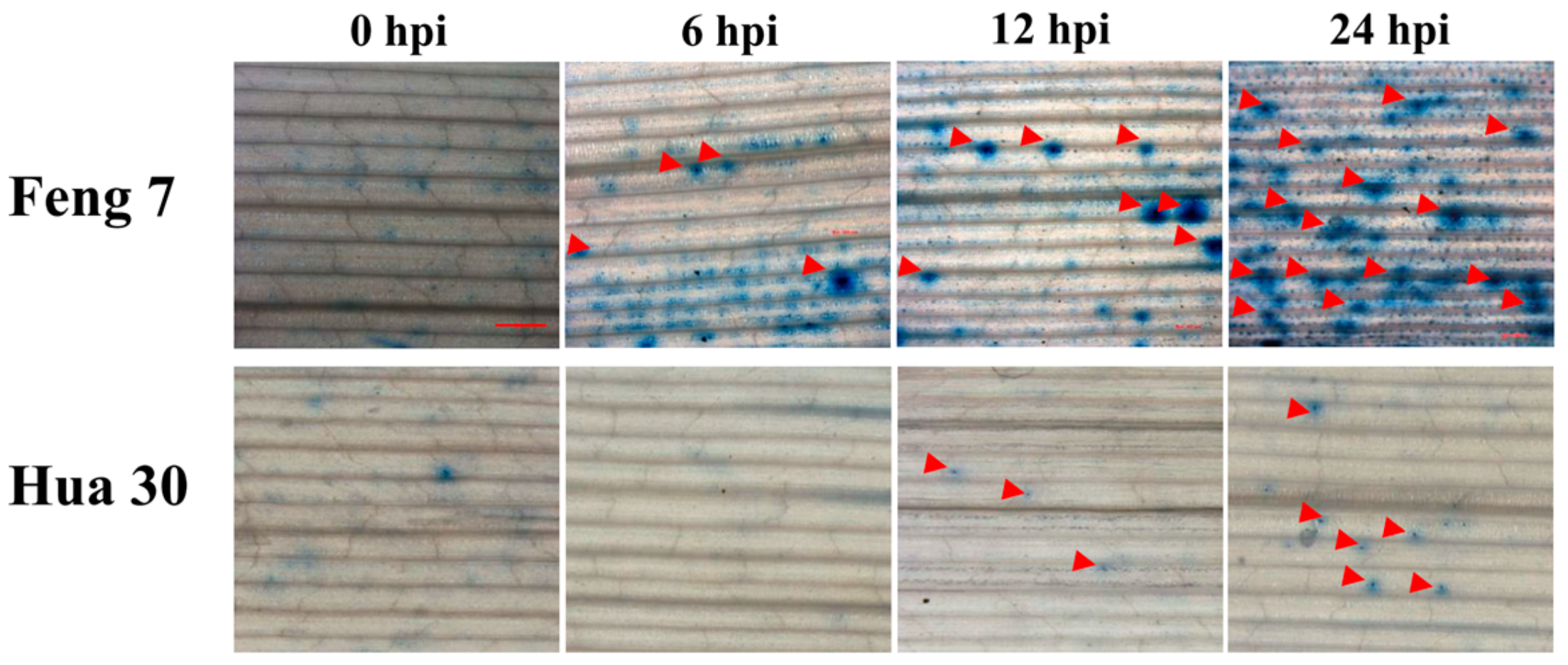
2.1. Resistance Determination of Barley Variety Feng 7 to Bgh Infection
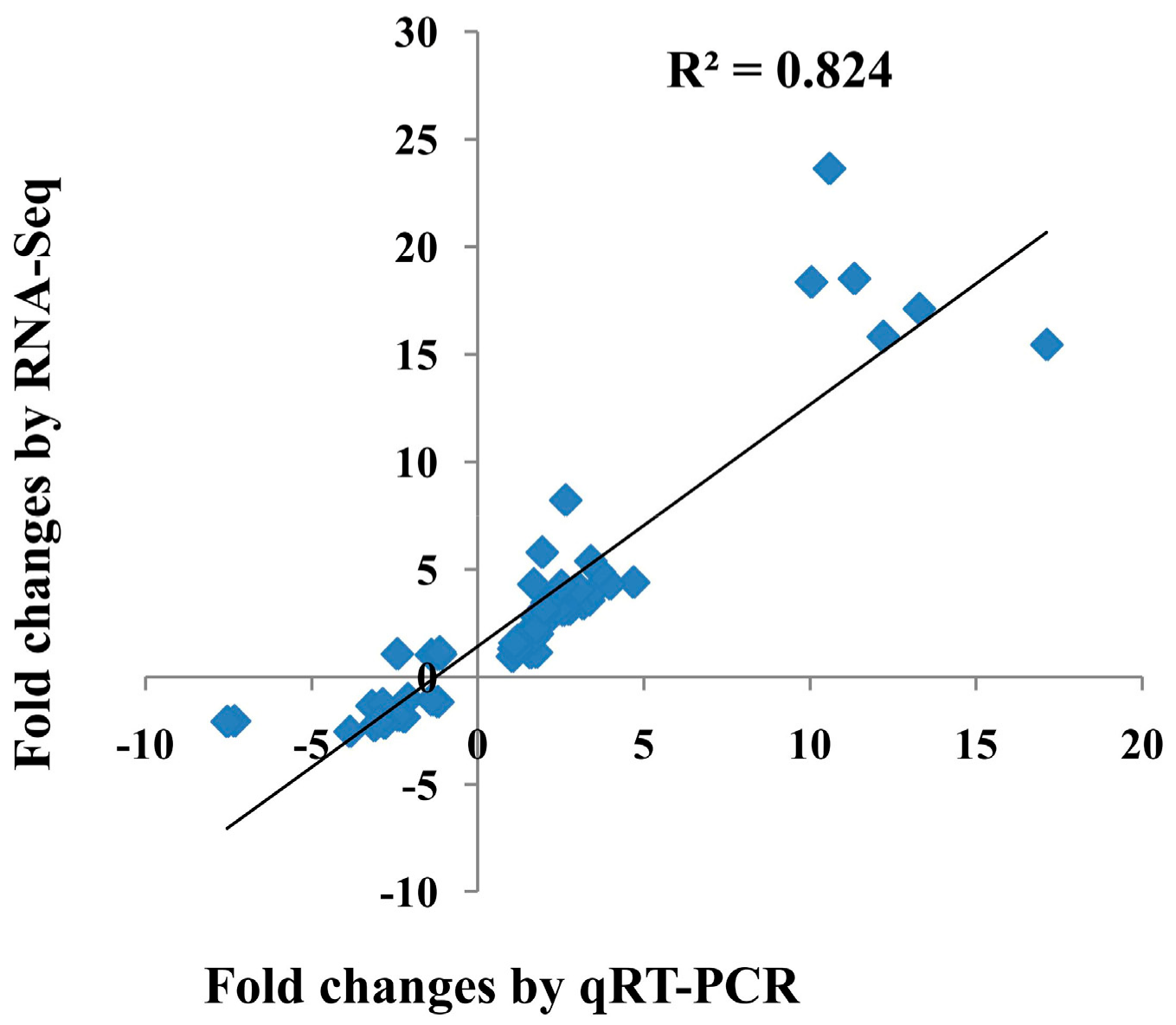
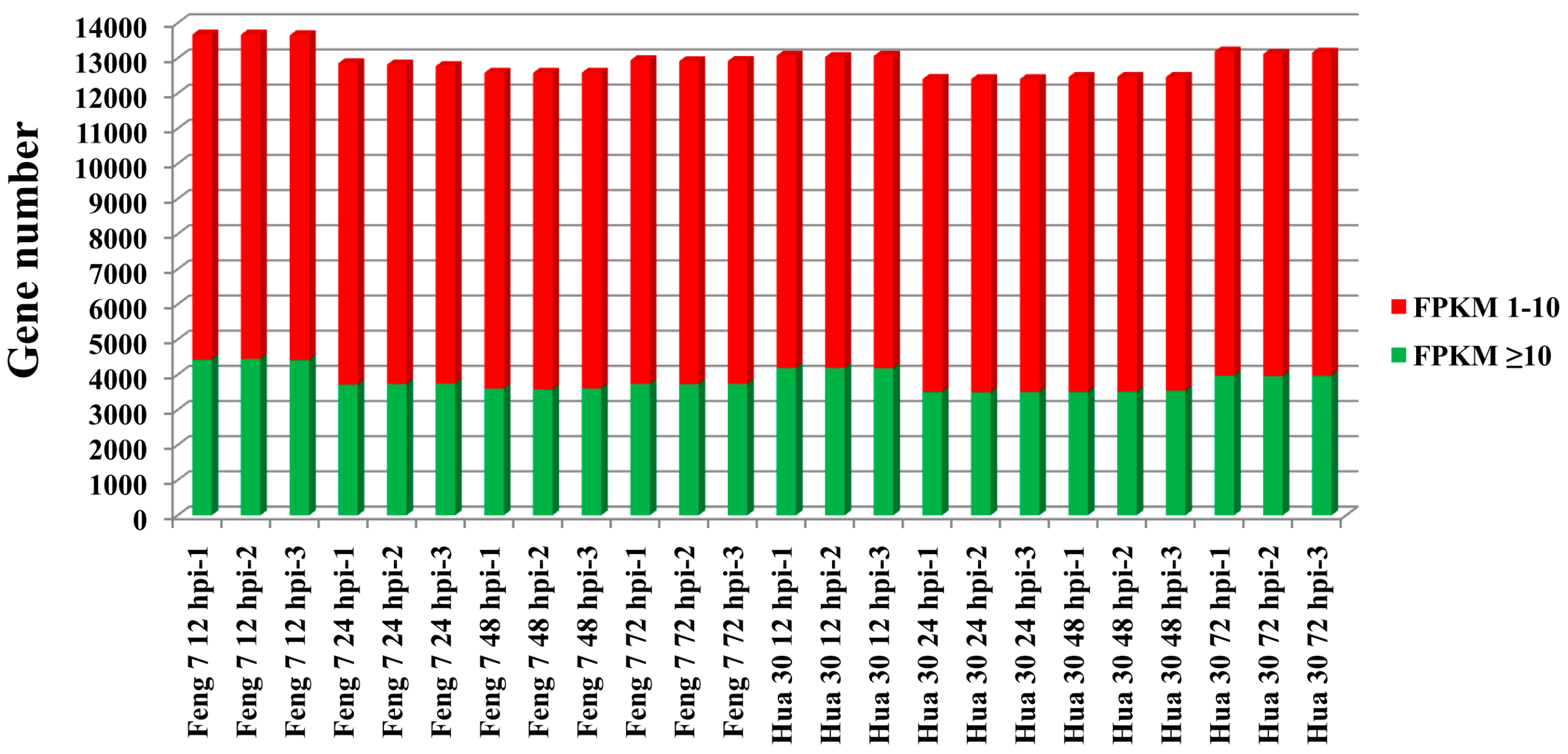
2.2. Illumina Sequence Analysis and Validation of Selected Differentially Expressed Genes (DEGs) Using qRT-PCR
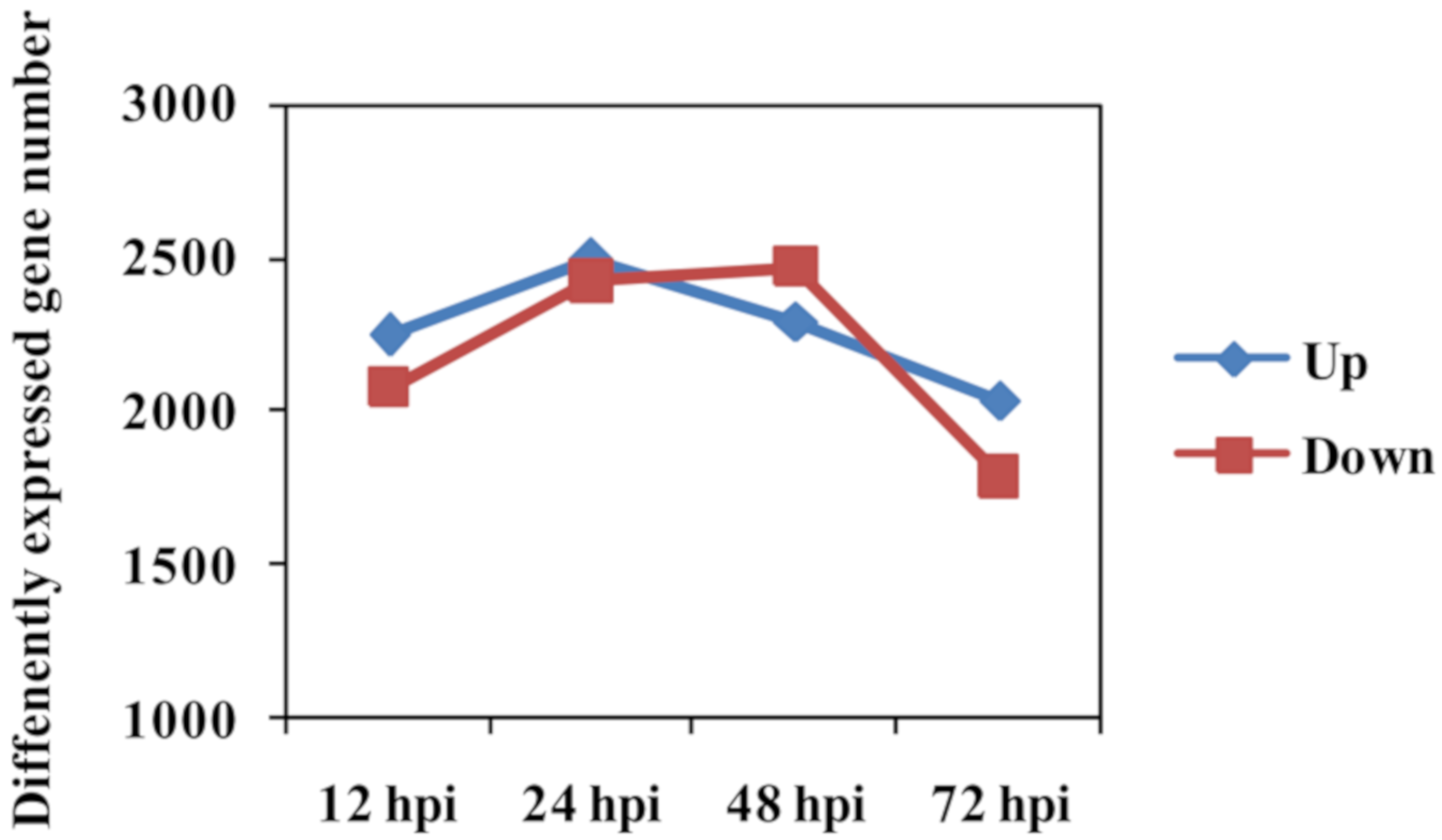
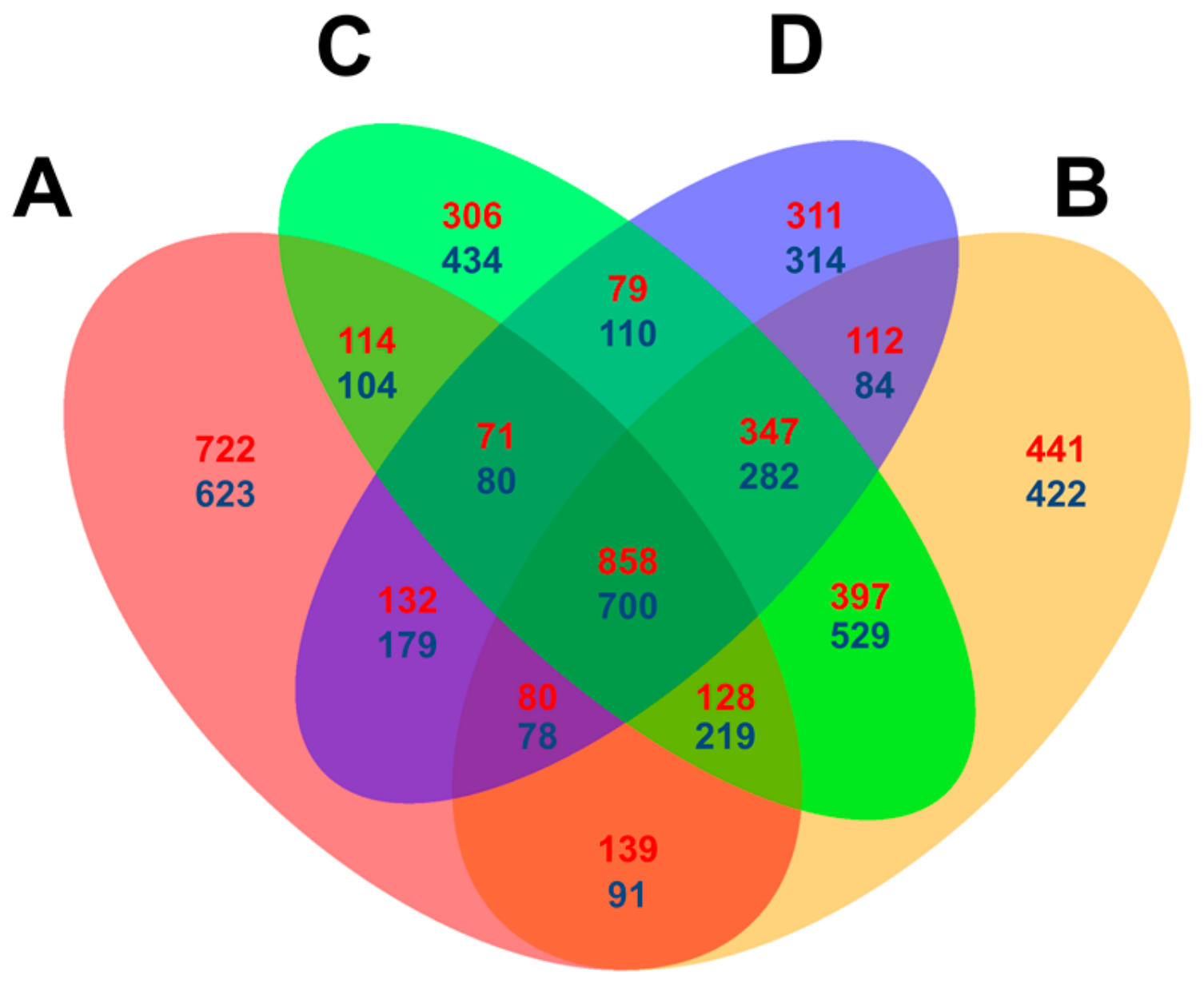
2.3. Comparative Analysis of Feng 7 and Hua 30 Transcriptomes
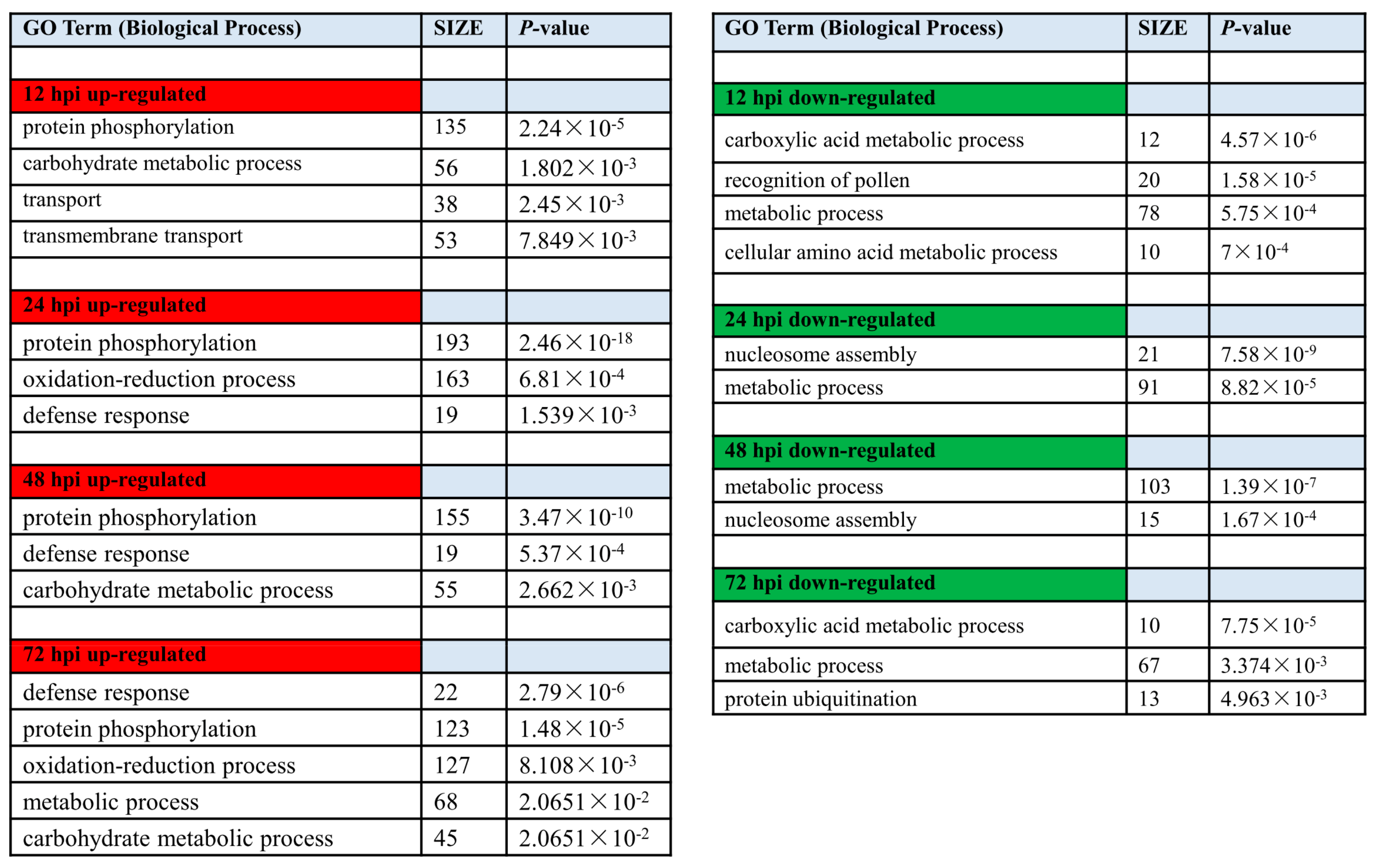
2.4. Gene Ontology (GO)-Based Analysis of the DEGs
2.5. KEGG Pathway Analysis of the DEGs
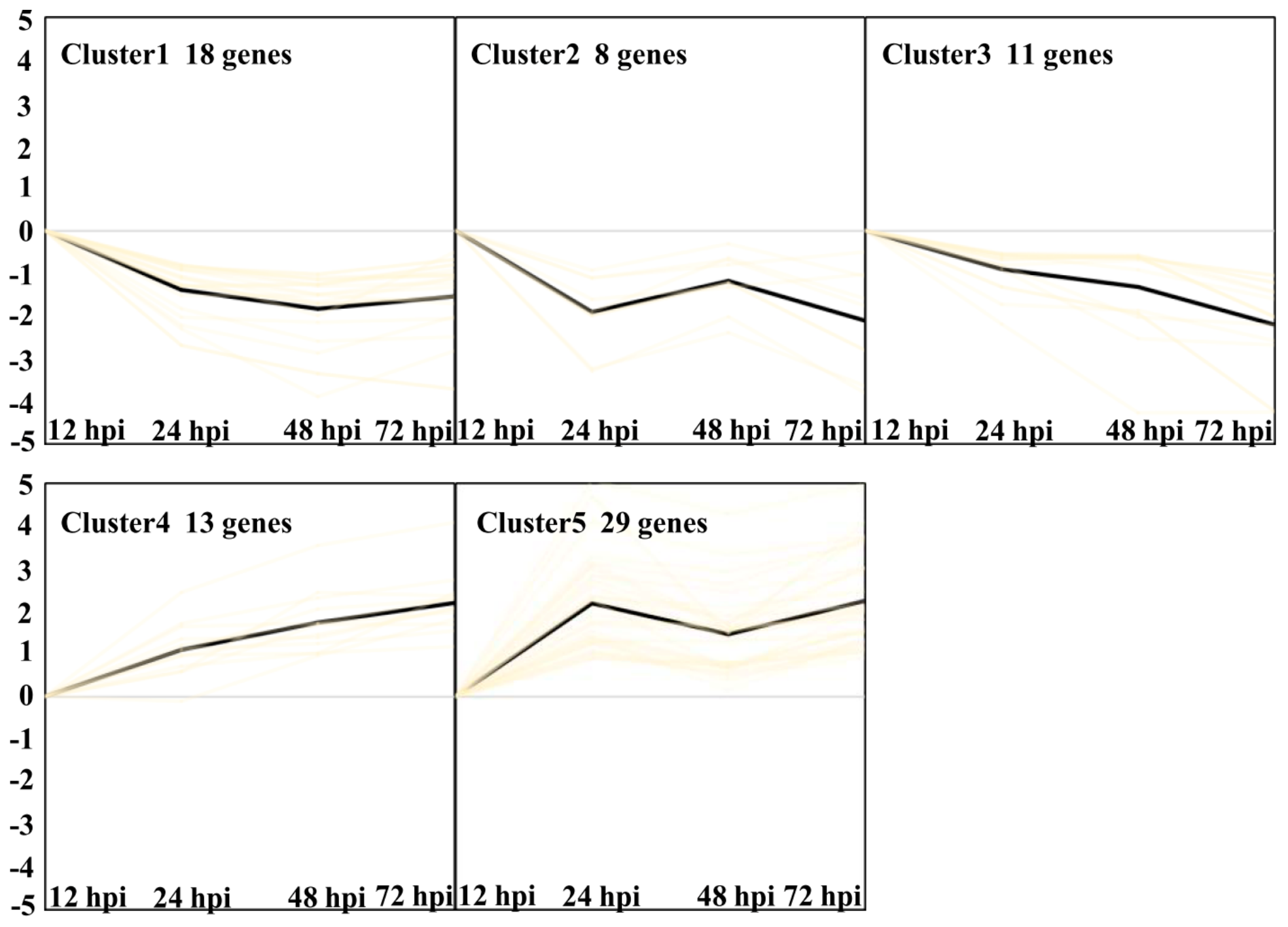
2.6. Clustering Analysis of DEGs to Compare Gene Expression Patterns
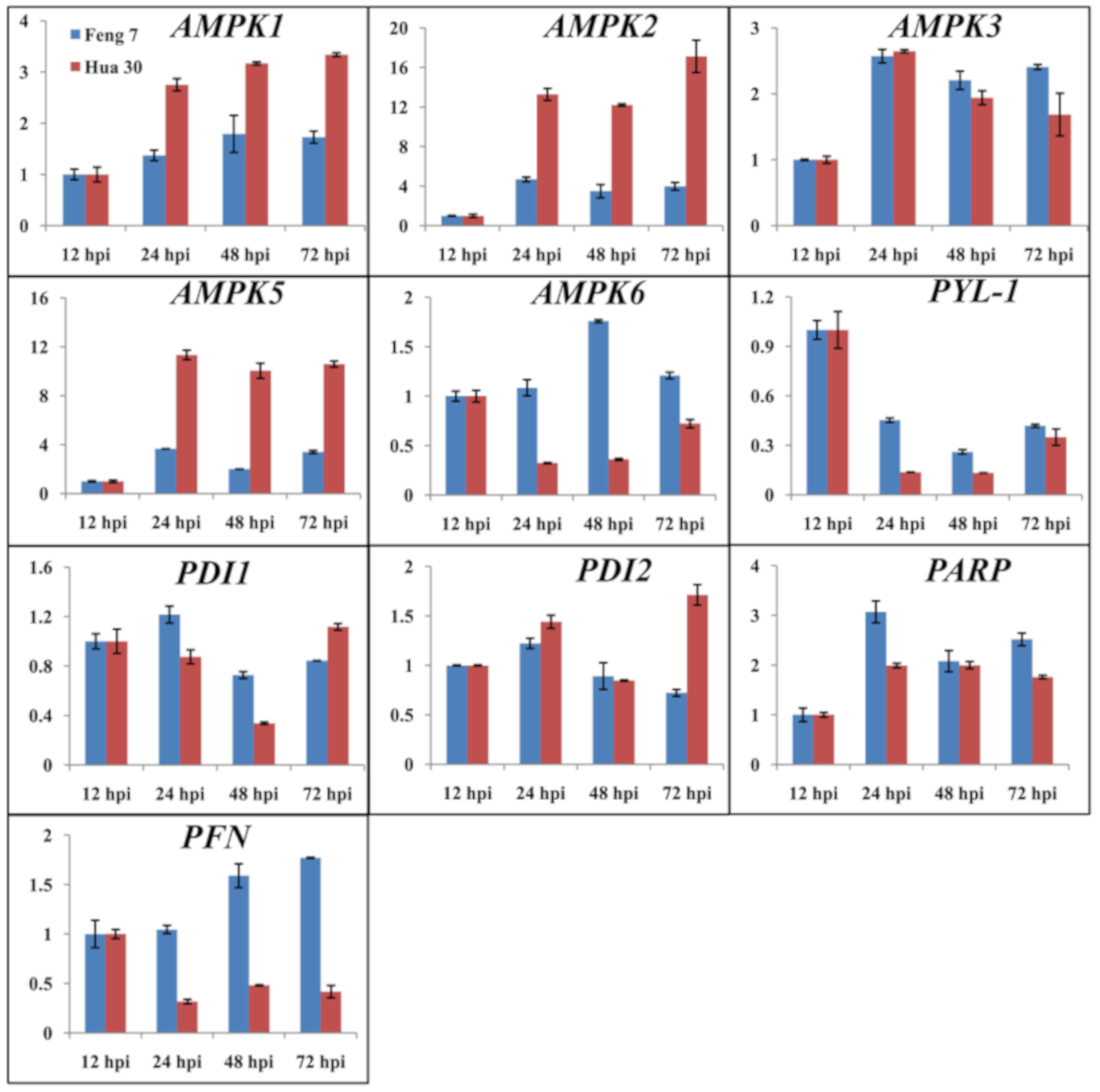
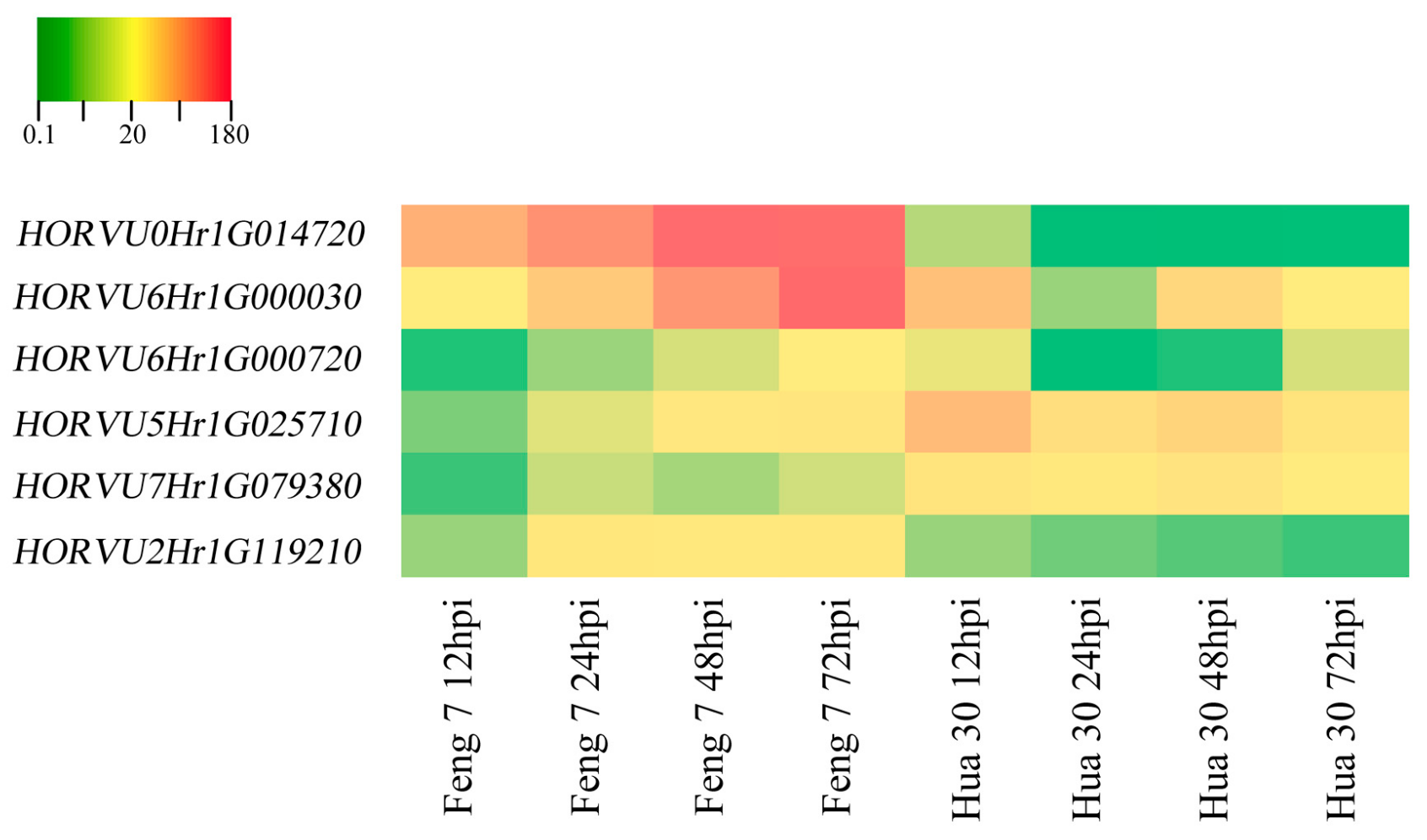
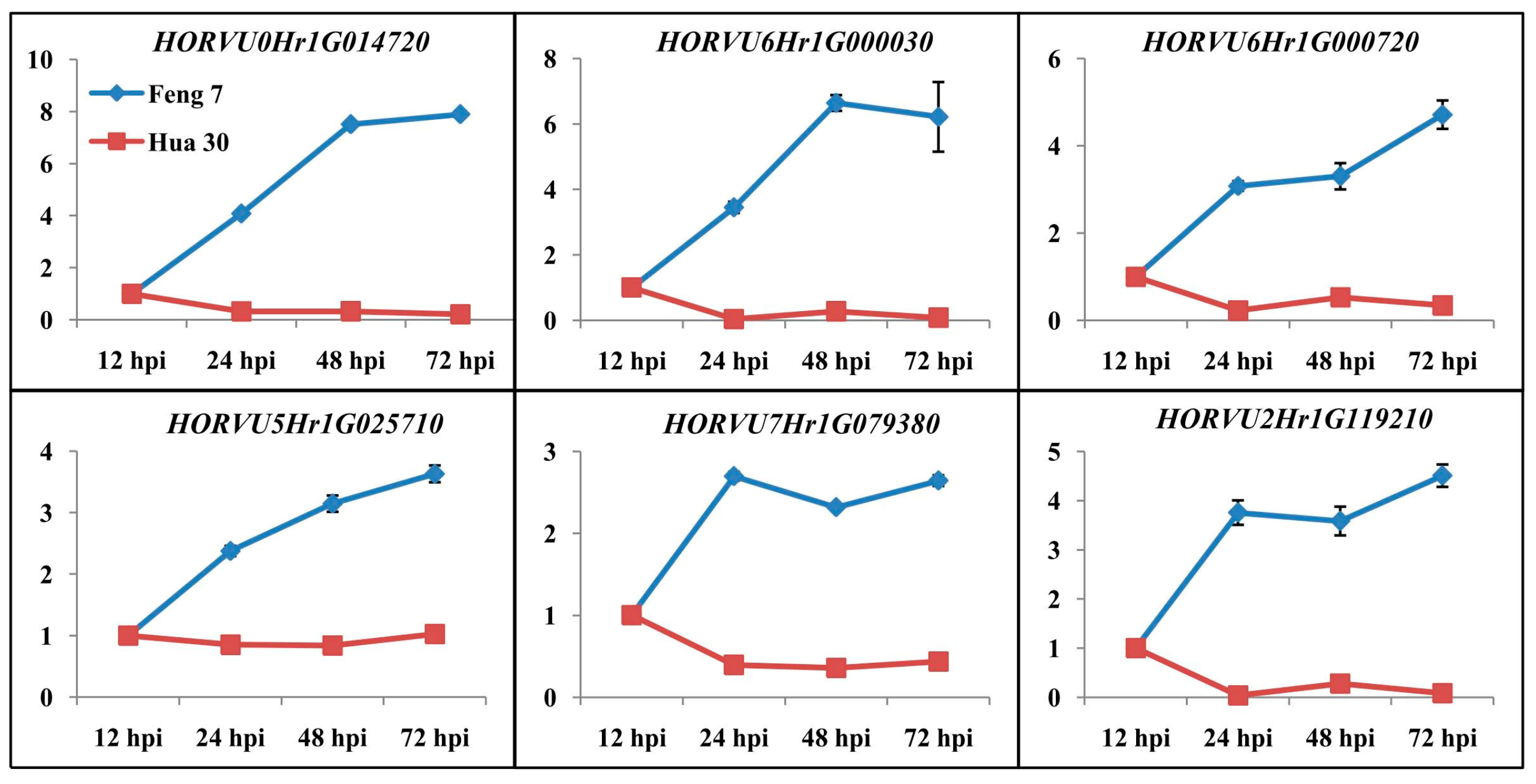
2.7. Identification of Candidate Genes that Were Related to Feng 7 Mediated Resistance
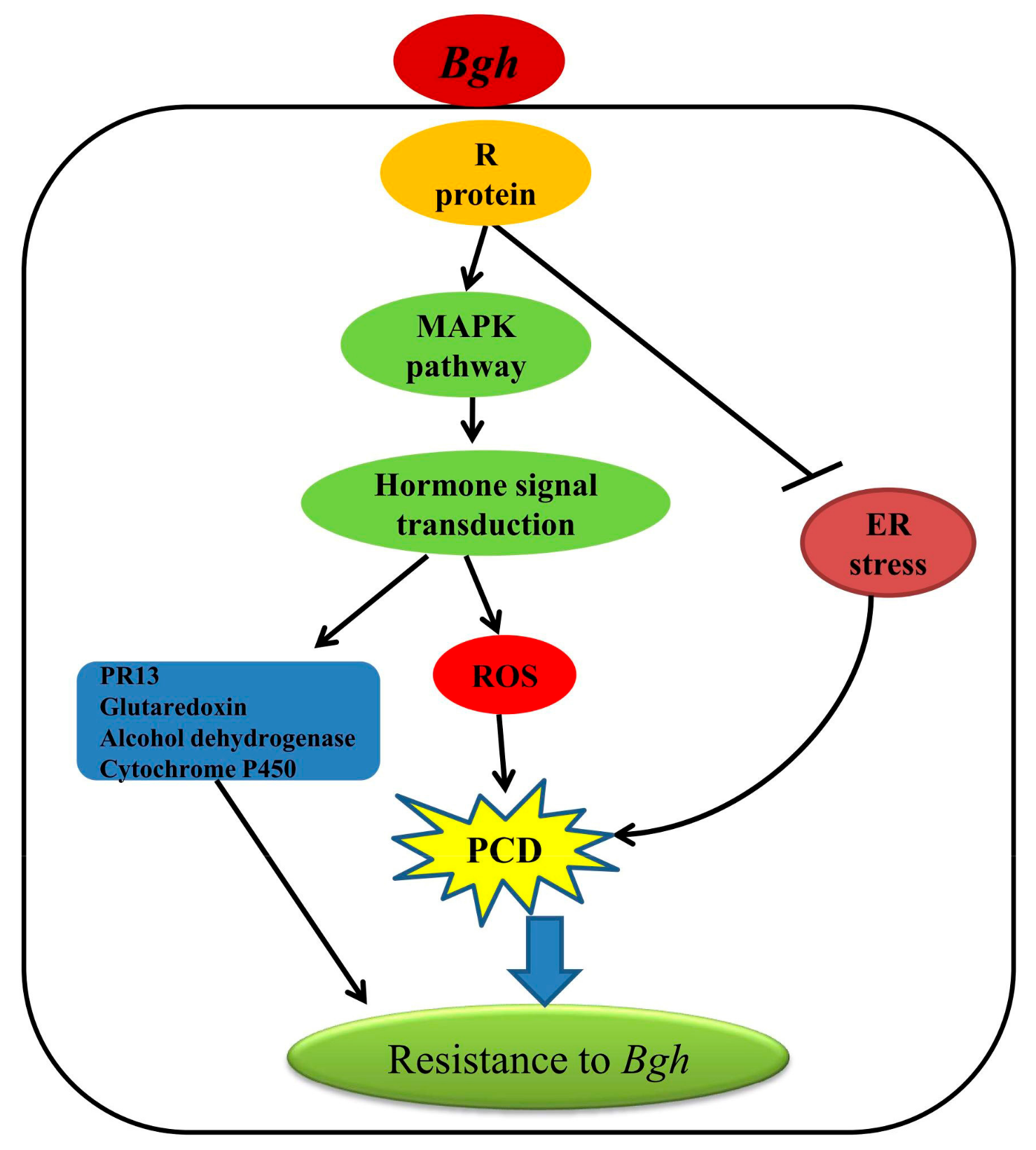
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Growth Conditions, Bgh Treatment
4.2. Trypan Blue Staining
4.3. Evaluation of Powdery Mildew Resistance
4.4. RNA Extraction and Preparation of cDNA Library
4.5. RNA Sequencing and Data Analysis
4.6. Quantitative RT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Bgh | Blumeria graminis f. sp. hordei |
| PAMPs | Pathogen-associated molecular patterns |
| (R) gene | Resistance gene |
| NGS | Next-generation sequencing |
| DGE | Digital gene expression |
| FPKM | Fragments Per Kilo bases per Million reads |
| DEGs | Differentially expressed genes |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia for Genes and Genomes |
| STEM | Short Time-series Expression Miner |
| MAPK | Mitogen-activated protein kinase |
| PR | Pathogenesis-related |
| ROS | Reactive oxygen species |
| PCD | Programmed cell death |
| HR | Hypersensitive response |
References
- Balkema-Boomstra, A.G.; Mastebroek, H.D. Effect of powdery mildew (Erysiphe graminis f. sp. hordei) on photosynthesis and grain yield of partially resistant genotypes of spring barley (Hordeum vulgare L.). Plant Breed. 1995, 114, 126–130. [Google Scholar]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.H.; Wolfe, M. Genetics of powdery mildew resistance in barley. Crit. Rev. Plant Sci. 1994, 13, 97–119. [Google Scholar] [CrossRef]
- Chełkowski, J.; Tyrka, M.; Sobkiewicz, A. Resistance genes in barley (Hordeum vulgare L.) and their identification with molecular markers. J. Appl. Genet. 2003, 44, 291–309. [Google Scholar] [PubMed]
- Zhou, F.; Kurth, J.; Wei, F.; Elliott, C.; Valè, G.; Yahiaoui, N.; Keller, B.; Somerville, S.; Wise, R.; Schulze-Lefert, P. Cell-autonomous expression of barley Mla1 confers race-specific resistance to the powdery mildew fungus via a Rar1-independent signaling pathway. Plant Cell 2001, 13, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Büschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; van Daelen, R.; van der Lee, T.; Diergaarde, P.; Groenendijk, J.; et al. The barley Mlo gene: A novel control element of plant pathogen resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Meng, Y.; Moscou, M.J.; Wise, R.P. Blufensin1 negatively impacts basal defense in response to barley powdery mildew. Plant Physiol. 2009, 149, 271–285. [Google Scholar] [CrossRef]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Bayer, M. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef]
- Caldo, R.A.; Nettleton, D.; Wise, R.P. Interaction-dependent gene expression in Mla-specified response to barley powdery mildew. Plant Cell 2004, 16, 2514–2528. [Google Scholar] [CrossRef]
- Louis, K.S.; Siegel, A.C. Cell viability analysis using trypan blue: Manual and automated methods. In Mammalian Cell Viability; Human Press: Totowa, NJ, USA, 2011; pp. 7–12. [Google Scholar]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.; Dixon, R.A. The oxidative burst in plant disease resistance. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 251–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Henderson, C.; Perfect, E.; Carver, T.L.W.; Thomas, B.J.; Skamnioti, P.; Gurr, S.J. Of genes and genomes, needles and haystacks: Blumeria graminis and functionality. Mol. Plant Pathol. 2005, 6, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Pitzschke, A.; Djamei, A.; Bitton, F.; Hirt, H. A major role of the MEKK1-MKK1/2-MPK4 pathway in ROS signalling. Mol. Plant 2009, 2, 120–137. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, S. MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Liu, J.X.; Howell, S.H. Endoplasmic reticulum protein quality control and its relationship to environmental stress responses in plants. Plant Cell 2010, 22, 2930–2942. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef]
- Ruberti, C.; Kim, S.J.; Stefano, G.; Brandizzi, F. Unfolded protein response in plants: One master, many questions. Curr. Opin. Plant Boil. 2015, 27, 59–66. [Google Scholar] [CrossRef]
- Shao, H.B.; Chu, L.Y.; Shao, M.A.; Jaleel, C.A.; Mei, H.M. Higher plant antioxidants and redox signaling under environmental stresses. C. R. Biol. 2008, 331, 433–441. [Google Scholar] [CrossRef]
- Coupe, S.A.; Palmer, B.G.; Lake, J.A.; Overy, S.A.; Oxborough, K.; Woodward, F.I.; Gray, G.E.; Quick, W.P. Systemic signalling of environmental cues in Arabidopsis leaves. J. Exp. Bot. 2005, 57, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kiddle, G.; Pastori, G.M.; Bernard, S.; Pignocchi, C.; Antoniw, J.; Verrier, P.J.; Foyer, C.H. Effects of leaf ascorbate content on defense and photosynthesis gene expression in Arabidopsis thaliana. Antioxid. Redox Signal. 2003, 5, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Borland, A.; Elliott, S.; Patterson, S.; Taybi, T.; Cushman, J.; Pater, B.; Barnes, J. Are the metabolic components of crassulacean acid metabolism up-regulated in response to an increase in oxidative burden? J. Exp. Bot. 2005, 57, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.D. Primary metabolism and plant defense-fuel for the fire. Mol. Plant Microbe Interact. 2009, 22, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sudisha, J.; Sharathchandra, R.G.; Amruthesh, K.N.; Kumar, A.; Shetty, H.S. Pathogenesis related proteins in plant defense response. In Plant Defence: Biologiacl Control; Springer: Dordrecht, The Netherlands, 2012; pp. 379–403. [Google Scholar]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial peptides from plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef]
- Epple, P.; Apel, K.; Bohlmann, H. An Arabidopsis-thaliana thionin gene is inducible via a signal-transduction pathway different from that for pathogenesis-related proteins. Plant Physiol. 1995, 109, 813–820. [Google Scholar] [CrossRef]
- Andresen, I.; Becker, W.; Schluter, K.; Burges, J.; Parthier, B.; Apel, K. The identification of leaf thionin as one of the main jasmonate-induced proteins of barley (Hordeum vulgare). Plant Mol. Biol. 1992, 19, 193–204. [Google Scholar] [CrossRef]
- Penninckx, I.A.; Eggermont, K.; Terras, F.R.; Thomma, B.P.; De Samblanx, G.W.; Buchala, A.; Metraux, J.P.; Manners, J.M.; Broekaert, W.F. Pathogen-induced systemic activation of a plant defensin gene in Arabidopsis follows a salicylic acid-independent pathway. Plant Cell 1996, 8, 2309–2323. [Google Scholar]
- Noctor, G.; Reichheld, J.P.; Foyer, C.H. ROS-related redox regulation and signaling in plants. Semin. Cell Dev. Biol. 2018, 80, 3–12. [Google Scholar] [CrossRef]
- Koornneef, A.; Pieterse, C.M. Cross talk in defense signaling. Plant Physiol. 2008, 146, 839–844. [Google Scholar] [CrossRef]
- Chang, C.; Meyerowitz, E.M. Molecular cloning and DNA sequence of the Arabidopsis thaliana alcohol dehydrogenase gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, W.; Yao, Y.; Wei, Y.; Chan, Z. Alcohol dehydrogenase 1 (ADH1) confers both abiotic and biotic stress resistance in Arabidopsis. Plant Sci. 2017, 262, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Rong, W.; Luo, M.; Shan, T.; Wei, X.; Du, L.; Xu, H.; Zhang, Z. A wheat cinnamyl alcohol dehydrogenase TaCAD12 contributes to host resistance to the sharp eyespot disease. Front. Plant Sci. 2016, 7, 1723. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kim, S.Y.; Paek, K.H.; Choi, D.; Park, J.M. Suppression of CaCYP1, a novel cytochrome P450 gene, compromises the basal pathogen defense response of pepper plants. Biochem. Biophys. Res. Commun. 2006, 345, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Peterhänsel, C.; Schulze-Lefert, P. Interaction analyses of genes required for resistance responses to powdery mildew in barley reveal distinct pathways leading to leaf cell death. Plant Cell 1997, 9, 1397–1409. [Google Scholar] [CrossRef]
- Sheng, B. Grades of resistance to powdery mildew classified by different phenotypes of response in the seedling stage of wheat. Plant Prot. 1988, 1, 49. [Google Scholar]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level-the DESeq Package. Available online: www.bioconductor.org/packages/devel/bioc/vignettes/DESeq/inst/doc/DESeq.pdf (accessed on 24 December 2019).
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Goodall, A.J.; Kumar, P.; Tobin, A.K. Identification and expression analyses of cytosolic glutamine synthetase genes in barley (Hordeum vulgare L.). Plant Cell Physiol. 2013, 54, 492–505. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Point | No. of Necrotic Spots/Leaf Segment | Average | t-Test (p < 0.05) |
|---|---|---|---|
| 0 hpi | 0 | 1 | |
| 2 | |||
| 1 | |||
| 6 hpi | 6 | 6.3 | * |
| 8 | |||
| 5 | |||
| 12 hpi | 12 | 11 | * |
| 10 | |||
| 11 | |||
| 24 hpi | 22 | 23.3 | * |
| 23 | |||
| 25 |
| F1 Generation | No. of Plants | No. of Resistance Plants (Grade 0, 0; 1) | No. of High Susceptible Plants (Grade 2, 3, 4) |
|---|---|---|---|
| Feng 7 × Hua 30 | 25 | 25 | 0 |
| Hua 30 × Feng 7 | 19 | 19 | 0 |
| Pathways | 12 hpi | 24 hpi | 48 hpi | 72 hpi | ||||
|---|---|---|---|---|---|---|---|---|
| ≥1 | ≤−1 | ≥1 | ≤−1 | ≥1 | ≤−1 | ≥1 | ≤−1 | |
| Galactose metabolism | 14 | 13 | 15 | 13 | ||||
| ABC transporters | 8 | 7 | 8 | 10 | 7 | |||
| Brassinosteroid biosynthesis | 3 | 4 | 3 | |||||
| Plant hormone signal transduction | 28 | 29 | ||||||
| Calcium signaling pathway | 9 | 8 | 9 | |||||
| MAPK signaling pathway | 22 | 25 | ||||||
| Steroid hormone biosynthesis | 7 | 8 | 7 | 6 | ||||
| Protein processing in endoplasmic reticulum | 38 | 28 | ||||||
| Photosynthesis-antenna proteins | 20 | 5 | 14 | |||||
| Glutathione metabolism | 23 | 18 | ||||||
| Phenylalanine metabolism | 13 | 11 | 13 | 8 | 13 | 9 | 8 | |
| Total | 60 | 56 | 99 | 64 | 67 | 22 | 52 | 64 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Guo, G.; Zhou, L.; Chen, Y.; Zong, Y.; Huang, J.; Lu, R.; Liu, C. Transcriptome Analysis Identifies Candidate Genes and Functional Pathways Controlling the Response of Two Contrasting Barley Varieties to Powdery Mildew Infection. Int. J. Mol. Sci. 2020, 21, 151. https://doi.org/10.3390/ijms21010151
Li Y, Guo G, Zhou L, Chen Y, Zong Y, Huang J, Lu R, Liu C. Transcriptome Analysis Identifies Candidate Genes and Functional Pathways Controlling the Response of Two Contrasting Barley Varieties to Powdery Mildew Infection. International Journal of Molecular Sciences. 2020; 21(1):151. https://doi.org/10.3390/ijms21010151
Chicago/Turabian StyleLi, Yingbo, Guimei Guo, Longhua Zhou, Yunyun Chen, Yingjie Zong, Jianhua Huang, Ruiju Lu, and Chenghong Liu. 2020. "Transcriptome Analysis Identifies Candidate Genes and Functional Pathways Controlling the Response of Two Contrasting Barley Varieties to Powdery Mildew Infection" International Journal of Molecular Sciences 21, no. 1: 151. https://doi.org/10.3390/ijms21010151
APA StyleLi, Y., Guo, G., Zhou, L., Chen, Y., Zong, Y., Huang, J., Lu, R., & Liu, C. (2020). Transcriptome Analysis Identifies Candidate Genes and Functional Pathways Controlling the Response of Two Contrasting Barley Varieties to Powdery Mildew Infection. International Journal of Molecular Sciences, 21(1), 151. https://doi.org/10.3390/ijms21010151