1. Introduction
Pluripotent stem cells (PSCs) have the potential for long-term self-renewal and expansion while maintaining their differentiated potential. PSCs can be manufactured in large quantities in an undifferentiated state and be cryopreserved as master cell banks (MCBs) and working cell banks (WCBs) under current good manufacturing procedures (cGMPs) [
1,
2]. This unique characteristic of PSCs makes them an attractive source for regenerative cell therapy. These banks can be subsequently thawed, expanded, and differentiated for clinical applications. Establishing appropriate MCB and WBC strategies would allow access to high-quality starting materials that could minimize potential variabilities during the development of final PSC derived cell therapy products. In some instances, these starting materials might need to stay cryopreserved for many years after the initial cell manufacture/banking. Therefore, long-term cryopreservation of quality-assured PSCs is required to ensure an adequate supply of these cells for clinical and commercial manufacturing.
PSCs are often maintained, passaged, and cryopreserved as clumps of cells. Prior observations suggest that different cryopreservation solutions, cell lines, and cryopreservation strategies affect the revival of the cells post thaw and may lead to inefficient and slow cell revival [
3]. Long-term storage of PSCs in LN
2 carries a risk of mycoplasma, bacterial, and viral infection [
4]. In addition, long-term storage may cause a loss of critical quality attributes (CQA) including identity, purity, potency, and sterility. Recently, several groups have raised concerns about critical cryoprotectant materials used in the stem cell cryopreservation such as Dimethyl sulphoxide (DMSO), fetal bovine serum (FBS), or serum replacement [
5]. DMSO has been demonstrated to be toxic to the tissues and cells depending on the exposure time [
3,
6]. DMSO is also reported to cause cryoinjury including chromosomal/telomere instability and epigenetic changes in embryonic stem cells (ESCs) [
7].
Owing to these concerns and in order to demonstrate the long-term stability of induced PSCs (iPSCs), we have evaluated the effect of long-term cryopreservation on the CQAs of human iPSC lines post thaw, including cell revival, 2D and 3D expansion, expression of pluripotency markers, spontaneous and directed lineage-specific differentiation, sterility, genomic stability, and changes in telomerase activity and telomere length. Our findings from this study can generate a more in-depth understanding of the impact of long-term cryopreservation on quality of human iPSC banks and their utility for the generation of high-quality clinical products.
3. Discussion
We have previously reported the development of a manufacturing process to generate cGMP-compliant human iPSC lines with detailed characterization of the generated cell lines [
1,
2]. Large-scale manufacturing of cGMP-iPSC banks is a key step towards the establishment of a reliable starting material for regenerative medicine products. It requires that these banked cells maintain their critical quality attributes post thaw and their ability to generate functional, therapeutically relevant cell products. The effectiveness of cryopreserved stem cells from different sources, including bone marrow and cord blood, has been demonstrated for several disorders that include, but are not limited to, graft versus host disease [
15,
16], Scleroderma [
17], Thalassemia [
18], and multiple sclerosis [
17,
19]. Implementing a successful cryopreservation strategy can stabilize the supply of critical therapeutic products and support centralized manufacturing operations.
To date, the primary focus of academic and industrial labs has been mainly on the characterization of undifferentiated human iPSC lines post-derivation and expansion rather than post-cryopreservation. Despite the implementation of cryopreservation as a routine and conventional method for preserving iPSCs long-term, there is limited knowledge on how the cryopreservation and thaw strategy affects the iPSC genomic integrity and differentiation capacity to desired lineages. Some groups have shown that freeze/thaw process may lead to DNA and chromosomal aberrations due to production of free radicals in some cell types [
20], but to the best of our knowledge, there is no such study on the long-term stability of cryopreserved iPSC MCBs and/or WCBs.
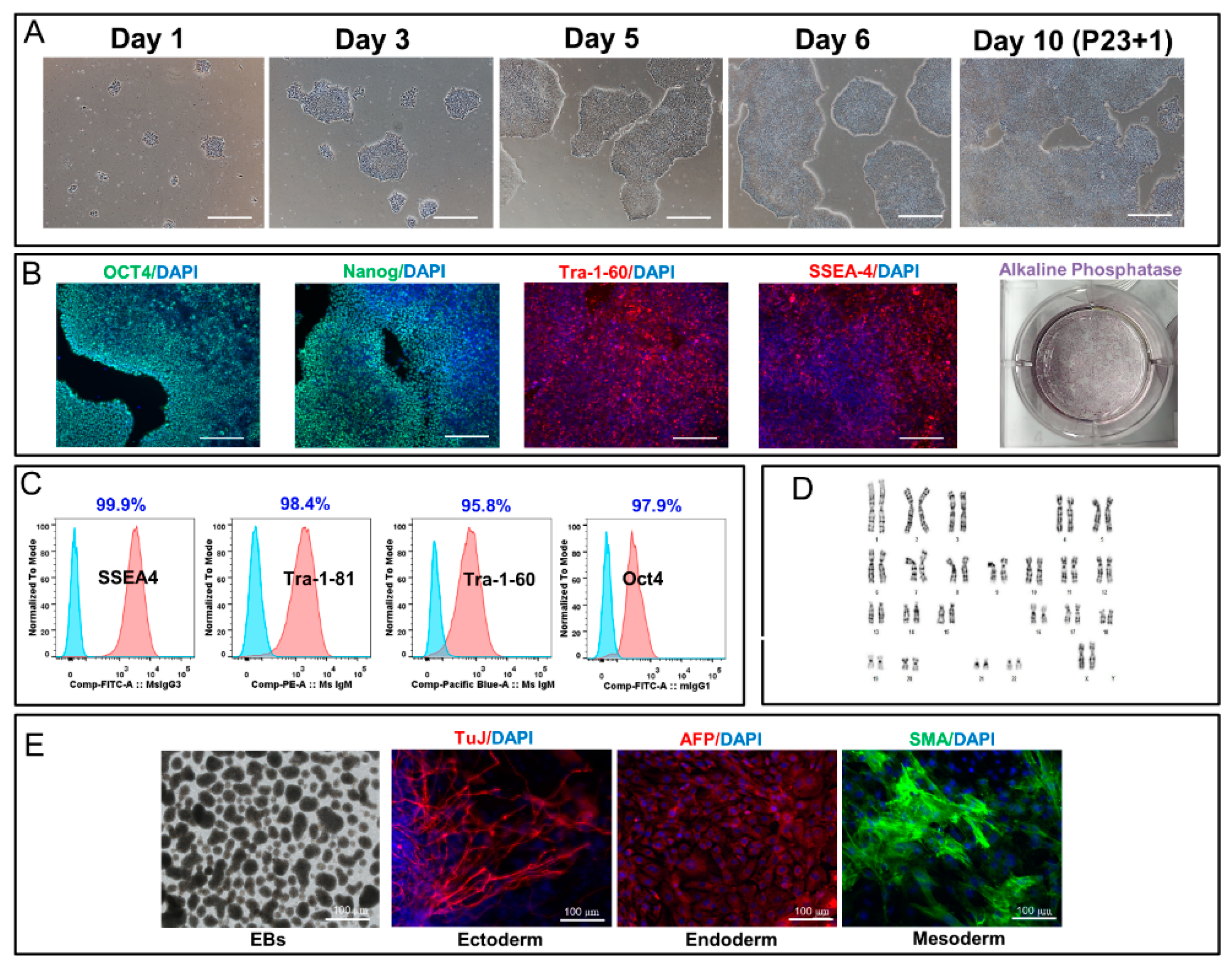
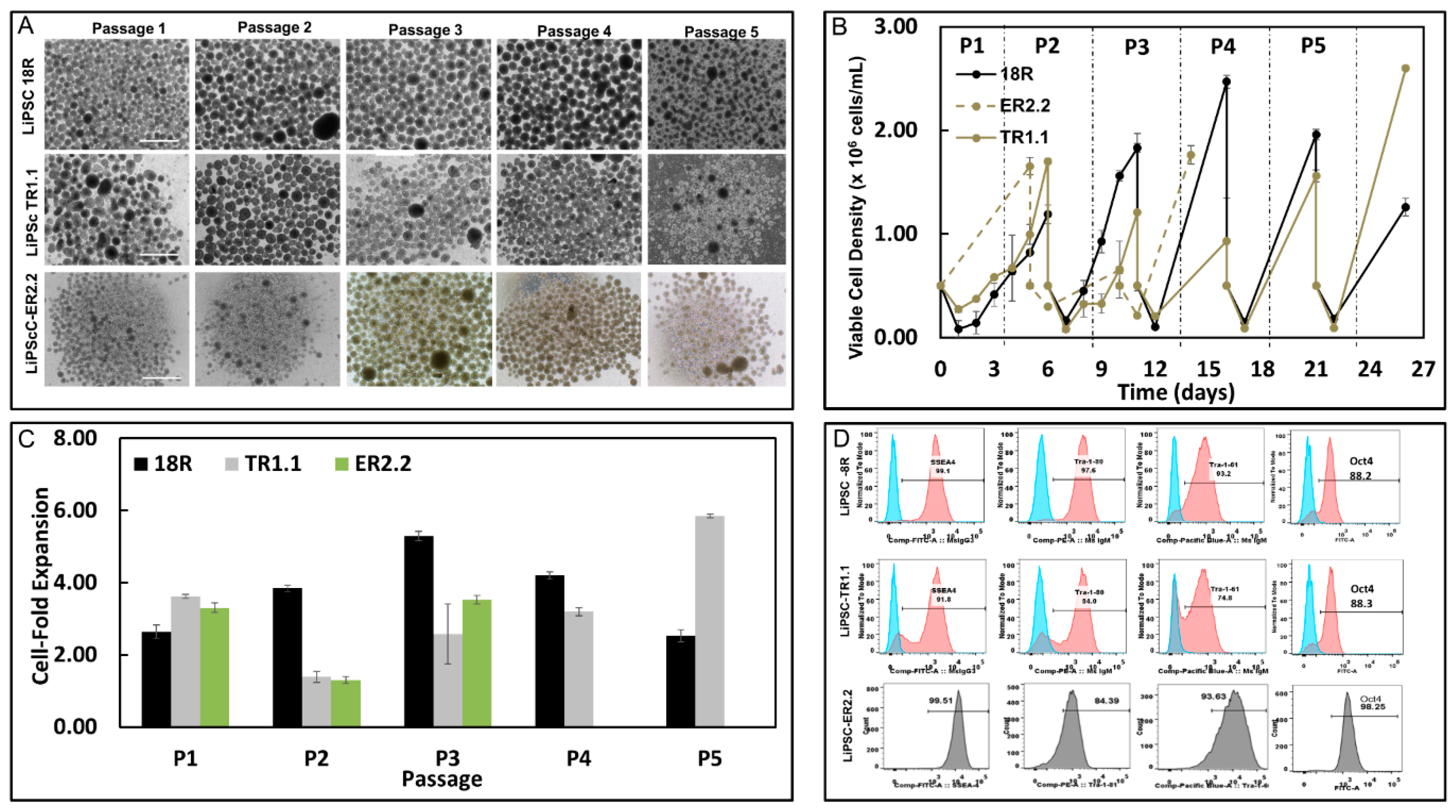
Our data showed that, after five years of cryopreservation, all three cGMP-manufactured cell lines demonstrated normal karyotypes post thaw. The lines maintained their genomic integrity for 15 passages in 2D culture environment and for 5 passages in 3D suspension culture. Several groups have demonstrated that cryopreservation and recovery of human ESCs trigger apoptosis, loss of pluripotency, and spontaneous differentiation [
21,
22]. The recovery of human ESCs decreases to 16%–23% during the freeze/thaw process, as measured by the number of attached colonies 9–14 days post thaw combined with a low growth rate and high percent of differentiation [
23,
24]. Our results indicated that all three lines could be thawed efficiently with high plating rate within 7–9 days with less than 5.0% differentiation observed. The plating rate was demonstrated by measuring the attachment efficiency (number of iPSC colonies attached after thaw/passaging) and detected through alkaline phosphatase staining. Although the viability of one line (ER2.2) was approximately 58.0% post thaw, all three lines exhibited a high level of attachment and formed typical PSCs colonies in 7–9 days prior to the first passage. Also, all three lines preserved their critical quality attributes (CQA) including spontaneous and directed differentiation potential to cells from the ectoderm, mesoderm, and endoderm lineages after prolonged cryopreservation. These lines were fully characterized for their CQA prior to the cryopreservation five years ago, as demonstrated in our previous reports [
1,
25]. As the directed differentiation has not been optimized for each line, the three lines showed variability in their differentiation capacity. Line-to-line differentiation variability may depend on several factors including chromatin status, DNA methylation signature (somatic memory), reprogramming method, and tissue source [
26,
27]. Stability of human iPSC lines to proliferate and expand in 3D suspension environment post-prolonged cryobanking is also one of the key factors for the generation of a large-number of cells to be derived into clinically-relevant cells in the regenerative medicine field [
28,
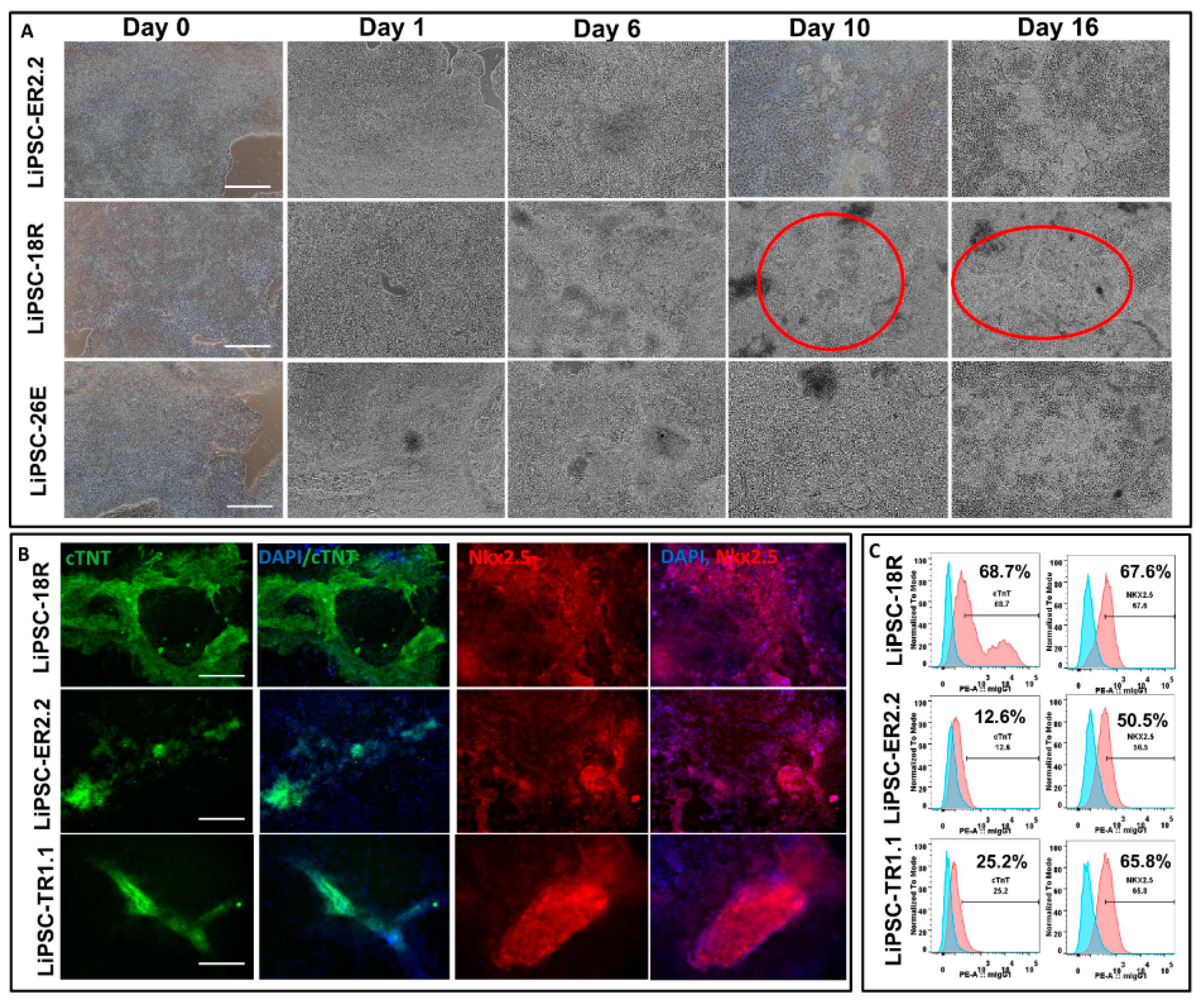
29]. We evaluated all three lines for their inherent potential to form free-floating aggregates (i.e., no microcarriers), self-renew, and have the ability to expand up to clinically-relevant cell density in stirred suspension bioreactors. Our results showed that long-term cryopreservation of these lines did not affect their expansion potential in 3D culture. Furthermore, all three lines maintained their CQA such as pluripotency and differentiation capacity and showed normal karyotype after five passages.
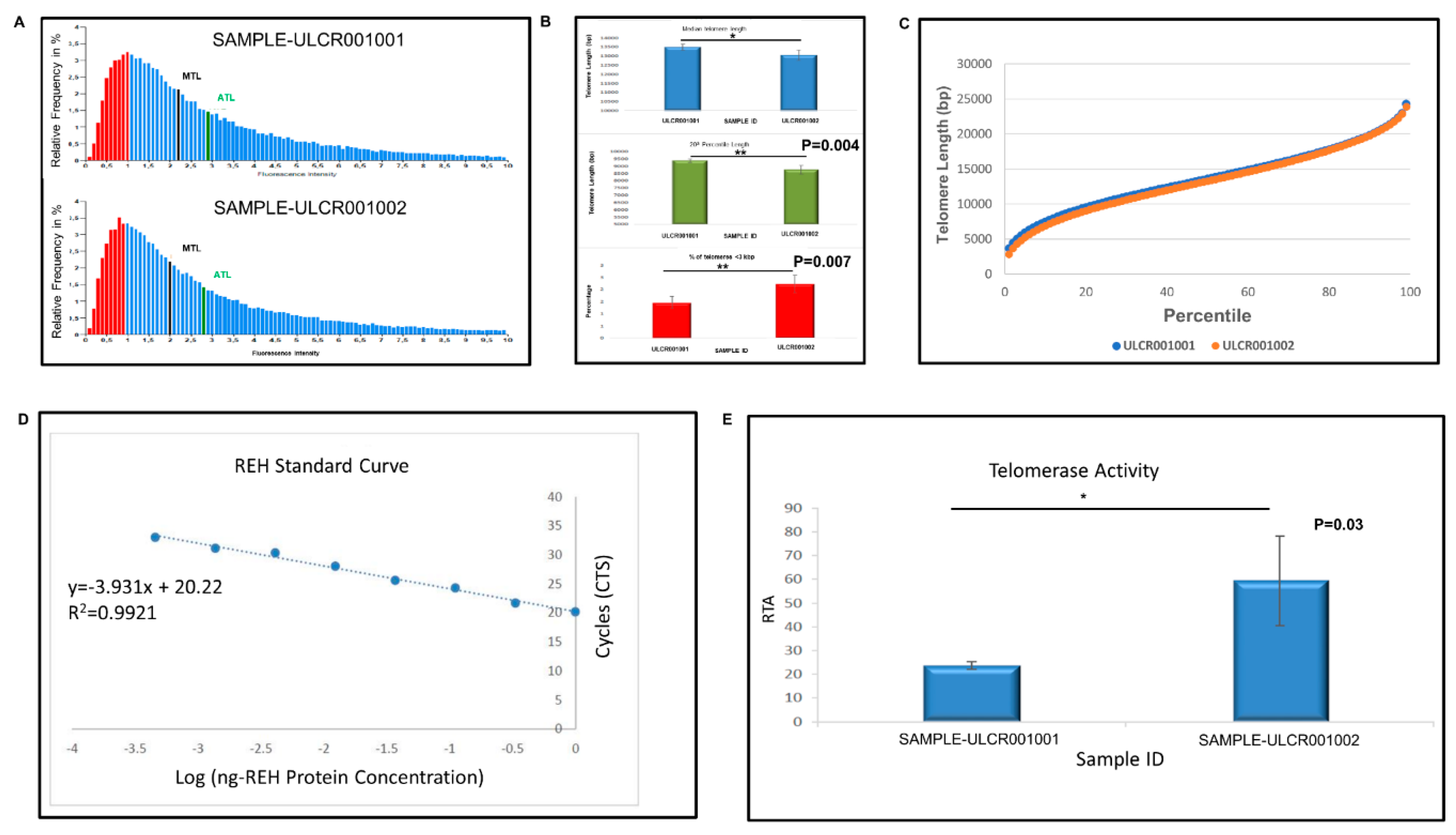
Pluripotent stem cells show high telomere length and appropriate telomerase activity to support their self-renewal and proliferation capacity [
30,
31]. Furthermore, telomere maintenance, regulation, and homeostasis is critical for hPSC long-term culture and its chromosomal stability [
31]. LiPSC-18R line (+15 passages) demonstrated higher telomerase expression (reverse transcriptase TERT and RNA template TERC) and lower telomere length compared with the freshly-thawed cells that were cryopreserved five years ago. The shorter telomere length observed after 15 passages may be caused by cell division during the early passages post thaw. To compensate for this shorter telomere length and to maintain the pluripotency and chromosomal integrity, human iPSCs induce the expression of telomerase to extend the telomere length as evident by the higher telomerase activity following 15 passages post thaw. In agreement, Zeng et al. demonstrated that telomeres were elongated during in vitro expansion and were then stabilized through a telomerase-dependent mechanism [
32]. Our 2D and 3D expansion and directed differentiation of the revived cells demonstrated stable expression of telomerase and telomere length, as several studies have reported a correlation between short telomeres and unstable directed differentiation in mouse and human pluripotent cells [
33,
34].
The effectiveness of human iPSCs in research and therapeutic applications highly relies on their genomic, phenotypic, and molecular stability during long-term passaging, expansion, and prolonged cryopreservation. This study addresses these aspects and the stability of cryopreserved GMP-manufactured iPSCs after storage in LN2 for five years. Considering the number of iPSC lines assessed (n = 3) in the current study, more lines that are generated with different reprogramming methods need to be evaluated in the future studies. In conclusion, this study suggests that iPSCs remain stable and therapeutically-efficacious after banking and that the current cryopreservation strategy can be utilized for the long-term storage of iPSCs.
4. Materials and Methods
4.1. Thawing and Cell Culture
A cGMP-compliant environment was used to generate the iPSC lines LiPSC-18R, LiPSC-TR1.1, and LiPSC-ER2, as described before [
2], and continuously maintained on the defined L7
TM hPSC matrix (Lonza, FP-5020) using L7™ hPSCs medium. The L7
TM hPSC medium consists of basal L7
TM hPSC medium (FP-5107) and L7
TM hPSC supplement (FP-5207). Per the manufacturer’s protocols, the cells were serially passaged using L7
TM hPSC passaging solution (FP-5013) and cultured in humidified a 37 °C incubator at 5% CO
2. These lines were cryopreserved under cGMP condition in CryoStor CS10 (BioLife Solutions, Bothell, WA) with a cell density of 1.0–2.0 × 10
6/mL (
Table 1) and maintained for five years in the vapor phase of LN
2. Briefly, three vials for each iPSC line were thawed in a 37 °C water bath for 3–4 min and the content of each vial was transferred to a 15 mL conical tube. Then, 9 mL of L7
TM hPSC medium was added drop by drop to the cells to minimize the osmotic shock to the cells. The cell suspension was centrifuged at 300×
g for 5 min. The supernatant of the cells was removed and the cells were resuspended in 2–3mL of L7
TM hPSC medium. The cells were plated on L7
TM matrix coated plates using L7
TM hPSC medium. Total cell count and viability was measured before plating using the NucleoCounter NC-200 (Chemometec, Denmark) with Via1-cassettes (two cassettes count with Solution 10). Vials from each line were pooled together and plated on at least six wells. All thawed cells were maintained undifferentiated for 15 passages in a 2D culture system followed by 5 passages in a 3D culture system. Cells in 2D culture were passaged as clumps in static culture every 4–6 days utilizing a ratio between 1:6 to 1:12 based on the confluency.
4.2. Post-Thaw Expansion of iPSCs in 3D Biott Bioreactor System
Each of the iPSC lines were harvested from the 2D cultures after 3–4 passages as single cells and inoculated in ABLE Biotts (Reprocell, Beltsville, MD) in either L7TM or Nutristem, in duplicate, at a cell density of 5.0 × 105 cells/mL in a volume of 30 mL. On Day 0, the culture medium was supplemented with 10 µm Y-27632. The Biotts were agitated at 60 rpm and placed in a humidified incubator operating at 37 °C and 5% CO2. Every day, a 50% medium change was performed by removing the Biotts from the incubator and allowing the aggregates to settle for 5 min. After 5 min, 50% of the medium was removed and centrifuged at 200× g for 3 min to collect any aggregates in suspension. After centrifugation, the supernatant was discarded, the collected aggregates were resuspended in fresh medium and added back into the appropriate bioreactor. The Biotts were fed until the cells reached between 1.5 × 106 and 2.0 × 106 cells/mL.
When the Biotts reached this cell density, the culture was removed from the incubator, and the cell aggregates were collected and centrifuged at 200× g for 3 min. The supernatant was discarded, and the aggregates were washed one time with 1 × phosphate buffer saline (PBS), and centrifuged again at 200× g for 3 min. The supernatant was discarded, and the aggregates were enzymatically dissociated to single cells in the presence of TrypLE. The TrypLE-cell aggregate solution was placed for 5 min in a 37 °C water bath, and the culture was triturated with a P1000 five times to break up the aggregates. The enzymatic reaction was diluted with culture medium and the cell suspension was centrifuged at 300× g for 5 min. Following centrifugation, the supernatant was discarded, the cells were resuspended in the appropriate culture medium + 10 µm Y27632 and re-inoculated at a density of 5.0 × 105 cells/mL. This process was repeated for a total of five passages, after which the cells were evaluated for spontaneous differentiation and karyotype.
4.3. Cell Counts
Cell counts were performed daily for passage 1 (P1) and P2 to determine the growth kinetics. For P3–5, cell counts were only done on day 1 and on harvest day (as determined by the counts during P1/2). To obtain the cell counts, a 1 mL sample was removed from the Biott and placed in a 15 mL conical tube and centrifuged at 200× g for 3 min. The supernatant was discarded and the cells were washed with 1 × PBS, and centrifuged at 200× g for 3 min. Following centrifugation, the supernatant was discarded and the cells were resuspended in TrypLE and placed in a 37 °C water bath. After 5 min, the cells were pipetted to break apart the aggregates, culture medium was added to the suspension to dilute the enzymatic reaction, and then centrifuged at 300× g for 5 min. The supernatant was discarded and the cells were then resuspened in culture medium and counted using the two-cassette method on the NC-200 (i.e., Solution 10 method).
4.4. Flow Cytometry
Flow cytometry was performed on the iPSCs when they reached approximately 70% to 80% confluency at P1 and P15 post thaw in 2D. Flow cytometry analysis was also performed on samples taken at five passages post thaw on iPSCs cultured in a 3D culture system. The iPSCs were dissociated into a single-cell using TrypLE solution (ThermoFisher). Then, 4% paraformaldehyde (PFA) and perm/wash buffer (Becton Dickinson) were used to fix and permeabilize the cells for intracellular staining respectively. Cells were incubated with Alexa Flour 488 anti-IgG isotype control and Alexa-488 anti-OCT3/4 (Cell Signaling, 5177S) post permeabilization. Prior to fixation, PE-conjugated antigen-specific antibodies and respective isotypes with manufacturer’s recommended concentrations were used to detect membrane antigens as below: anti-SSEA4 (Becton Dickinson, 560128), anti-TRA-1-60 (Becton Dickinson, 560193), anti-TRA-1-81 (Becton Dickinson, 560161), anti-IgM isotype (Becton Dickinson, 555584), and anti-IgG3 isotype (Becton Dickinson, 556659). FACSCanto™ II flow cytometer (Becton Dickinson) was used to process the samples. BD FACS Diva and Flowjo 7.6 softwares were used to acquire flow data and analyze the results.
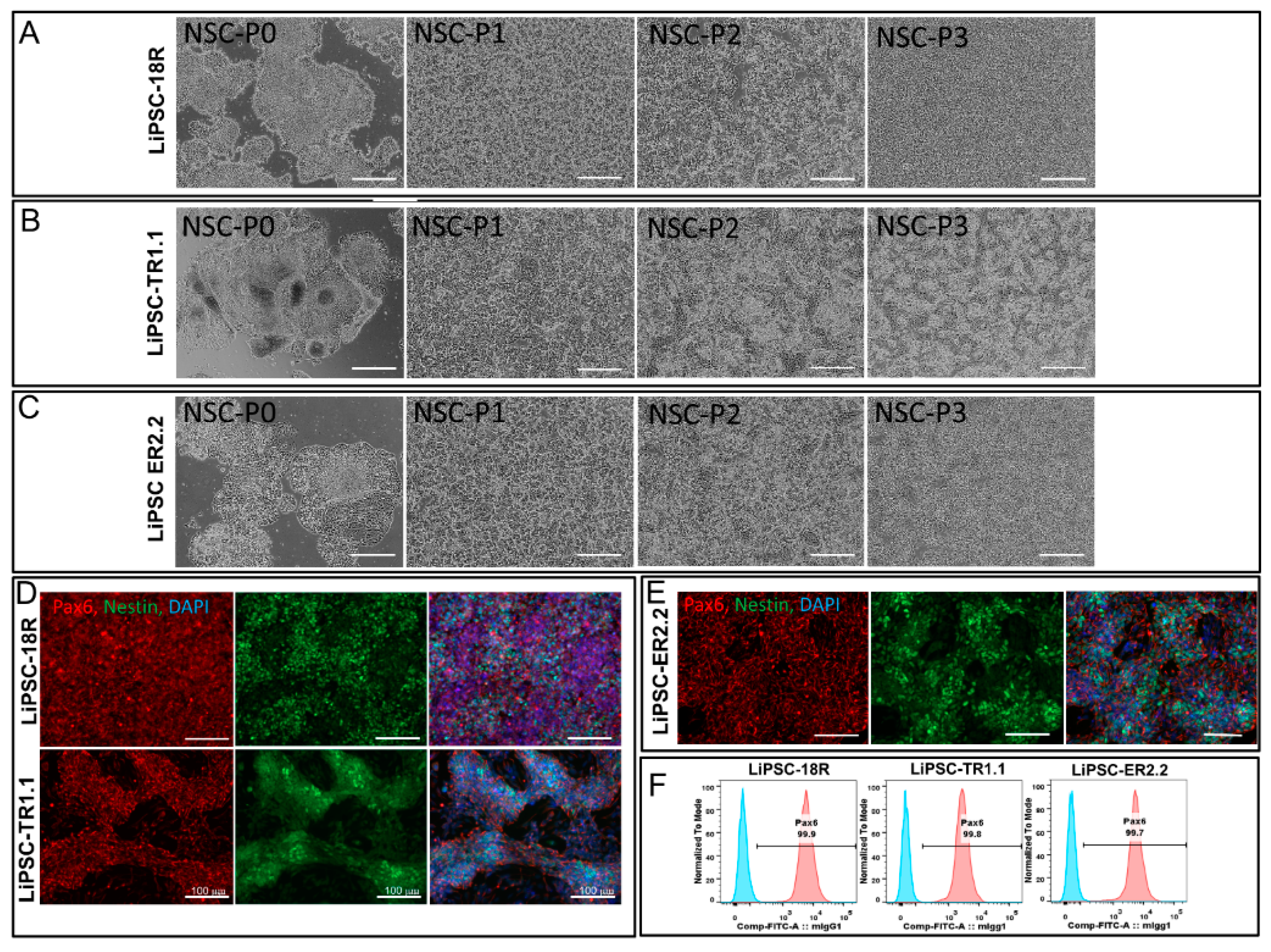
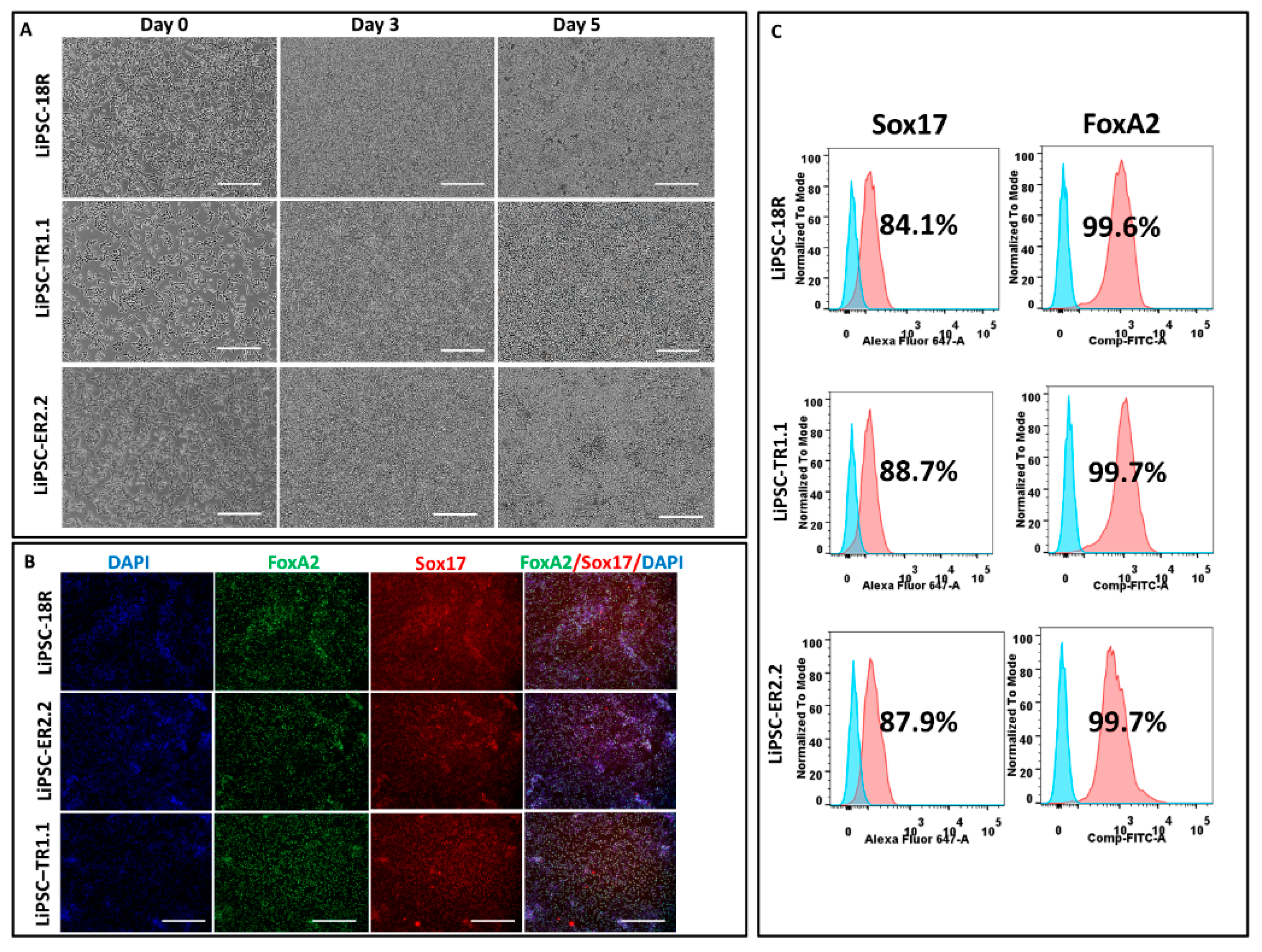
iPSC-derived differentiated cells were harvested and dissociated to single cells. The cells are stained as follows: PBS (−/−) and Accutase solution (Millipore, SCR005) were used to wash and dissociate iPSC-derived neural stem cells (NSCs) into single cells, respectively, as per the manufacturer’s instruction (6 min at 37 °C). To quench the activity of Accutase, an equal volume of neural medium, supplemented with 10 µm Y-27632 (Abcam, ab120129) was added to the cells. Cells were fixed and permeabilized for intracellular staining with 4% PFA and perm/wash buffer (Becton Dickinson, 554723) for post cell count and viability measurement, respectively. Alexa Fluor 488 mouse anti-Pax6 (BD, PN561664) or respective Alexa Fluor 488 anti-IgG2a isotype control (BD, PN558055) were used to stain permeabilized cells. iPSC-derived DE cells were harvested as single cells using TrypLE and stained with Sox17 and FoxA2, as described above.
A mixture of Liberase (2.5 mg or 13 units/mL) and TrypLE solution (50:1) was used to digest iPSC-derived cardiomyocytes for 15 min at 37 °C post PBS (−/−) wash. The solution was quenched with the same volume of cardiac differentiation medium. The cells were filtered using 100 µm cell strainer after 3–4 times trituration, and were fixed and permeabilized with the 4% PFA and perm/wash buffer, respectively, for intracellular staining. Anti-cTnT (ab8295) or respective anti-IgG isotype control (abcam, ab91353) were used to stained the permeabilized cells. For the second antibody, PE goat anti-mouse IgG (ThermoFisher, P21129) was used (ThermoFisher, P21129) to stain the cells post wash. The positively stained cells were then captured through a FACSCanto™ II flow cytometer (Becton Dickinson). Flow data were acquired using BD FACS Diva software and then analyzed with Flowjo 7.6 software.
4.5. Immunocytochemistry
iPSCs and derived-differentiated cells were stained as per the following method: the cells were washed twice with 1 × Dulbecco’s phosphate buffered saline (DPBS) (Lonza Biosciences, 17-513F) post culture medium aspiration. The washed cells were then fixed with 4% PFA (Electron Microscopy Sciences, 15710) for 20 min. The cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, T9284) for 15 min and blocked with 10% goat serum in PBS (−/−) in 0.1% Triton X-100 at RT for 15 min. The primary antibodies were prepared in 1% bovine serum albumin (BSA) in 1 × DPBS and the cells were incubated overnight at 4 °C. Primary antibodies against NANOG (R&D Systems, AF1997; 6.7µg/mL) and OCT4 (Abcam, ab19857; 1:350), Desmin (Dako, 1:300), Actin (Sigma, A111, 1:500), cTnT (abcam, ab8295, 1:200), FoxA2 (abcam, ab108422, 1:300), MYL2 (Sigma, HPA019763, 1:200), Pax6 (BioLegend, 901301, 1:300), Sox17 (R&D systems, MAB19241), and Nestin (R&D systems, MAB1259, 1:50) were used in combination with the related secondary antibodies Alexa Fluor 594 Goat anti mouse (ThermoFisher, A11005, 1:1000) and Alexa Fluor 488 Goat anti Rabbit (ThermoFisher, A11070, 1:1000). Cells were then rinsed three times with 1% BSA in 1x DPBS. All cells were incubated with 5 µg/mL 4’, 6-diamidino-2-phenylindole (DAPI) (ThermoFisher, D1306) in 1% BSA in 1x DPBS and secondary antibodies for 1 h at room temperature (RT) in the dark. PSCs (both iPSCs and ESCs as control) were treated with primary antibodies detecting surface markers TRA-1-81 (Stemgent, 09-0011; 1:100), TRA-1-60 (Millipore, MAB4360; 1:100), and SSEA4 (Millipore, MAB4304; 1:100) overnight at 4 °C post blocking step and prior to being permeabilized. All fluorescence images were captured using a Zeiss Observer.Z1 microscope equipped with ZEN software.
4.6. Embryoid Body (EB) Differentiation
iPSCs cultures were dissociated using L7TM hPSC passaging solution as small aggregates. EB formation medium consisting of DMEM/F12 (Life Technologies, 11330-032) and 10 μM Y27632 (Millipore, SCM075) was used to resuspend the cell aggregates. Cell clumps were allowed to settle down by gravity in a conical tube. The supernatant was then removed and the cells were resuspended in fresh EB medium. Cell aggregates were plated on ultra low attachment plates (Corning, YO-01835-24) with a split ratio of 1:1 and incubated for 12 to 24 h. Large cell aggregates were collected into a conical tube after the incubation time and allowed to settle down by gravity. The medium was then replaced with differentiation medium (80% DMEM High Glucose (Life Technologies, 11965-092), 55 μM β-Mercaptoethanol (Life Technologies, 21985-023), 2 mM L-glutamine (Cellgro/Mediatech, 25-005-CI), 20% defined fetal bovine serum (FBS, Hyclone, SH30070.03), and 1x non-essential amino acids (NEAA, Life Technologies, 11140-050). The cell aggregates were then cultured in ultra low attachment plates using a split ratio of 1:1 in 0.4 mL differentiation medium/cm2. The differentiation culture medium was changed every other day for six days. EBs were seeded on gelatin-coated plates (EmbryoMax® ES Cell Qualified Gelatin Solution (Millipore, ES006-B) on the seventh day at approximately 10 EBs/cm2. The EBs were allowed to attach to the coated dishes for two days. The medium was changed every other day afterward with 0.4 mL/cm2 differentiation medium. On day 14, spontaneously differentiated iPSCs were fixed and permeabilized with 4% PFA and 0.1% Triton X-100 PBS solution, respectively, as described above. The cells were incubated with DPBS containing 10% goat serum (Life Technologies, 10000C) for 2 h at RT post rinsing with PBS/Tween solution. Primary antibodies detecting beta-III-tubulin (Millipore, MAB1637; 1:400), smooth muscle actin (DAKO, M0851; 1:500), and alpha-1 fetoprotein (Abcam, ab3980; 1:200 or R&D systems, MAB1369, 1:100) were added to blocked cultures and incubated overnight at 2–8 °C. The cells were washed twice with PBS/Tween solution, and the secondary antibodies including Alexa Fluor 488-conjugated goat anti-mouse IgG(H+L) (Life Technologies, A11001; 1:1000) and Alexa Fluor 494-conjugated goat anti-mouse IgG (H+L) (Life Technologies, A-11032; 1:1000) and DAPI solution were added and incubated for at least 2 h at RT. The cultures were then rinsed three times (5 min each) in 1x DPBS and maintained in 50% glycerol for analysis.
4.7. Coating Protocol for Culturing NSCs
In order to prepare the vessels for the NSCs differentiation, each well of a six well plate was coated with 1 mL/well of poly-L-Ornithine (Sigma, P4957) diluted solution (20 µg/mL final concentration). The plates were incubated at 37 °C for 2 h or at 4 °C overnight. The wells were then rinsed twice with DPBS (−/−). Human recombinant Laminin (Biolamina, LN521) solution was added to each well (1 mL/well) at 15 µg/mL concentration and incubated at 37 °C for at least for 1 h, but not for more than 6 h. The Laminin solution was removed prior to plating the iPSCs.
4.8. Neural Stem Cell Differentiation
Cells were passaged from 2D culture using L7TM passaging solution in order to generate small clumps. Cells were plated with L7TM medium onto L7TM matrix coated plates at a 1:10 to 1:20 ratio depending on the harvest cell density. After 16–20 h, the medium was switched to Lonza neural induction medium (3 mL/well of a six well plate, day 0) consisting of primary neuron basal medium (PNBM, Lonza, CC-3256), 1x B27 (ThermoFisher, 17504044), 1x Glutamax (ThermoFisher, 35050-061), 4 µm CHIR99021 (Tocris, 4423), 3 µm SB43152 (Stemgent, 04-0010), and 10 ng/mL human leukemia inhibitory factor (hLIF, Millipore, LIF1010). The media was changed every other day until day 7. The cells were passaged by exposing cells to Accutase for 8–10 min. Y27632 (5 µm/mL) was added to the neural induction medium after plating. The cells were then plated at 1.0 × 106/well in a six well plate that was pre-coated with poly-L-ornithine/Laminin (NSCs-P1). On the following day, the medium was changed using neural induction medium (no Y27632). The medium was changed every other day until cells reached 95%–100% confluency (4–5 days). The cells were then passaged as described above and re-inoculated into new pre-coated (Poly-L-Ornithine/Laminin) wells of a six well plate. Further expansion of the cells was conducted to enable flow cytometry and immunostaining at P3 and cryopreserved at P4.
4.9. Definitive Endoderm Differentiation
iPSCs were induced to differentiate to definitive endoderm (DE) for five days using the STEMdiff Definitive Endoderm kit (Stem Cell Technologies, 05110) with some modifications to the manufacturer’s protocol: (1) All iPSC lines were cultured for 3–4 passages using L7TM medium + matrix prior to differentiation. (2) The gentle cell dissociation reagent (Stem Cell Technologies, Cat#07174) was replaced with TrypLE to prepare the single cell suspension for day 0 of the protocol. (3) The iPSCs were plated at a density of 2.0 × 106 cells/well onto pre-coated plates with L7TM matrix to have a confluency of near 100% on day 1. The manufacturer’s instructions were followed for the rest of the protocol.
4.10. Differentiation of iPSCs to Cardiomyocytes
With small modifications to protocols described by Burridge et al. (Burridge, Matsa et al. 2014), iPSCs were induced to differentiate into cardiomyocytes (CMs). Prior to the initiation of differentiation, the iPSCs were first maintained in Lonza L7TM medium + L7TM matrix for 2–3 passages. On day 0 of differentiation, the cells were at approximately 95%–100% confluency in six well plates with minimal spontaneous differentiation (i.e., <5%) before the start of CM induction. On day 0 of the CM induction, the medium was changed to cardiac differentiation media (CDM) consisting of RPMI1640 basal media (Lonza, 12-702F). The basal media was supplemented with 213 μg/mL l-ascorbic acid 2-phosphate (Sigma, A8960-5mg), 500 μg/mL O. Sativa-derived recombinant human albumin (Sigma, A0237) and 6 µm CHIR99021 (Tocris, 4423) to complete the differentiation media. The cells were placed in a humidified incubator operating at 37 °C and 5% CO2. On day 1, the medium was completely exchanged with the same fresh medium containing CHIR99021. On day 2, the medium was replaced with CDM supplemented with 2 µm Wnt-C59 (Tocris, 5148), and a complete medium exchange was performed on day 3 with the same medium (i.e., CDM + Wnt-C59). On day 5, 6, and every other day up to day 14, a complete medium exchange was performed with CDM (3 mL/well on day 5 and 6; 2 mL/well from day 8 to 14). Spontaneous contractile activity first appeared around day 6–7 on different areas of the cell culture depending on the cell line. Representative images and videos were taken using Nikon Eclipse Ti inverted microscope using NIS Elements software.
4.11. Alkaline Phosphatase Staining
Alkaline phosphatase (AP) staining was performed using the Millipore kit (Cat#SCR066) according to manufacturer’s protocol.
4.12. Karyotype Analysis
Karyotype and Short Tandem Repeat (STR) analyses were performed by a qualified service provider (LabCorp, Santa Fe, MN) using standard G-banding methods.
4.13. Mycoplasma and Sterility Test
Mycoplasma testing was performed using MycoAlert Mycoplasma detection Kit (Lonza, LT07–118) as per manufacturer’s protocol. The supernatant of the cells was examined one day post thaw and after five passages in the 2D culture system. The sterility test was performed using Bac-T Alert sterility test following the manufacturer’s protocol.
4.14. Telomere Size and Telomerase Activity
Telomerase relative activity was measured by quantitative telomeric repeat amplification protocol (Q-TRAP) (Life Length, Madrid, Spain). Briefly, protein extracts were prepared by lysing the cell pellets and quantifying the protein concentration form the resulting lysates. Telomerase protein extracts were then incubated with a specific oligonucleotide substrate to allow the enzymatic addition of telomeric DNA repeats by the endogenous telomerase. Following the enzymatic reaction, telomerase extension products were amplified and quantified by real-time quantitative PCR (qPCR). In real time PCR, a positive reaction was detected by accumulation of a fluorescent signal. The Ct (cycle threshold) was defined as the number of cycles required for fluorescence to cross the threshold (i.e., exceeds background levels). The telomerase-positive standard dilution series was plotted against the telomere protein concentration (r2 > 0.9) as a standard curve of Ct values. The assay was performed in triplicate. The mean and standard deviation (SD) from each triplicate was calculated, which include both positive and negative controls. Data were reported as RTA (relative telomerase activity).
Telomere median length was determined by telomere analysis technology (TAT) using a high throughput Q-FISH (fluorescence in situ hybridization) technique (Life Length, Madrid, Spain). This method was modified for cells in interphase. In brief, telomeres were hybridized with a fluorescent peptide nucleic acid probe (PNA) that recognizes three telomere repeats (sequence: Alexa488-OO-CCCTAACCCTAACCCTAA, Panagene). Using a high-content screen system, the images of the nuclei and telomeres were captured. The length of the telomere was proportional to the intensity of the fluorescent signal from the PNA probe. Using a control cell line with known telomere length, the fluorescence intensities were translated to base pairs through a standard regression curve.
Sample preparation and Q-FISH: the samples and control cell lines were removed from liquid nitrogen storage and thawed in a water bath at 37 °C and counted to determine the cell number and viability.
Cells were seeded at a density of 15,000 cells/well in clear bottom black-walled 384-well plates. Replicates of each sample (five replicates) and each control cell line (five replicates) were seeded and fixed with methanol/acetic acid (3/1, vol/vol).
Once the cells are fixed onto the plate, they were treated with pepsin to digest the cytoplasm and the resulting nuclei are processed for in situ hybridization with the PNA probe. Following washing of the hybridized nuclei, the DNA was stained using a standard DAPI incubation, and the wells were subsequently filled with mounting medium and the plate was stored overnight at 4 °C.
HT Microscopy: the Acapella software, Version 1.8 (Perkin Elmer), was used to acquire and analyze quantitative images on a high content screening opera system (Perkin Elmer). The images for analysis were captured using a 40 × 0.95 NA water immersion objective. DAPI was detected using UV wavelengths and Alexa Flour 488 signals were detected using 488 nm wavelengths. With constant exposure settings, 15 independent images were captured at different positions for each well. Next, the nuclei images were used to define the region of interest for each cell, and the telomere fluorescence intensity of the Alexa Flour 488 image in all nuclei is then measured. The results were then exported to the Columbus 2.4 software (Perkin Elmer) for data analysis. Telomere length distribution and median telomere length were calculated by Life Length’s proprietary algorithms. Statistical analysis of the data was performed using Student’s t-test.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}