Inhibition of Calcium/Calmodulin-Dependent Protein Kinase Kinase β Is Detrimental in Hypoxia–Ischemia Neonatal Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
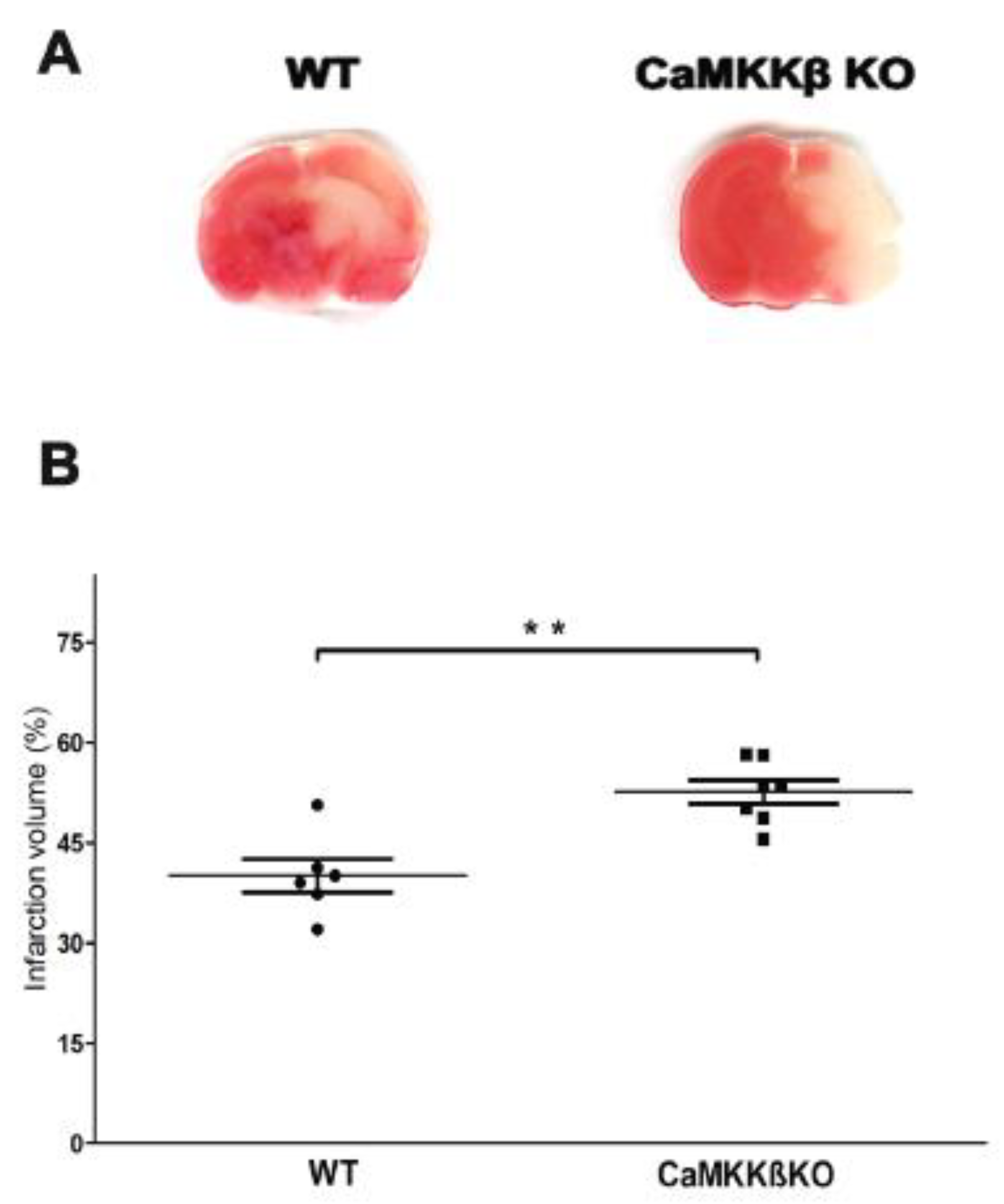
2.1. Deletion of CaMKK β Increased Infarct Volume in Neonatal Mice Assessed 24 h after HI
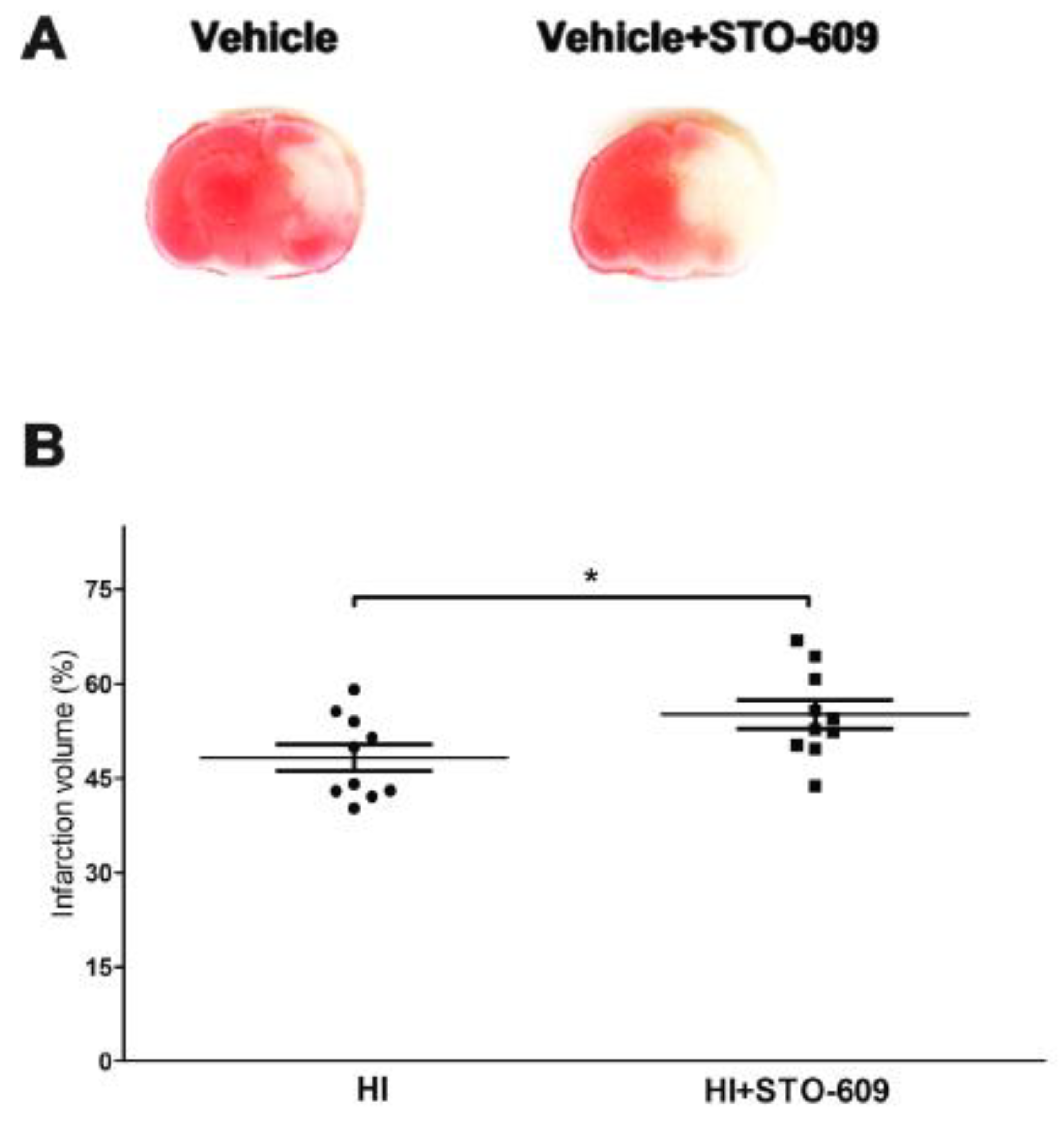
2.2. Inhibition of CaMKK Produced Larger Infarct Volume in Wild-Type (WT) Mice at 24 h Post-HI
2.3. STO-609 Suppressed Functional Recovery at 3 Weeks after HI
2.4. Inhibition of CaMKK Increased HI-Induced Tissue Loss after Long-Term Survival
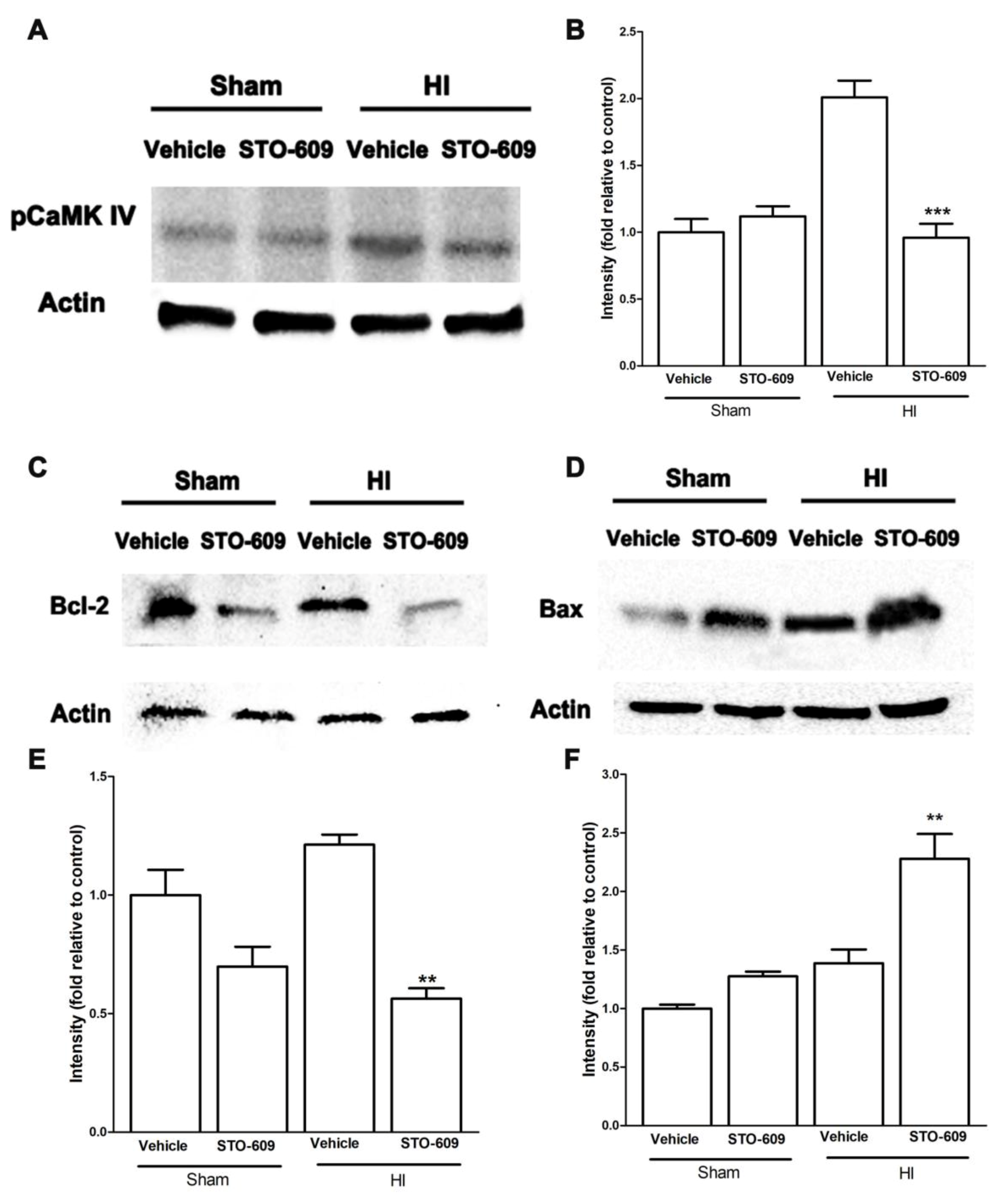
2.5. Inhibition of CaMKK Reduced Levels of pCaMK IV, Bcl-2, Collagen IV and Claudin-5 and Increased Levels of Bax at 6 h after HI
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Animals Model of Hypoxic–Ischemic Encephalopathy (HIE)
4.3. Infarct Volume Qualification
4.4. Neurobehavioral Tests
4.5. Brain Atrophy Measurement
4.6. Western Blotting
4.7. Data Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HI | Hypoxia-Ischemia |
| CaMKK | Calcium/calmodulin-dependent protein kinase kinase |
| WT | Wild type |
| KO | Knockout |
| Bcl-2 | B-cell lymphoma 2 |
| BBB | Blood–brain barrier |
| HDAC4 | Histone deacetylase 4 |
| CREB | cAMP response element-binding protein |
| MMPs | Matrix metalloproteinases |
References
- Min, J.W.; Hu, J.J.; He, M.; Sanchez, R.M.; Huang, W.X.; Liu, Y.Q.; Bsoul, N.B.; Han, S.; Yin, J.; Liu, W.H.; et al. Vitexin reduces hypoxia-ischemia neonatal brain injury by the inhibition of HIF-1alpha in a rat pup model. Neuropharmacology 2015, 99, 38–50. [Google Scholar] [CrossRef]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. New Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ling, E.A. Periventricular white matter damage in the hypoxic neonatal brain: Role of microglial cells. Prog. Neurobiol. 2009, 87, 264–280. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Limburg, M. Calcium antagonists for ischemic stroke: A systematic review. Stroke 2001, 32, 570–576. [Google Scholar] [CrossRef]
- Zundorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- McCullough, L.D.; Tarabishy, S.; Liu, L.; Benashski, S.; Xu, Y.; Ribar, T.; Means, A.; Li, J. Inhibition of calcium/calmodulin-dependent protein kinase kinase beta and calcium/calmodulin-dependent protein kinase IV is detrimental in cerebral ischemia. Stroke 2013, 44, 2559–2566. [Google Scholar] [CrossRef]
- Soderling, T.R. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem. Sci. 1999, 24, 232–236. [Google Scholar] [CrossRef]
- Means, A.R. The Year in Basic Science: Calmodulin kinase cascades. Mol. Endocrinol. 2008, 22, 2759–2765. [Google Scholar] [CrossRef]
- Bolger, T.A.; Yao, T.P. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 2005, 25, 9544–9553. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Denton, K.; Liu, L.; Li, X.J.; Benashski, S.; McCullough, L.; Li, J. Nuclear translocation of histone deacetylase 4 induces neuronal death in stroke. Neurobiol. Dis. 2016, 91, 182–193. [Google Scholar] [CrossRef]
- Durukan, A.; Marinkovic, I.; Strbian, D.; Pitkonen, M.; Pedrono, E.; Soinne, L.; Abo-Ramadan, U.; Tatlisumak, T. Post-ischemic blood-brain barrier leakage in rats: One-week follow-up by MRI. Brain Res. 2009, 1280, 158–165. [Google Scholar] [CrossRef]
- Sun, P.; Bu, F.; Min, J.W.; Munshi, Y.; Howe, M.D.; Liu, L.; Koellhoffer, E.C.; Qi, L.; McCullough, L.D.; Li, J. Inhibition of calcium/calmodulin-dependent protein kinase kinase (CaMKK) exacerbates impairment of endothelial cell and blood-brain barrier after stroke. Eur. J. Neurosci. 2019, 49, 27–39. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. 2008, 32, S55–S59. [Google Scholar] [CrossRef]
- Rousset, C.I.; Leiper, F.C.; Kichev, A.; Gressens, P.; Carling, D.; Hagberg, H.; Thornton, C. A dual role for AMP-activated protein kinase (AMPK) during neonatal hypoxic-ischaemic brain injury in mice. J. Neurochem. 2015, 133, 242–252. [Google Scholar] [CrossRef]
- Wayman, G.A.; Lee, Y.S.; Tokumitsu, H.; Silva, A.J.; Soderling, T.R. Calmodulin-kinases: Modulators of neuronal development and plasticity. Neuron 2008, 59, 914–931. [Google Scholar] [CrossRef]
- Liu, L.; McCullough, L.; Li, J. Genetic deletion of calcium/calmodulin-dependent protein kinase kinase beta (CaMKK beta) or CaMK IV exacerbates stroke outcomes in ovariectomized (OVXed) female mice. BMC Neurosci. 2014, 15, 118. [Google Scholar] [CrossRef]
- Chen, S.D.; Lin, T.K.; Lin, J.W.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 3144–3154. [Google Scholar]
- Sasaki, T.; Takemori, H.; Yagita, Y.; Terasaki, Y.; Uebi, T.; Horike, N.; Takagi, H.; Susumu, T.; Teraoka, H.; Kusano, K.; et al. SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB. Neuron 2011, 69, 106–119. [Google Scholar] [CrossRef]
- Latour, L.L.; Kang, D.W.; Ezzeddine, M.A.; Chalela, J.A.; Warach, S. Early blood-brain barrier disruption in human focal brain ischemia. Ann. Neurol. 2004, 56, 468–477. [Google Scholar] [CrossRef]
- Gaines, P.; Lamoureux, J.; Marisetty, A.; Chi, J.; Berliner, N. A cascade of Ca(2+)/calmodulin-dependent protein kinases regulates the differentiation and functional activation of murine neutrophils. Exp. Hematol. 2008, 36, 832–844. [Google Scholar] [CrossRef]
- Mirza, M.A.; Ritzel, R.; Xu, Y.; McCullough, L.D.; Liu, F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J. Neuroinflamm. 2015, 12, 32. [Google Scholar] [CrossRef]
- Rice, J.E., 3rd; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Comi, A.M.; Weisz, C.J.; Highet, B.H.; Johnston, M.V.; Wilson, M.A. A new model of stroke and ischemic seizures in the immature mouse. Pediatric Neurol. 2004, 31, 254–257. [Google Scholar] [CrossRef]
- Kadam, S.D.; Mulholland, J.D.; Smith, D.R.; Johnston, M.V.; Comi, A.M. Chronic brain injury and behavioral impairments in a mouse model of term neonatal strokes. Behav. Brain Res. 2009, 197, 77–83. [Google Scholar] [CrossRef]
- Shibata, M.; Yamasaki, N.; Miyakawa, T.; Kalaria, R.N.; Fujita, Y.; Ohtani, R.; Ihara, M.; Takahashi, R.; Tomimoto, H. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 2007, 38, 2826–2832. [Google Scholar] [CrossRef]
- Kotoda, M.; Ishiyama, T.; Mitsui, K.; Hishiyama, S.; Matsukawa, T. Neuroprotective effects of amiodarone in a mouse model of ischemic stroke. BMC Anesthesiol. 2017, 17, 168. [Google Scholar] [CrossRef]
- Al Mamun, A.; Yu, H.; Romana, S.; Liu, F. Inflammatory Responses are Sex Specific in Chronic Hypoxic-Ischemic Encephalopathy. Cell Transplant. 2018, 27, 1328–1339. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, J.-W.; Bu, F.; Qi, L.; Munshi, Y.; Kim, G.S.; Marrelli, S.P.; McCullough, L.D.; Li, J. Inhibition of Calcium/Calmodulin-Dependent Protein Kinase Kinase β Is Detrimental in Hypoxia–Ischemia Neonatal Brain Injury. Int. J. Mol. Sci. 2019, 20, 2063. https://doi.org/10.3390/ijms20092063
Min J-W, Bu F, Qi L, Munshi Y, Kim GS, Marrelli SP, McCullough LD, Li J. Inhibition of Calcium/Calmodulin-Dependent Protein Kinase Kinase β Is Detrimental in Hypoxia–Ischemia Neonatal Brain Injury. International Journal of Molecular Sciences. 2019; 20(9):2063. https://doi.org/10.3390/ijms20092063
Chicago/Turabian StyleMin, Jia-Wei, Fan Bu, Li Qi, Yashasvee Munshi, Gab Seok Kim, Sean P. Marrelli, Louise D. McCullough, and Jun Li. 2019. "Inhibition of Calcium/Calmodulin-Dependent Protein Kinase Kinase β Is Detrimental in Hypoxia–Ischemia Neonatal Brain Injury" International Journal of Molecular Sciences 20, no. 9: 2063. https://doi.org/10.3390/ijms20092063
APA StyleMin, J.-W., Bu, F., Qi, L., Munshi, Y., Kim, G. S., Marrelli, S. P., McCullough, L. D., & Li, J. (2019). Inhibition of Calcium/Calmodulin-Dependent Protein Kinase Kinase β Is Detrimental in Hypoxia–Ischemia Neonatal Brain Injury. International Journal of Molecular Sciences, 20(9), 2063. https://doi.org/10.3390/ijms20092063