Metabolic Inhibition Induces Transient Increase of L-type Ca2+ Current in Human and Rat Cardiac Myocytes



Abstract
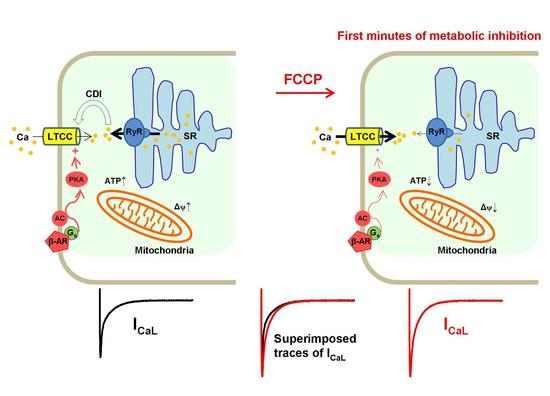
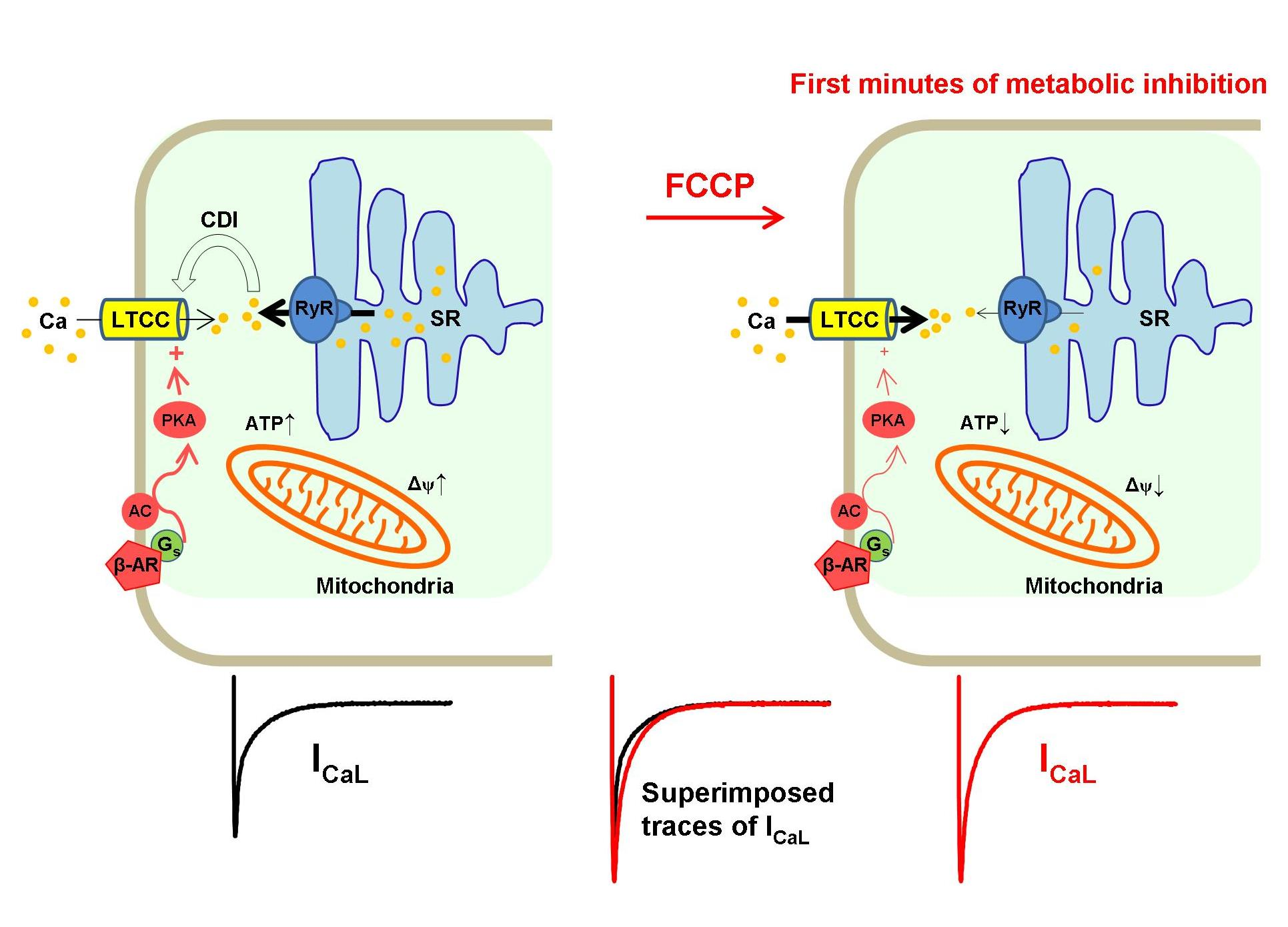{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
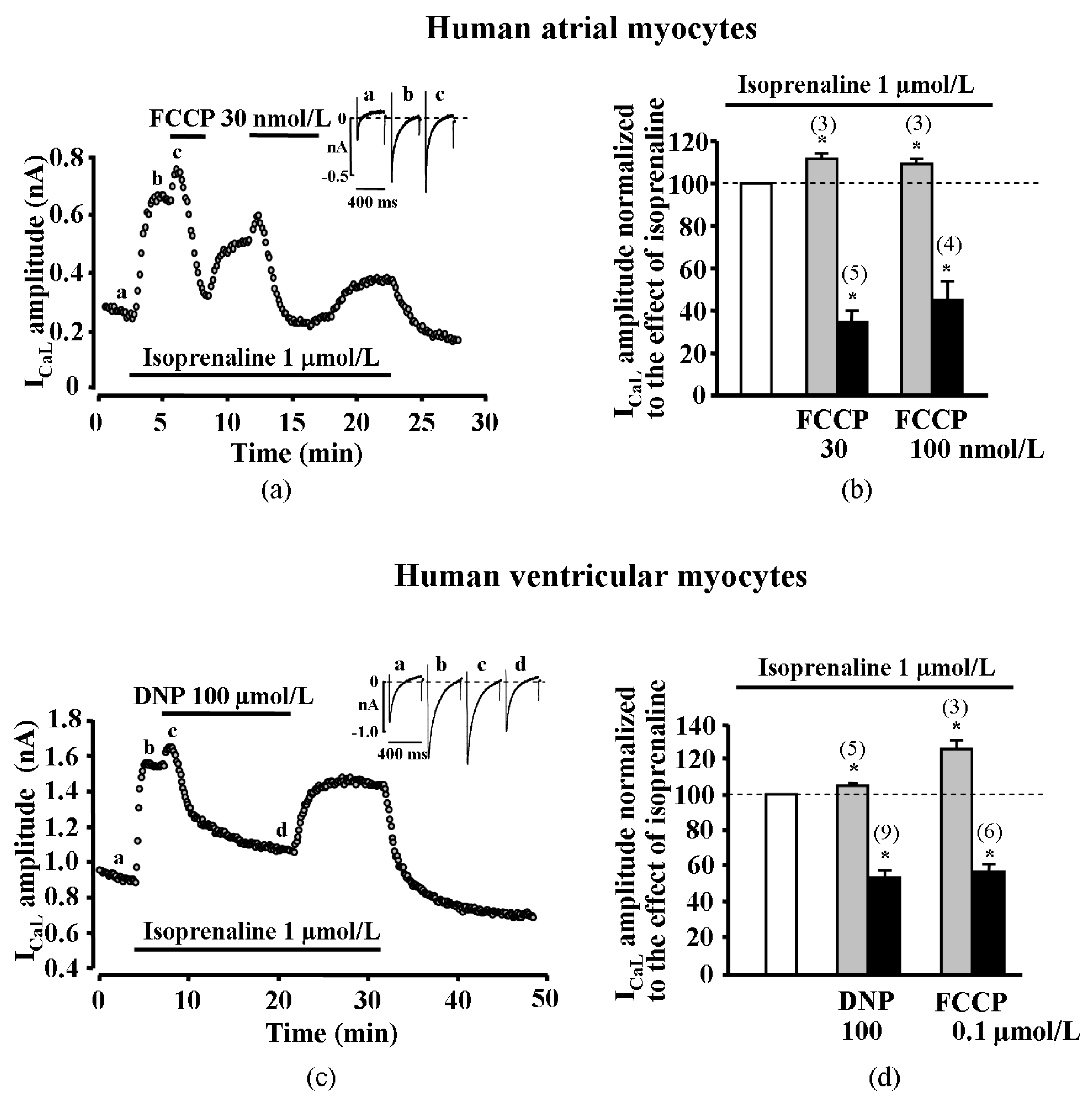
2.1. Effect of Metabolic Inhibition on ICaL in Human Atrial Myocytes
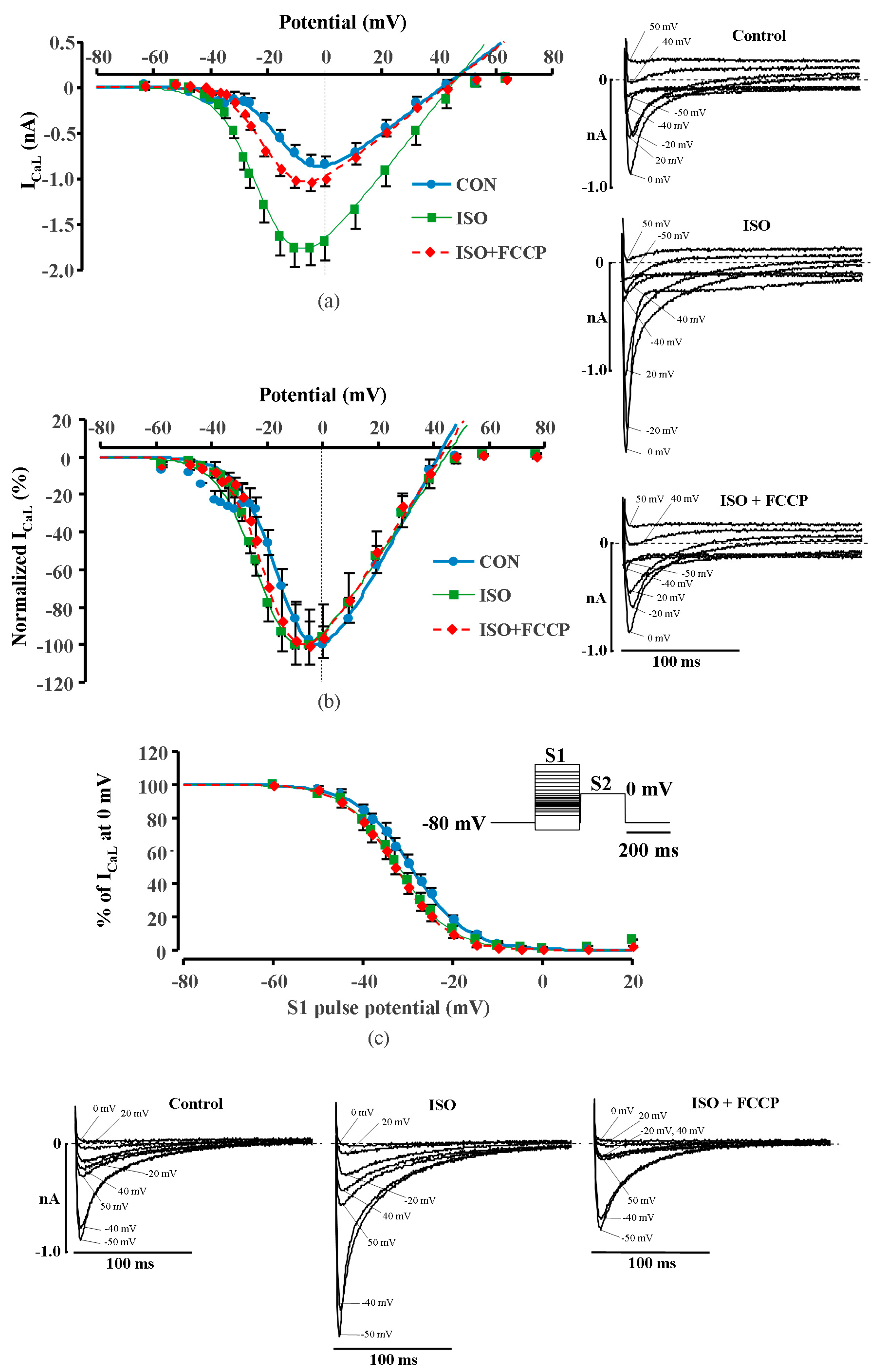
2.2. Effect of Metabolic Inhibition on ICaL in Human Ventricular Myocytes
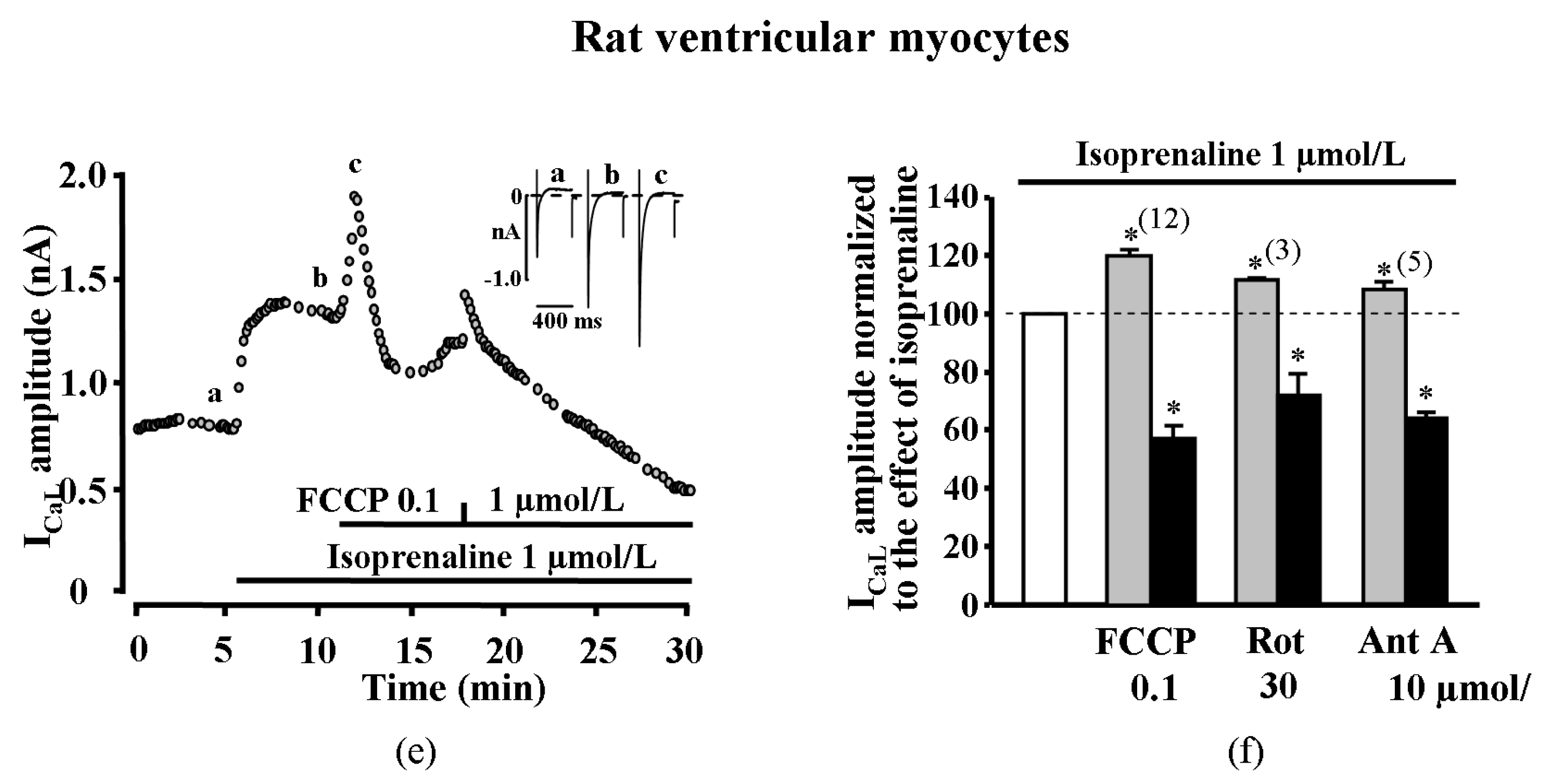
2.3. Transient Increase of ICaL is Also Elicited in Rat Cardiac Myocytes
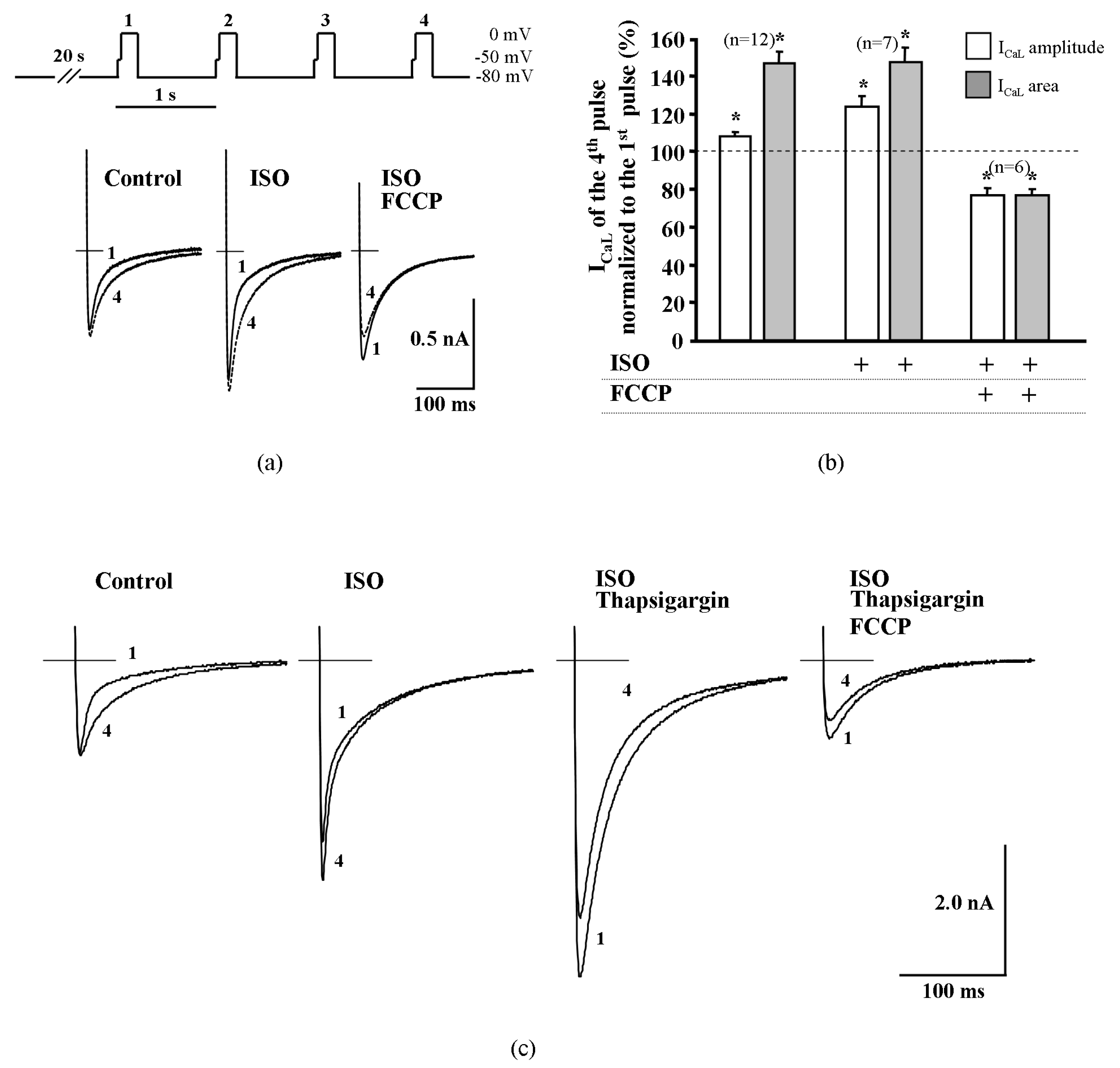
2.4. Effect of Metabolic Inhibition on ICaL Kinetics
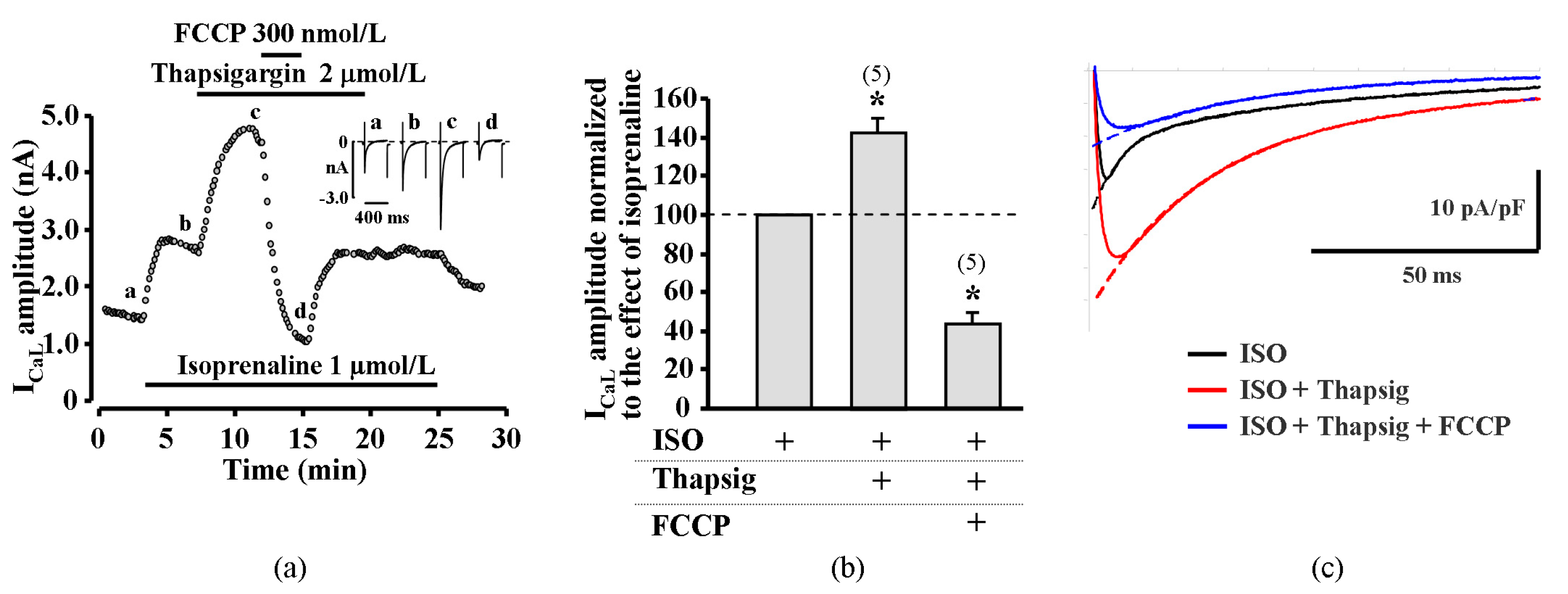
2.5. Suppression of Cytosolic Ca2+ Release Abolish Transient Increase of ICaL
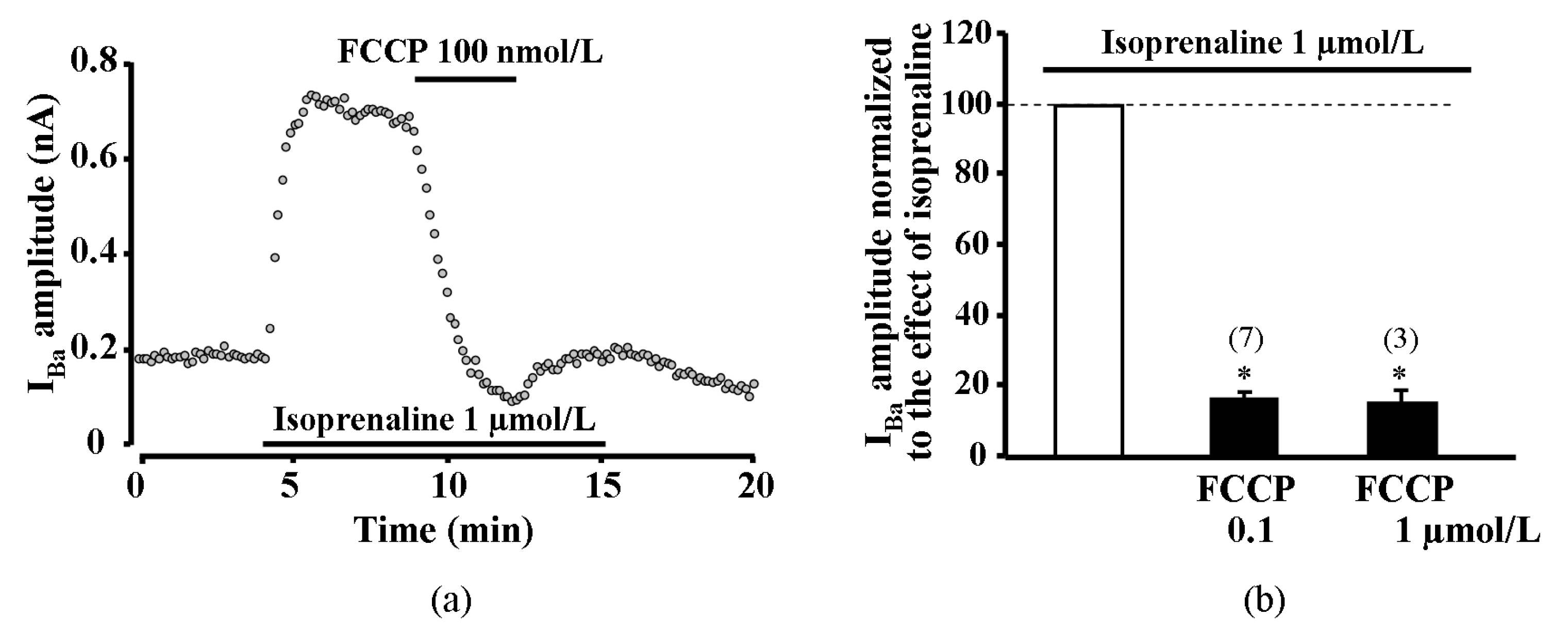
2.6. Effect of Metabolic Inhibition on Ba2+ Current (IBa) through L-Type Calcium Channels
3. Discussion
4. Materials and Methods
4.1. Isolation of Cardiomyocytes
4.1.1. Human Atrial and Ventricular Myocytes
4.1.2. Rat Ventricular Myocytes
4.1.3. Electrophysiology
4.1.4. Chemicals and Stock Solutions
4.1.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LTCC | L-type Ca2+ channel |
| SR | Sarcoplasmic reticulum |
| CDI | Calcium dependent inactivation |
| RyRs | Ryanodine receptors |
| ISO | Isoprenaline |
| FCCP | Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
References
- Bers, D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006, 21, 380–387. [Google Scholar] [CrossRef]
- Sham, J.S. Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes: Evidence for local Ca2+ signalling. J. Physiol. 1997, 500 Pt 2, 285–295. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Perrier, E.; Fauconnier, J.; Perrier, R.; Pereira, L.; Gomez, A.M.; Benitah, J.P. ‘Ca(2+)-induced Ca(2+) entry’ or how the L-type Ca(2+) channel remodels its own signalling pathway in cardiac cells. Prog. Biophys. Mol. Biol. 2006, 90, 118–135. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Veldkamp, M.W.; van Ginneken, A.C.; Bouman, L.N. Biphasic response of action potential duration to metabolic inhibition in rabbit and human ventricular myocytes: Role of transient outward current and ATP-regulated potassium current. J. Mol. Cell. Cardiol. 1996, 28, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Mačianskienė, R.; Martišienė, I.; Navalinskas, A.; Treinys, R.; Jurevičius, J. Mechanism of action potential prolongation during metabolic inhibition in the whole rabbit heart. Front. Physiol. 2018, 9, 1077. [Google Scholar] [CrossRef]
- Chantawansri, C.; Huynh, N.; Yamanaka, J.; Garfinkel, A.; Lamp, S.T.; Inoue, M.; Bridge, J.H.; Goldhaber, J.I. Effect of metabolic inhibition on couplon behavior in rabbit ventricular myocytes. Biophys. J. 2008, 94, 1656–1666. [Google Scholar] [CrossRef]
- Fukumoto, G.H.; Lamp, S.T.; Motter, C.; Bridge, J.H.; Garfinkel, A.; Goldhaber, J.I. Metabolic inhibition alters subcellular calcium release patterns in rat ventricular myocytes: Implications for defective excitation-contraction coupling during cardiac ischemia and failure. Circ. Res. 2005, 96, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Ferrier, G.R.; Howlett, S.E. Changes in excitation-contraction coupling in an isolated ventricular myocyte model of cardiac stunning. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H800–H810. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Pabbidi, M.R.; Lipsius, S.L.; Blatter, L.A. Effects of mitochondrial uncoupling on Ca(2+) signaling during excitation-contraction coupling in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H983–H993. [Google Scholar] [CrossRef]
- Kanaporis, G.; Treinys, R.; Fischmeister, R.; Jurevicius, J. Metabolic inhibition reduces cardiac L-type Ca2+ channel current due to acidification caused by ATP hydrolysis. PLoS ONE 2017, 12, e0184246. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Beta-adrenergic receptors signaling and heart failure in mice, rabbits and humans. J. Mol. Cell. Cardiol. 2006, 41, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Tiaho, F.; Piot, C.; Nargeot, J.; Richard, S. Regulation of the frequency-dependent facilitation of L-type Ca2+ currents in rat ventricular myocytes. J. Physiol. 1994, 477 Pt 2, 237–251. [Google Scholar] [CrossRef]
- Tiaho, F.; Nargeot, J.; Richard, S. Voltage-dependent regulation of L-type cardiac Ca channels by isoproterenol. Pflugers Arch. 1991, 419, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Tijskens, P.; Meissner, G.; Franzini-Armstrong, C. Location of ryanodine and dihydropyridine receptors in frog myocardium. Biophys. J. 2003, 84, 1079–1092. [Google Scholar] [CrossRef]
- Janczewski, A.M.; Lakatta, E.G. Thapsigargin inhibits Ca2+ uptake, and Ca2+ depletes sarcoplasmic reticulum in intact cardiac myocytes. Am. J. Physiol. 1993, 265, H517–H522. [Google Scholar] [CrossRef]
- McHugh, D.; Beech, D.J. Modulation of Ca2+ channel activity by ATP metabolism and internal Mg2+ in guinea-pig basilar artery smooth muscle cells. J. Physiol. 1996, 492 Pt 2, 359–376. [Google Scholar] [CrossRef]
- Cens, T.; Rousset, M.; Leyris, J.P.; Fesquet, P.; Charnet, P. Voltage- and calcium-dependent inactivation in high voltage-gated Ca(2+) channels. Prog. Biophys. Mol. Biol. 2006, 90, 104–117. [Google Scholar] [CrossRef]
- Overend, C.L.; Eisner, D.A.; O’Neill, S.C. Altered cardiac sarcoplasmic reticulum function of intact myocytes of rat ventricle during metabolic inhibition. Circ. Res. 2001, 88, 181–187. [Google Scholar] [CrossRef]
- Hanninen, S.L.; Ronkainen, J.J.; Leskinen, H.; Tavi, P. Mitochondrial uncoupling downregulates calsequestrin expression and reduces SR Ca2+ stores in cardiomyocytes. Cardiovasc. Res. 2010, 88, 75–82. [Google Scholar] [CrossRef]
- Fauconnier, J.; Bedut, S.; Le Guennec, J.Y.; Babuty, D.; Richard, S. Ca2+ current-mediated regulation of action potential by pacing rate in rat ventricular myocytes. Cardiovasc. Res. 2003, 57, 670–680. [Google Scholar] [CrossRef]
- Takamatsu, H.; Nagao, T.; Ichijo, H.; Adachi-Akahane, S. L-type Ca2+ channels serve as a sensor of the SR Ca2+ for tuning the efficacy of Ca2+-induced Ca2+ release in rat ventricular myocytes. J. Physiol. 2003, 552 Pt 2, 415–424. [Google Scholar] [CrossRef]
- Zhang, H.B.; Li, R.C.; Xu, M.; Xu, S.M.; Lai, Y.S.; Wu, H.D.; Xie, X.J.; Gao, W.; Ye, H.; Zhang, Y.Y.; et al. Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc. Res. 2013, 98, 269–276. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Bito, V.; Volders, P.G.; Antoons, G.; Mubagwa, K.; Sipido, K.R. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ. Res. 2002, 91, 1023–1030. [Google Scholar] [CrossRef]
- Zhao, Z.; Gordan, R.; Wen, H.; Fefelova, N.; Zang, W.J.; Xie, L.H. Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PLoS ONE 2013, 8, e80574. [Google Scholar] [CrossRef]
- Kirstein, M.; Rivet-Bastide, M.; Hatem, S.; Benardeau, A.; Mercadier, J.J.; Fischmeister, R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. J. Clin. Investig. 1995, 95, 794–802. [Google Scholar] [CrossRef]
- Grundy, D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J. Physiol. 2015, 593, 2547–2549. [Google Scholar] [CrossRef]
- Verde, I.; Vandecasteele, G.; Lezoualc’h, F.; Fischmeister, R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br. J. Pharmacol. 1999, 127, 65–74. [Google Scholar] [CrossRef]
- Treinys, R.; Bogdelis, A.; Rimkute, L.; Jurevicius, J.; Skeberdis, V.A. Differences in the control of basal L-type Ca(2+) current by the cyclic AMP signaling cascade in frog, rat, and human cardiac myocytes. J. Physiol. Sci. 2016, 66, 327–336. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Fontanaud, P.; Noble, P.J.; Noble, D.; Benkemoun, H.; Nargeot, J.; Richard, S. Facilitation of the L-type calcium current in rabbit sino-atrial cells: Effect on cardiac automaticity. Cardiovasc. Res. 2000, 48, 375–392. [Google Scholar] [CrossRef]
- Richard, S.; Charnet, P.; Nerbonne, J.M. Interconversion between distinct gating pathways of the high threshold calcium channel in rat ventricular myocytes. J. Physiol. 1993, 462, 197–228. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Treinys, R.; Kanaporis, G.; Fischmeister, R.; Jurevičius, J. Metabolic Inhibition Induces Transient Increase of L-type Ca2+ Current in Human and Rat Cardiac Myocytes. Int. J. Mol. Sci. 2019, 20, 1501. https://doi.org/10.3390/ijms20061501
Treinys R, Kanaporis G, Fischmeister R, Jurevičius J. Metabolic Inhibition Induces Transient Increase of L-type Ca2+ Current in Human and Rat Cardiac Myocytes. International Journal of Molecular Sciences. 2019; 20(6):1501. https://doi.org/10.3390/ijms20061501
Chicago/Turabian StyleTreinys, Rimantas, Giedrius Kanaporis, Rodolphe Fischmeister, and Jonas Jurevičius. 2019. "Metabolic Inhibition Induces Transient Increase of L-type Ca2+ Current in Human and Rat Cardiac Myocytes" International Journal of Molecular Sciences 20, no. 6: 1501. https://doi.org/10.3390/ijms20061501
APA StyleTreinys, R., Kanaporis, G., Fischmeister, R., & Jurevičius, J. (2019). Metabolic Inhibition Induces Transient Increase of L-type Ca2+ Current in Human and Rat Cardiac Myocytes. International Journal of Molecular Sciences, 20(6), 1501. https://doi.org/10.3390/ijms20061501