Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Androgen Receptor Signaling in Prostate Cancer
3. Wnt/β-Catenin Signaling in PCa
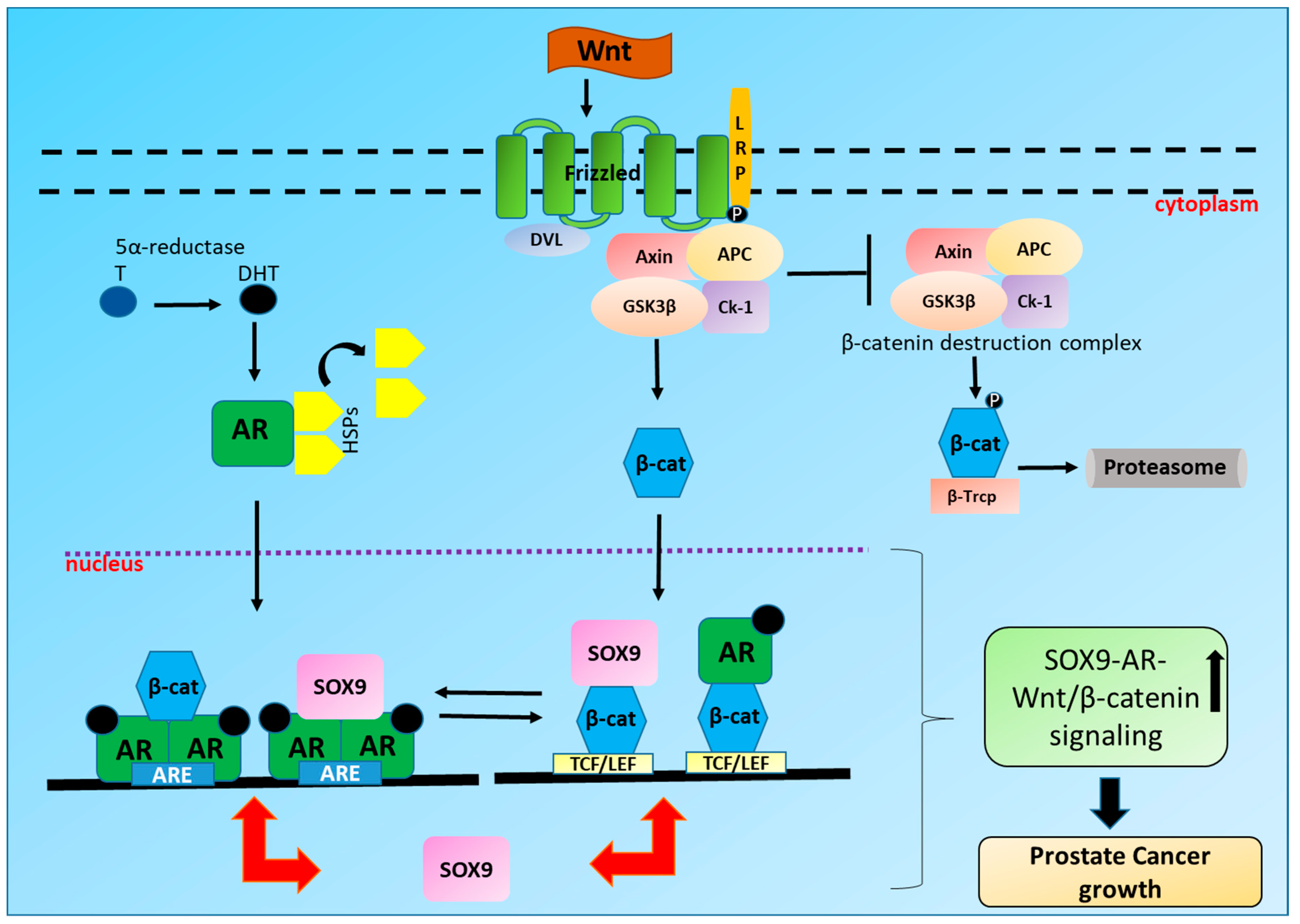
4. Interplay between AR and Wnt/β-Catenin Signaling in PCa
5. SOX9
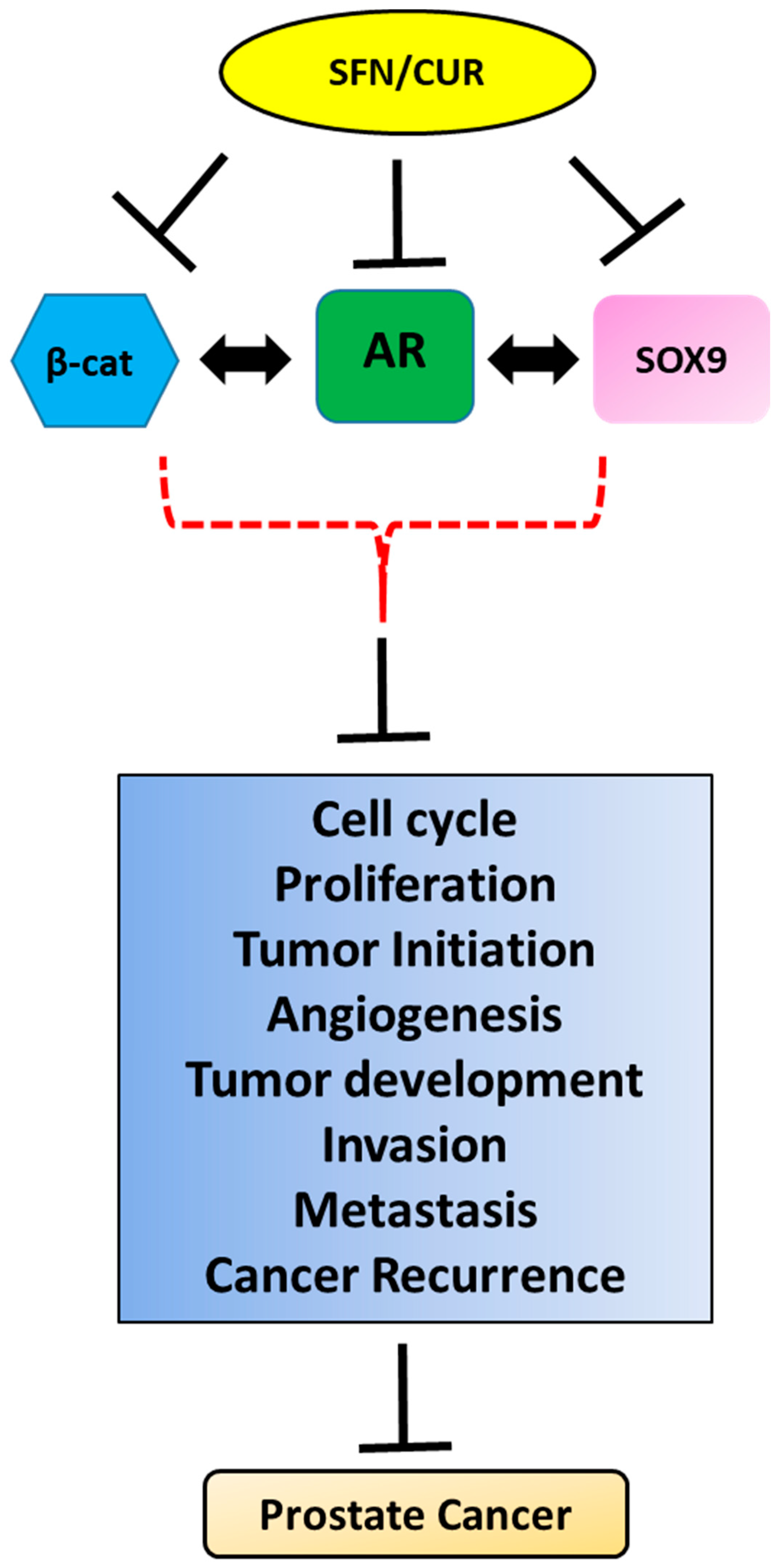
6. SOX9 and AR Signaling
7. SOX9 and Wnt/β-Catenin Signaling
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| PCa | Prostate cancer |
| CRPC | Castration-resistant prostate cancer |
| LBD | Ligand binding domain |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Brawer, M.K.; Chetner, M.P.; Beatie, J.; Buchner, D.M.; Vessella, R.L.; Lange, P.H. Screening for prostatic carcinoma with prostate specific antigen. J. Urol. 1992, 147, 841–845. [Google Scholar] [CrossRef]
- Gordetsky, J.; Epstein, J. Grading of prostatic adenocarcinoma: Current state and prognostic implications. Diagn. Pathol. 2016, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Garnick, M.B. Prostate cancer: Screening, diagnosis, and management. Ann. Intern. Med. 1993, 118, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Partin, A.W.; Kattan, M.W.; Subong, E.N.P.; Walsh, P.C.; Wojno, K.J.; Oesterling, J.E.; Scardino, P.T.; Pearson, J.D. Combination of Prostate-Specific Antigen, Clinical Stage, and Gleason Score to Predict Pathological Stage of Localized Prostate Cancer. JAMA 1997, 277, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Kohli, M.; Tindall, D.J. New Developments in the Medical Management of Prostate Cancer. Mayo Clin. Proc. 2010, 85, 77–86. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharm. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Zhou, Y.; Bolton, E.C.; Jones, J.O. Androgens and androgen receptor signaling in prostate tumorigenesis. J. Mol. Endocrinol. 2015, 54, R15–R29. [Google Scholar] [CrossRef]
- Hodgson, M.C.; Bowden, W.A.; Agoulnik, I.U. Androgen receptor footprint on the way to prostate cancer progression. World J. Urol. 2012, 30, 279–285. [Google Scholar] [CrossRef]
- Eder, I.E.; Culig, Z.; Putz, T.; Nessler-Menardi, C.; Bartsch, G.; Klocker, H. Molecular Biology of the Androgen Receptor: From Molecular Understanding to the Clinic. Eur. Urol. 2001, 40, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Whitbread, A.K.; Veveris-Lowe, T.L.; Lawrence, M.G.; Nicol, D.L.; Clements, J.A. The role of kallikrein-related peptidases in prostate cancer: Potential involvement in an epithelial to mesenchymal transition. Biol. Chem. 2006, 387, 707–714. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar] [PubMed]
- Debes, J.D.; Tindall, D.J. The role of androgens and the androgen receptor in prostate cancer. Cancer Lett. 2002, 187, 1–7. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef]
- Suzuki, H.; Ueda, T.; Ichikawa, T.; Ito, H. Androgen receptor involvement in the progression of prostate cancer. Endocr. Relat. Cancer 2003, 10, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Brooke, G.; Bevan, C. The Role of Androgen Receptor Mutations in Prostate Cancer Progression. Curr. Genom. 2009, 10, 18–25. [Google Scholar] [CrossRef]
- Yuan, X.; Cai, C.; Chen, S.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014, 33, 2815–2825. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. The role of the androgen receptor in prostate cancer. Crit. Rev. Eukaryot. Gene Expr. 2002, 12, 193–207. [Google Scholar] [CrossRef]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.T.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Ryan, C.J. Androgen Receptor Directed Therapies in Castration-Resistant Metastatic Prostate Cancer. Curr. Treat. Options Oncol. 2012, 13, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. AFFIRM Investigators Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Cheng, M.L. Abiraterone acetate for the treatment of prostate cancer. Expert Opin. Pharmacother. 2013, 14, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yeh, S.; Niu, Y.; Li, G.; Zheng, J.; Li, L.; Chang, C. Targeting androgen receptor versus targeting androgens to suppress castration resistant prostate cancer. Cancer Lett. 2017, 397, 133–143. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS ONE 2011, 6, e27970. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Z.; Xiao, W.; Yan, L.; Guan, W.; Hu, Z.; Wu, L.; Huang, Q.; Wang, J.; Xu, H.; et al. Androgen-receptor splice variant-7-positive prostate cancer: A novel molecular subtype with markedly worse androgen-deprivation therapy outcomes in newly diagnosed patients. Mod. Pathol. 2018, 31, 198–208. [Google Scholar] [CrossRef]
- Dong, Y.; Sartor, O. Androgen receptor variant-7: An important predictive biomarker in castrate resistant prostate cancer. Asian J. Androl. 2015, 17, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Welti, J.; Blagg, J.; De Bono, J.S. Targeting Androgen Receptor Aberrations in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4280–4282. [Google Scholar] [CrossRef]
- Khurana, N.; Talwar, S.; Chandra, P.K.; Sharma, P.; Abdel-Mageed, A.B.; Mondal, D.; Sikka, S.C. Sulforaphane increases the efficacy of anti-androgens by rapidly decreasing androgen receptor levels in prostate cancer cells. Int. J. Oncol. 2016, 49, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Kim, H.; Chandra, P.K.; Talwar, S.; Sharma, P.; Abdel-Mageed, A.B.; Sikka, S.C.; Mondal, D. Multimodal actions of the phytochemical sulforaphane suppress both AR and AR-V7 in 22Rv1 cells: Advocating a potent pharmaceutical combination against castration-resistant prostate cancer. Oncol. Rep. 2017, 38, 2774–2786. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. Androgen receptor cross-talk with cell signalling pathways. Growth Factors 2004, 22, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Sikka, S. Targeting Crosstalk between Nrf-2, NF-κB and Androgen Receptor Signaling in Prostate Cancer. Cancers 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. THE WNT SIGNALING PATHWAY IN DEVELOPMENT AND DISEASE. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Logan, S.K. Revisiting the role of Wnt/β-catenin signaling in prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 3–8. [Google Scholar] [CrossRef]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/β-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Wodarz, A.; Nusse, R. MECHANISMS OF WNT SIGNALING IN DEVELOPMENT. Annu. Rev. Cell Dev. Biol. 1998, 14, 59–88. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Hennighausen, L. Beta-catenin: A transforming actor on many stages. Breast Cancer Res. 2003, 5, 63–68. [Google Scholar] [CrossRef]
- Moon, R.T.; Kohn, A.D.; De Ferrari, G.V.; Kaykas, A. WNT and β-catenin signalling: Diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y.; Jiang, M.; Bierie, B.; Roy-Burman, P.; Shen, M.M.; Taketo, M.M.; Wills, M.; Matusik, R.J. Activation of β-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate 2009, 69, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar] [CrossRef] [PubMed]
- Beildeck, M.E.; Gelmann, E.P.; Byers, S.W. Cross-regulation of signaling pathways: An example of nuclear hormone receptors and the canonical Wnt pathway. Exp. Cell Res. 2010, 316, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Chesire, D.R.; Isaacs, W.B. Beta-catenin signaling in prostate cancer: An early perspective. Endocr. Relat. Cancer 2003, 10, 537–560. [Google Scholar] [CrossRef]
- Kypta, R.M.; Waxman, J. Wnt/β-catenin signalling in prostate cancer. Nat. Rev. Urol. 2012, 9, 418–428. [Google Scholar] [CrossRef]
- Nowicki, A.; Sporny, S.; Duda-Szymańska, J. β-catenin as a prognostic factor for prostate cancer (PCa). Cent. Eur. J. Urol. 2012, 65, 119–123. [Google Scholar] [CrossRef]
- Chesire, D.R.; Ewing, C.M.; Sauvageot, J.; Bova, G.S.; Isaacs, W.B. Detection and analysis of beta-catenin mutations in prostate cancer. Prostate 2000, 45, 323–334. [Google Scholar] [CrossRef]
- Voeller, H.J.; Truica, C.I.; Gelmann, E.P. Beta-catenin mutations in human prostate cancer. Cancer Res. 1998, 58, 2520–2523. [Google Scholar]
- Gerstein, A.V.; Almeida, T.A.; Zhao, G.; Chess, E.; Shih, I.-M.; Buhler, K.; Pienta, K.; Rubin, M.A.; Vessella, R.; Papadopoulos, N. APC/CTNNB1 (beta-catenin) pathway alterations in human prostate cancers. Genes. Chromosomes Cancer 2002, 34, 9–16. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Y.; DeGraff, D.J.; Wills, M.L.; Matusik, R.J. Wnt/β-Catenin activation promotes prostate tumor progression in a mouse model. Oncogene 2011, 30, 1868–1879. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; Yu, X.; De Marzo, A.M.; Spiering, T.J.; Matusik, R.J.; Williams, B.O. Activation of Wnt/β-catenin signaling in a subpopulation of murine prostate luminal epithelial cells induces high grade prostate intraepithelial neoplasia. Prostate 2014, 74, 1506–1520. [Google Scholar] [CrossRef]
- Chesire, D.R.; Ewing, C.M.; Gage, W.R.; Isaacs, W.B. In vitro evidence for complex modes of nuclear β-catenin signaling during prostate growth and tumorigenesis. Oncogene 2002, 21, 2679–2694. [Google Scholar] [CrossRef]
- De la Taille, A.; Rubin, M.A.; Chen, M.-W.; Vacherot, F.; de Medina, S.G.-D.; Burchardt, M.; Buttyan, R.; Chopin, D. Beta-catenin-related anomalies in apoptosis-resistant and hormone-refractory prostate cancer cells. Clin. Cancer Res. 2003, 9, 1801–1807. [Google Scholar]
- Patriarca, C.; Petrella, D.; Campo, B.; Colombo, P.; Giunta, P.; Parente, M.; Zucchini, N.; Mazzucchelli, R.; Montironi, R. Elevated E-cadherin and alpha/beta-catenin expression after androgen deprivation therapy in prostate adenocarcinoma. Pathol. Res. Pr. 2003, 199, 659–665. [Google Scholar] [CrossRef]
- Chen, G.; Shukeir, N.; Potti, A.; Sircar, K.; Aprikian, A.; Goltzman, D.; Rabbani, S.A. Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma. Cancer 2004, 101, 1345–1356. [Google Scholar] [CrossRef]
- Murillo-Garzón, V.; Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 2017, 14, 683–696. [Google Scholar] [CrossRef]
- Pawlowski, J.E.; Ertel, J.R.; Allen, M.P.; Xu, M.; Butler, C.; Wilson, E.M.; Wierman, M.E. Liganded Androgen Receptor Interaction with β-Catenin: Nuclear co-localization and modulation of transcriptional activity in neuronal cells. J. Biol. Chem. 2002, 277, 20702–20710. [Google Scholar] [CrossRef] [PubMed]
- Song, L.-N.; Herrell, R.; Byers, S.; Shah, S.; Wilson, E.M.; Gelmann, E.P. Beta-catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N-terminal domain and TIF2 on ligand-dependent transcription. Mol. Cell. Biol. 2003, 23, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, X.; Sharma, M.; Sasaki, C.Y.; Longo, D.L.; Lim, B.; Sun, Z. Linking β-Catenin to Androgen-signaling Pathway. J. Biol. Chem. 2002, 277, 11336–11344. [Google Scholar] [CrossRef]
- Masiello, D.; Chen, S.-Y.; Xu, Y.; Verhoeven, M.C.; Choi, E.; Hollenberg, A.N.; Balk, S.P. Recruitment of β-Catenin by Wild-Type or Mutant Androgen Receptors Correlates with Ligand-Stimulated Growth of Prostate Cancer Cells. Mol. Endocrinol. 2004, 18, 2388–2401. [Google Scholar] [CrossRef]
- Yumoto, F.; Nguyen, P.; Sablin, E.P.; Baxter, J.D.; Webb, P.; Fletterick, R.J. Structural basis of coactivation of liver receptor homolog-1 by β-catenin. Proc. Natl. Acad. Sci. USA 2012, 109, 143–148. [Google Scholar] [CrossRef]
- Truica, C.I.; Byers, S.; Gelmann, E.P. Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000, 60, 4709–4713. [Google Scholar] [PubMed]
- Yang, X.; Chen, M.-W.; Terry, S.; Vacherot, F.; Bemis, D.L.; Capodice, J.; Kitajewski, J.; de la Taille, A.; Benson, M.C.; Guo, Y.; et al. Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene 2006, 25, 3436–3444. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Yang, X.; Chen, M.-W.; Vacherot, F.; Buttyan, R. Multifaceted interaction between the androgen and Wnt signaling pathways and the implication for prostate cancer. J. Cell. Biochem. 2006, 99, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, L.; Rizzo, C.A.; Spires, T.E.; Platero, J.S.; Wu, Q.; Lin, T.-A.; Gottardis, M.M.; Attar, R.M. The androgen receptor can signal through Wnt/β-Catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol. 2008, 9, 4. [Google Scholar] [CrossRef]
- Wan, X.; Liu, J.; Lu, J.-F.; Tzelepi, V.; Yang, J.; Starbuck, M.W.; Diao, L.; Wang, J.; Efstathiou, E.; Vazquez, E.S.; et al. Activation of β-Catenin Signaling in Androgen Receptor-Negative Prostate Cancer Cells. Clin. Cancer Res. 2012, 18, 726–736. [Google Scholar] [CrossRef]
- Rajan, P.; Sudbery, I.M.; Villasevil, M.E.M.; Mui, E.; Fleming, J.; Davis, M.; Ahmad, I.; Edwards, J.; Sansom, O.J.; Sims, D.; et al. Next-generation Sequencing of Advanced Prostate Cancer Treated with Androgen-deprivation Therapy. Eur. Urol. 2014, 66, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.J.; Oh, S.; Lee, G.T.; Chung, J.; Min, K.; Yoon, J.; Kim, W.; Ryu, D.S.; Kim, I.Y.; Kang, D., II. Clinical Significance of Wnt/β-Catenin Signalling and Androgen Receptor Expression in Prostate Cancer. World J. Mens. Health 2013, 31, 36. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Luong, R.; Johnson, D.T.; Cunha, G.R.; Rivina, L.; Gonzalgo, M.L.; Sun, Z. Androgen signaling is a confounding factor for β-catenin-mediated prostate tumorigenesis. Oncogene 2016, 35, 702–714. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, L.-N.; Gelmann, E.P. Interaction of β-Catenin and TIF2/GRIP1 in Transcriptional Activation by the Androgen Receptor. J. Biol. Chem. 2005, 280, 37853–37867. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kim, J.H.; Koh, S.S.; Stallcup, M.R. Synergistic Effects of Coactivators GRIP1 and β-Catenin on Gene Activation CROSS-TALK BETWEEN ANDROGEN RECEPTOR AND Wnt SIGNALING PATHWAYS*. J. Biol. Chem. 2004, 279, 4212–4220. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M.; Zhu, C.; Sun, J.; Weis, W.I.; Sun, Z. The β-Catenin Binding Protein ICAT Modulates Androgen Receptor Activity. Mol. Endocrinol. 2011, 25, 1677–1688. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clark, E.L.; Coulson, A.; Dalgliesh, C.; Rajan, P.; Nicol, S.M.; Fleming, S.; Heer, R.; Gaughan, L.; Leung, H.Y.; Elliott, D.J.; et al. The RNA Helicase p68 Is a Novel Androgen Receptor Coactivator Involved in Splicing and Is Overexpressed in Prostate Cancer. Cancer Res. 2008, 68, 7938–7946. [Google Scholar] [CrossRef]
- Clark, E.L.; Hadjimichael, C.; Temperley, R.; Barnard, A.; Fuller-Pace, F.V.; Robson, C.N. p68/DdX5 Supports β-Catenin & RNAP II during Androgen Receptor Mediated Transcription in Prostate Cancer. PLoS ONE 2013, 8, e54150. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Chen, K.; Haendler, B.; McDonald, T.V.; Bian, J.-S. Stimulation of N-Terminal Truncated Isoform of Androgen Receptor Stabilizes Human Ether-á-go-go-Related Gene-Encoded Potassium Channel Protein via Activation of Extracellular Signal Regulated Kinase 1/2. Endocrinology 2008, 149, 5061–5069. [Google Scholar] [CrossRef]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef]
- Francis, J.C.; Thomsen, M.K.; Taketo, M.M.; Swain, A. β-Catenin Is Required for Prostate Development and Cooperates with Pten Loss to Drive Invasive Carcinoma. PLoS Genet. 2013, 9, e1003180. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Cheng, H.; Reid, K.; Rennie, P.S.; Nelson, C.C. The Androgen Receptor Can Promote β-Catenin Nuclear Translocation Independently of Adenomatous Polyposis Coli. J. Biol. Chem. 2002, 277, 17933–17943. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Madar, A.; David, G.; Garabedian, M.J.; DasGupta, R.; Logan, S.K. Inhibition of androgen receptor and β-catenin activity in prostate cancer. Proc. Natl. Acad. Sci. 2013, 110, 15710–15715. [Google Scholar] [CrossRef]
- Koh, S.S.; Li, H.; Lee, Y.-H.; Widelitz, R.B.; Chuong, C.-M.; Stallcup, M.R. Synergistic Coactivator Function by Coactivator-associated Arginine Methyltransferase (CARM) 1 and β-Catenin with Two Different Classes of DNA-binding Transcriptional Activators. J. Biol. Chem. 2002, 277, 26031–26035. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. β-catenin signaling and cancer. Bioessays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Kuefer, R.; Ethier, S.P.; Day, M.L. Cleavage of β-Catenin by Calpain in Prostate and Mammary Tumor Cells. Cancer Res. 2004, 64, 7237–7240. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, A.S.L.; Tsang, D.P.; Li, M.S.; Go, M.Y.; Cheung, Y.S.; Zhao, G.; Ng, S.S.; Lin, M.C.; Yu, J.; et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives β-catenin/T cell factor-dependent hepatocarcinogenesis. J. Clin. Investig. 2011, 121, 3159–3175. [Google Scholar] [CrossRef] [PubMed]
- Awuah, P.K.; Monga, S.P. Cell cycle-related Kinase links androgen receptor & β-catenin signaling in HCC: Why men are at a loss? Hepatology 2012, 55, 970. [Google Scholar] [CrossRef]
- Lin, C.; Yin, Y.; Stemler, K.; Humphrey, P.; Kibel, A.S.; Mysorekar, I.U.; Ma, L. Constitutive β-Catenin Activation Induces Male-Specific Tumorigenesis in the Bladder Urothelium. Cancer Res. 2013, 73, 5914–5925. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Y.; Izumi, K.; Ishiguro, H.; Ye, B.; Li, F.; Miyamoto, H. Androgen activates β-catenin signaling in bladder cancer cells. Endocr. Relat. Cancer 2013, 20, 293–304. [Google Scholar] [CrossRef]
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty pairs of sox: Extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev. Cell 2002, 3, 167–170. [Google Scholar] [CrossRef]
- Thomsen, M.K.; Francis, J.C.; Swain, A. The role of Sox9 in prostate development. Differentiation 2008, 76, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.K.; Butler, C.M.; Shen, M.M.; Swain, A. Sox9 is required for prostate development. Dev. Biol. 2008, 316, 302–311. [Google Scholar] [CrossRef]
- Wang, H.; McKnight, N.C.; Zhang, T.; Lu, M.L.; Balk, S.P.; Yuan, X. SOX9 Is Expressed in Normal Prostate Basal Cells and Regulates Androgen Receptor Expression in Prostate Cancer Cells. Cancer Res. 2007, 67, 528–536. [Google Scholar] [CrossRef]
- Wang, H.; Leav, I.; Ibaragi, S.; Wegner, M.; Hu, G.-f.; Lu, M.L.; Balk, S.P.; Yuan, X. SOX9 Is Expressed in Human Fetal Prostate Epithelium and Enhances Prostate Cancer Invasion. Cancer Res. 2008, 68, 1625–1630. [Google Scholar] [CrossRef]
- Schaeffer, E.M.; Marchionni, L.; Huang, Z.; Simons, B.; Blackman, A.; Yu, W.; Parmigiani, G.; Berman, D.M. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene 2008, 27, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Qin, G.; Dai, Q.; Han, Z.; Chen, S.; Ling, X.; Fu, X.; Cai, C.; Chen, J.; Chen, X.; et al. SOXs in human prostate cancer: Implication as progression and prognosis factors. BMC Cancer 2012, 12, 248. [Google Scholar] [CrossRef]
- Huang, Z.; Hurley, P.J.; Simons, B.W.; Marchionni, L.; Berman, D.M.; Ross, A.E.; Schaeffer, E.M. Sox9 is required for prostate development and prostate cancer initiation. Oncotarget 2012, 3, 651–663. [Google Scholar] [CrossRef]
- Francis, J.C.; Capper, A.; Ning, J.; Knight, E.; de Bono, J.; Swain, A. SOX9 is a driver of aggressive prostate cancer by promoting invasion, cell fate and cytoskeleton alterations and epithelial to mesenchymal transition. Oncotarget 2018, 9, 7604–7615. [Google Scholar] [CrossRef]
- Xi, M.; Wan, S.; Hua, W.; Zhou, Y.; Jiang, W.; Hu, J. Correlation of SOX9 and NM23 genes with the incidence and prognosis of prostate cancer. Oncol. Lett. 2018, 17, 2296–2302. [Google Scholar] [CrossRef]
- Thomsen, M.K.; Ambroisine, L.; Wynn, S.; Cheah, K.S.E.; Foster, C.S.; Fisher, G.; Berney, D.M.; Møller, H.; Reuter, V.E.; Scardino, P.; et al. Transatlantic Prostate Group SOX9 Elevation in the Prostate Promotes Proliferation and Cooperates with PTEN Loss to Drive Tumor Formation. Cancer Res. 2010, 70, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, R.S.; Werner, L.; Regan, M.M.; Yannucci, J.; Ko, Y.-J.; Wang, H.-Y.; Rosen, S.; Genega, E.; Morrissey, M.-E.; Duggan, S.; et al. Possible Risk Factors Associated with Relapse in Patients Treated with Neoadjuvant Chemohormonal Therapy for High Risk Prostate Cancer. Open Prostate Cancer J. 2011, 4, 6–13. [Google Scholar] [CrossRef]
- Wang, G.; Lunardi, A.; Zhang, J.; Chen, Z.; Ala, U.; Webster, K.A.; Tay, Y.; Gonzalez-Billalabeitia, E.; Egia, A.; Shaffer, D.R.; et al. Zbtb7a suppresses prostate cancer through repression of a Sox9-dependent pathway for cellular senescence bypass and tumor invasion. Nat. Genet. 2013, 45, 739–746. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef]
- Cai, C.; Wang, H.; He, H.H.; Chen, S.; He, L.; Ma, F.; Mucci, L.; Wang, Q.; Fiore, C.; Sowalsky, A.G.; et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Investig. 2013, 123, 1109–1122. [Google Scholar] [CrossRef]
- Correa, R.G.; Krajewska, M.; Ware, C.F.; Gerlic, M.; Reed, J.C. The NLR-related protein NWD1 is associated with prostate cancer and modulates androgen receptor signaling. Oncotarget 2014, 5, 1666–1682. [Google Scholar] [CrossRef]
- Kormish, J.D.; Sinner, D.; Zorn, A.M. Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev. Dyn. 2010, 239, 56–68. [Google Scholar] [CrossRef]
- Blache, P.; van de Wetering, M.; Duluc, I.; Domon, C.; Berta, P.; Freund, J.-N.; Clevers, H.; Jay, P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J. Cell Biol. 2004, 166, 37–47. [Google Scholar] [CrossRef]
- Ma, F.; Ye, H.; He, H.H.; Gerrin, S.J.; Chen, S.; Tanenbaum, B.A.; Cai, C.; Sowalsky, A.G.; He, L.; Wang, H.; et al. SOX9 drives WNT pathway activation in prostate cancer. J. Clin. Investig. 2016, 126, 1745–1758. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Carver, B.S.; Li, E.; Wang, Y.; Fazli, L.; Gleave, M.; Chen, Z. Crosstalk between nuclear MET and SOX9/β-catenin correlates with castration-resistant prostate cancer. Mol. Endocrinol. 2014, 28, 1629–1639. [Google Scholar] [CrossRef]
- Akiyama, H.; Lyons, J.P.; Mori-Akiyama, Y.; Yang, X.; Zhang, R.; Zhang, Z.; Deng, J.M.; Taketo, M.M.; Nakamura, T.; Behringer, R.R.; et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004, 18, 1072–1087. [Google Scholar] [CrossRef]
- Li, X.M.; Piao, Y.J.; Sohn, K.-C.; Ha, J.-M.; Im, M.; Seo, Y.-J.; Whang, K.U.; Lee, J.-H.; Lee, Y.; Kim, C.D. Sox9 is a β-catenin-regulated transcription factor that enhances the colony-forming activity of squamous cell carcinoma cells. Mol. Med. Rep. 2016, 14, 337–342. [Google Scholar] [CrossRef]
- Guo, Y.-Z.; Xie, X.-L.; Fu, J.; Xing, G.-L. SOX9 regulated proliferation and apoptosis of human lung carcinoma cells by the Wnt/β-catenin signaling pathway. Eur. Rev. Med. Pharm. Sci. 2018, 22, 4898–4907. [Google Scholar] [CrossRef]
- Santos, J.C.; Carrasco-Garcia, E.; Garcia-Puga, M.; Aldaz, P.; Montes, M.; Fernandez-Reyes, M.; de Oliveira, C.C.; Lawrie, C.H.; Araúzo-Bravo, M.J.; Ribeiro, M.L.; et al. SOX9 Elevation Acts with Canonical WNT Signaling to Drive Gastric Cancer Progression. Cancer Res. 2016, 76, 6735–6746. [Google Scholar] [CrossRef]
- Panza, A.; Pazienza, V.; Ripoli, M.; Benegiamo, G.; Gentile, A.; Valvano, M.R.; Augello, B.; Merla, G.; Prattichizzo, C.; Tavano, F.; et al. Interplay between SOX9, β-catenin and PPARγ activation in colorectal cancer. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1853–1865. [Google Scholar] [CrossRef]
- Ullah, M.F. Sulforaphane (SFN): An Isothiocyanate in a Cancer Chemoprevention Paradigm. Medcines 2015, 2, 141–156. [Google Scholar] [CrossRef]
- Traka, M.H.; Melchini, A.; Mithen, R.F. Sulforaphane and prostate cancer interception. Drug Discov. Today 2014, 19, 1488–1492. [Google Scholar] [CrossRef]
- Teiten, M.-H.; Gaascht, F.; Eifes, S.; Dicato, M.; Diederich, M. Chemopreventive potential of curcumin in prostate cancer. Genes Nutr. 2010, 5, 61–74. [Google Scholar] [CrossRef]
- Dorai, T.; Diouri, J.; O’Shea, O.; Doty, S.B. Curcumin Inhibits Prostate Cancer Bone Metastasis by Up-Regulating Bone Morphogenic Protein-7 in Vivo. J. Cancer 2014, 5, 369–386. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Korkaya, H.; Liu, S.; Lee, H.F.; Newman, B.; Yu, Y.; Clouthier, S.G.; Schwartz, S.J.; Wicha, M.S.; et al. Sulforaphane, a Dietary Component of Broccoli/Broccoli Sprouts, Inhibits Breast Cancer Stem Cells. Clin. Cancer Res. 2010, 16, 2580–2590. [Google Scholar] [CrossRef]
- Li, Q.; Yao, Y.; Eades, G.; Liu, Z.; Zhang, Y.; Zhou, Q. Downregulation of miR-140 promotes cancer stem cell formation in basal-like early stage breast cancer. Oncogene 2014, 33, 2589–2600. [Google Scholar] [CrossRef]
- Nakamura, K.; Yasunaga, Y.; Segawa, T.; Ko, D.; Moul, J.W.; Srivastava, S.; Rhim, J.S. Curcumin down-regulates AR gene expression and activation in prostate cancer cell lines. Int. J. Oncol. 2002, 21, 825–830. [Google Scholar] [CrossRef]
- Tarapore, R.S.; Siddiqui, I.A.; Mukhtar, H. Modulation of Wnt/ β-catenin signaling pathway by bioactive food components. Carcinogenesis 2012, 33, 483–491. [Google Scholar] [CrossRef]
- Yu, C.-C.; Tsai, L.-L.; Wang, M.-L.; Yu, C.-H.; Lo, W.-L.; Chang, Y.-C.; Chiou, G.-Y.; Chou, M.-Y.; Chiou, S.-H. miR145 Targets the SOX9/ADAM17 Axis to Inhibit Tumor-Initiating Cells and IL-6-Mediated Paracrine Effects in Head and Neck Cancer. Cancer Res. 2013, 73, 3425–3440. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khurana, N.; Sikka, S.C. Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2066. https://doi.org/10.3390/ijms20092066
Khurana N, Sikka SC. Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(9):2066. https://doi.org/10.3390/ijms20092066
Chicago/Turabian StyleKhurana, Namrata, and Suresh C. Sikka. 2019. "Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer" International Journal of Molecular Sciences 20, no. 9: 2066. https://doi.org/10.3390/ijms20092066
APA StyleKhurana, N., & Sikka, S. C. (2019). Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences, 20(9), 2066. https://doi.org/10.3390/ijms20092066