The Crosstalk Between Cell Adhesion and Cancer Metabolism



Abstract
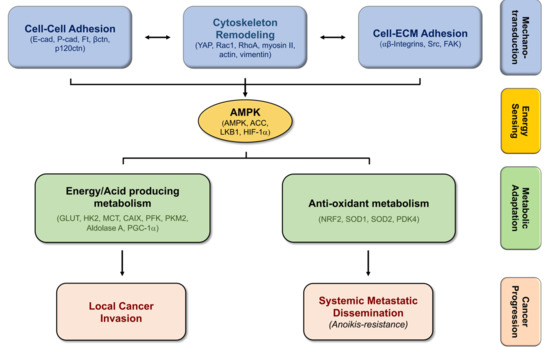
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Metabolic Rewiring During Cancer Progression
3. EMT, Cancer Stemness, and Metabolic Plasticity
4. Biomechanics, Tissue Stiffness, and Energetic Needs Regulate Cancer Cell Metabolism
5. Alterations in Cell-Matrix Adhesion Modulates Metabolic Pathways
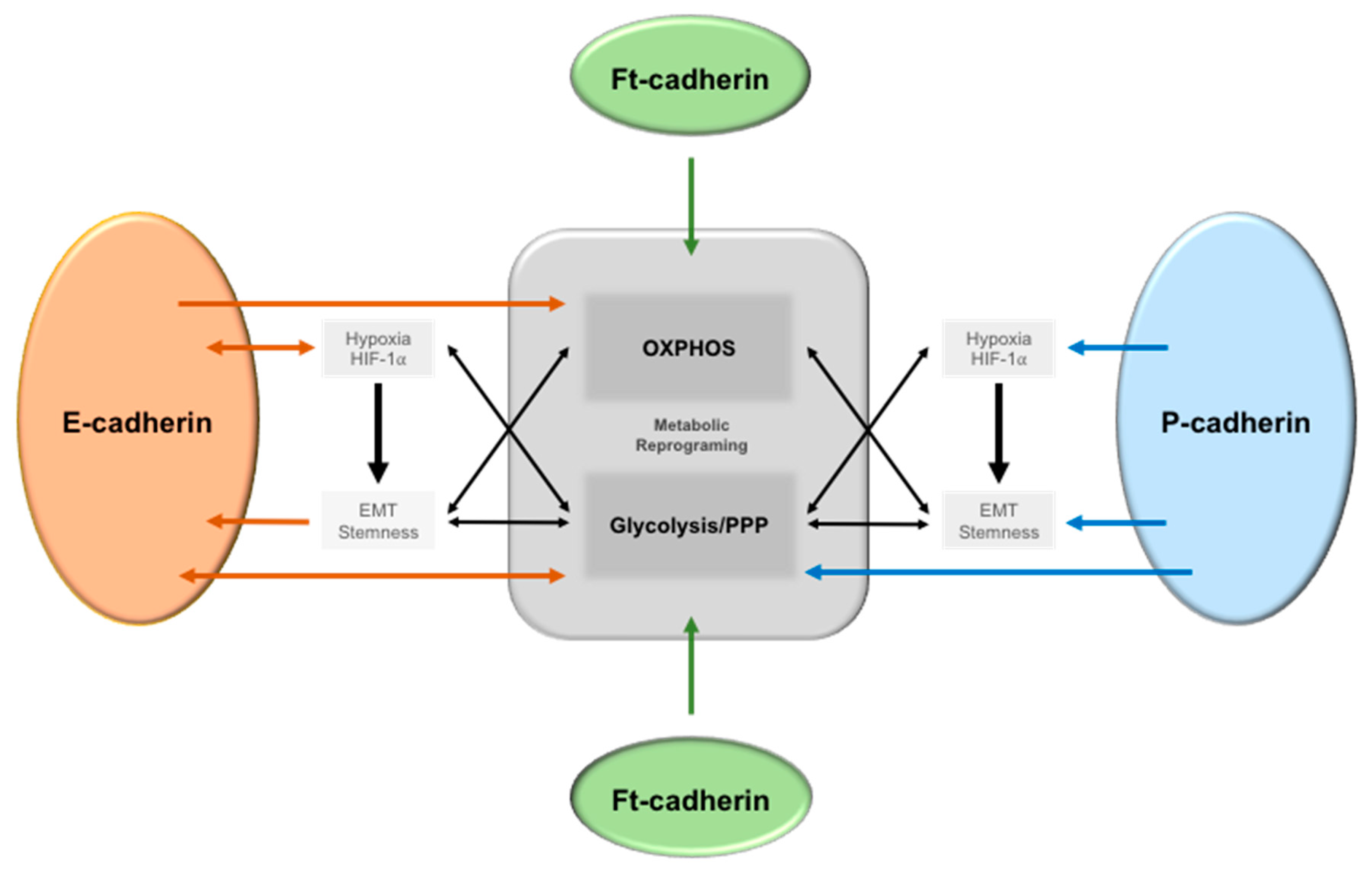
6. Modifications in Cell–Cell Adhesion Affects Cancer Metabolism
6.1. Epithelial-Cadherin (E-Cadherin)
6.2. Placental Cadherin (P-Cadherin)
6.3. Fat (ft)-like Cadherin
7. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC1/ACC2 | acetyl-CoA carboxylase 1/2 |
| ADP | Adenosine diphosphate |
| AKT | Protein kinase B (PKB) |
| ALDH | Aldehyde Dehydrogenase |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| Bad | BCL2 Associated Agonist of Cell Death |
| BCSC | Breast Cancer Stem Cells |
| BNIP3 | BCL2 Interacting Protein 3 |
| CAIX | Carbonic Anhydrase IX |
| CD147 | Cluster of Differentiation 147 |
| E-cadherin | Epithelial cadherin |
| ECM | Extracellular Matrix |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial Mesenchymal Transition |
| ERK | Extracellular-signal-Regulated Kinase |
| ETC | Electron Transport Chain |
| FAK | Focal Adhesion Kinase |
| FAO | Fatty Acid Oxidation |
| FAS | Fatty Acid Synthesis |
| FBP1 | Fructose-1,6-bisphosphatase 1 |
| Ft | Fat |
| Ftmito | Mitochondrial Fat Soluble fragment |
| GLUT1/4 | Glucose Transporter ¼ |
| GTP | Guanosine-5′-triphosphate |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HIF-1α | Hipoxia Inducible Factor-1α |
| HK2 | Hexokinase 2 |
| LKB1 | Large Kinase B1 |
| MAPK | Mitogen-Activated Protein Kinase |
| MCT | Monocarboxylate Transporters |
| MET | Mesenchymal Epithelial Transition |
| MMP | Matrix Metalloproteinase |
| mTOR | mammalian target of rapamycin |
| MYC | Myelocytomatosis oncogene |
| NAC | N-acetyl Cysteine |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| Ndufv2 | NADH dehydrogenase ubiquinone flavo-protein 2 |
| NF-κB | Nuclear Factor Kappa B |
| OXPHOS | Oxidative Phosphorylation |
| P-cadherin | Placental-cadherin |
| PDK4 | Pyruvate Dehydrogenase kinase 4 |
| PFK | Phosphofructokinase |
| PGC-1α | PPARg co-activator 1α |
| PGK1 | Phosphoglycerate kinase 1 |
| PI(3)K | Phosphoinositide 3-kinase |
| PKM2 | Pyruvate Kinase isozyme Muscle 1 |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PPP | Pentose Phosphate Pathway |
| Rac | Ras-related C3 botulinum toxin substrate 1 |
| REDD1 | Regulated in Development and DNA Damage Response 1 |
| ROS | Reactive Oxygen Species |
| SFK | Src family Kinase |
| SMC | smooth muscle cell |
| sP-cad | soluble P-cadherin fragment |
| STRAD | STe20 Related ADapter |
| TSG | Tumor Suppressor Gene |
| VEGF | Vascular Endothelial Growth Factor |
| YAP | Yes-associated protein |
References
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Doglioni, G.; Fendt, S.M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Caldieri, G.; Giacchetti, G.; van Horssen, R.; Wieringa, B.; Buccione, R. Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol. 2011, 90, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5, e19375. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Payen, V.L.; Porporato, P.E.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell. Mol. Life Sci. 2016, 73, 1333–1348. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Rodrigues, M.F.; Obre, E.; de Melo, F.H.; Santos, G.C., Jr.; Galina, A.; Jasiulionis, M.G.; Rossignol, R.; Rumjanek, F.D.; Amoedo, N.D. Enhanced OXPHOS, glutaminolysis and beta-oxidation constitute the metastatic phenotype of melanoma cells. Biochem. J. 2016, 473, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. Erratum: The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Biol. 2017, 19, 873. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef]
- Chiarugi, P.; Pani, G.; Giannoni, E.; Taddei, L.; Colavitti, R.; Raugei, G.; Symons, M.; Borrello, S.; Galeotti, T.; Ramponi, G. Reactive oxygen species as essential mediators of cell adhesion: The oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 2003, 161, 933–944. [Google Scholar] [CrossRef]
- Frisch, S.M. Tumor suppression activity of adenovirus E1a protein: Anoikis and the epithelial phenotype. Adv. Cancer Res. 2001, 80, 39–49. [Google Scholar]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef]
- Chen, J.; Lee, H.-J.; Wu, X.; Huo, L.; Kim, S.-J.; Xu, L.; Wang, Y.; He, J.; Bollu, L.R.; Gao, G.; et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015, 75, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R.; Felding-Habermann, B. Adaptation of Energy Metabolism in Breast Cancer Brain Metastases. Cancer Res. 2007, 67, 1472. [Google Scholar] [CrossRef]
- Kim, H.M.; Jung, W.H.; Koo, J.S. Site-specific metabolic phenotypes in metastatic breast cancer. J. Transl. Med. 2014, 12, 354. [Google Scholar] [CrossRef]
- Cha, Y.J.; Jung, W.H.; Koo, J.S. Differential Site-Based Expression of Pentose Phosphate Pathway-Related Proteins among Breast Cancer Metastases. Dis. Markers 2017, 2017, 7062517. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schafer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef]
- Brabletz, T. To differentiate or not--routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Sousa, B.; Ribeiro, A.S.; Paredes, J. Heterogeneity and Plasticity of Breast Cancer Stem Cells. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; in press. [Google Scholar]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69.e6–86.e6. [Google Scholar] [CrossRef] [PubMed]
- Tavares, S.; Vieira, A.F.; Taubenberger, A.V.; Araujo, M.; Martins, N.P.; Bras-Pereira, C.; Polonia, A.; Herbig, M.; Barreto, C.; Otto, O.; et al. Actin stress fiber organization promotes cell stiffening and proliferation of pre-invasive breast cancer cells. Nat. Commun. 2017, 8, 15237. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Keely, P.J. The role of focal adhesion kinase in tumor initiation and progression. Cell Adh. Migr. 2009, 3, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Kallergi, G.; Agelaki, S.; Markomanolaki, H.; Georgoulias, V.; Stournaras, C. Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell. Physiol. Biochem. 2007, 20, 977–986. [Google Scholar] [CrossRef]
- Guan, J.-L. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 2010, 62, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; Deberardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef]
- Mouw, J.K.; Yui, Y.; Damiano, L.; Bainer, R.O.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M.; et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, R.W.; Cowan, C.R.; Mih, J.D.; Koryakina, Y.; Gioeli, D.; Slack-Davis, J.K.; Blackman, B.R.; Tschumperlin, D.J.; Parsons, J.T. Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS ONE 2010, 5, e12905. [Google Scholar] [CrossRef] [PubMed]
- Goo, C.K.; Lim, H.Y.; Ho, Q.S.; Too, H.P.; Clement, M.V.; Wong, K.P. PTEN/Akt signaling controls mitochondrial respiratory capacity through 4E-BP1. PLoS ONE 2012, 7, e45806. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Chen, Q.K.; Lui, C.; Cichon, M.A.; Radisky, D.C.; Nelson, C.M. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.L.; Molish, I.R.; Robkin, L.; Holmsen, H. Nucleotide exchange between cytosolic ATP and F-actin-bound ADP may be a major energy-utilizing process in unstimulated platelets. Eur. J. Biochem. 1986, 156, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci. 2003, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Bays, J.L.; Campbell, H.K.; Heidema, C.; Sebbagh, M.; DeMali, K.A. Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nat. Cell Biol. 2017, 19, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Koh, H.; Kim, M.; Kim, Y.; Lee, S.Y.; Karess, R.E.; Lee, S.H.; Shong, M.; Kim, J.M.; Kim, J.; et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 2007, 447, 1017–1020. [Google Scholar] [CrossRef]
- Zheng, B.; Cantley, L.C. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 819–822. [Google Scholar] [CrossRef]
- Schaffer, B.E.; Levin, R.S.; Hertz, N.T.; Maures, T.J.; Schoof, M.L.; Hollstein, P.E.; Benayoun, B.A.; Banko, M.R.; Shaw, R.J.; Shokat, K.M.; et al. Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab. 2015, 22, 907–921. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef]
- Chang, M.Y.; Huang, D.Y.; Ho, F.M.; Huang, K.C.; Lin, W.W. PKC-dependent human monocyte adhesion requires AMPK and Syk activation. PLoS ONE 2012, 7, e40999. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Mogilner, A.; Oster, G. Cell motility driven by actin polymerization. Biophys. J. 1996, 71, 3030–3045. [Google Scholar] [CrossRef]
- Salvi, A.M.; DeMali, K.A. Mechanisms linking mechanotransduction and cell metabolism. Curr. Opin. Cell Biol. 2018, 54, 114–120. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Georgiadou, M.; Lilja, J.; Jacquemet, G.; Guzman, C.; Rafaeva, M.; Alibert, C.; Yan, Y.; Sahgal, P.; Lerche, M.; Manneville, J.B.; et al. AMPK negatively regulates tensin-dependent integrin activity. J. Cell Biol. 2017, 216, 1107–1121. [Google Scholar] [CrossRef]
- Rantala, J.K.; Pouwels, J.; Pellinen, T.; Veltel, S.; Laasola, P.; Mattila, E.; Potter, C.S.; Duffy, T.; Sundberg, J.P.; Kallioniemi, O.; et al. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat. Cell Biol. 2011, 13, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Blume, C.; Benz, P.M.; Walter, U.; Ha, J.; Kemp, B.E.; Renne, T. AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J. Biol. Chem. 2007, 282, 4601–4612. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.; Figueiredo, J.; Albergaria, A.; Oliveira, P.; Carvalho, J.; Ribeiro, A.S.; Caldeira, J.; Costa, A.M.; Simoes-Correia, J.; Oliveira, M.J.; et al. Epithelial E- and P-cadherins: Role and clinical significance in cancer. Biochim. Biophys. Acta 2012, 1826, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Toret, C.P.; D’Ambrosio, M.V.; Vale, R.D.; Simon, M.A.; Nelson, W.J. A genome-wide screen identifies conserved protein hubs required for cadherin-mediated cell–cell adhesion. J. Cell Biol. 2014, 204, 265–279. [Google Scholar] [CrossRef]
- van Roy, F.; Berx, G. The cell–cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Cancela Paredes, J.; Milanezi, F.; Schmitt, F.C. Clinicopathologic implications of E-cadherin reactivity in patients with lobular carcinoma in situ of the breast. Cancer 2002, 94, 2114–2115. [Google Scholar] [CrossRef]
- Carneiro, F.; Machado, J.C.; Seruca, R.; Sobrinho-Simoes, M. E-cadherin changes in gastric carcimona. Histopathology 1999, 35, 477–478. [Google Scholar] [CrossRef]
- Chen, J.; Imanaka, N.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Cha, Y.H.; Lee, J.; Lee, S.H.; Yang, J.H.; Yun, J.S.; Cho, E.S.; Zhang, X.; Nam, M.; Kim, N.; et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 2017, 8, 14374. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731. [Google Scholar] [CrossRef]
- Chu, K.; Boley, K.M.; Moraes, R.; Barsky, S.H.; Robertson, F.M. The paradox of E-cadherin: Role in response to hypoxia in the tumor microenvironment and regulation of energy metabolism. Oncotarget 2013, 4, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Bergin, E.; Levine, J.S.; Koh, J.S.; Lieberthal, W. Mouse proximal tubular cell–cell adhesion inhibits apoptosis by a cadherin-dependent mechanism. Am. J. Physiol. Renal Physiol. 2000, 278, F758–F768. [Google Scholar] [CrossRef]
- Kantak, S.S.; Kramer, R.H. E-cadherin regulates anchorage-independent growth and survival in oral squamous cell carcinoma cells. J. Biol. Chem. 1998, 273, 16953–16961. [Google Scholar] [CrossRef]
- Park, S.Y.; Shin, J.H.; Kee, S.H. E-cadherin expression increases cell proliferation by regulating energy metabolism through nuclear factor-kappaB in AGS cells. Cancer Sci. 2017, 108, 1769–1777. [Google Scholar] [CrossRef]
- Sebbagh, M.; Santoni, M.J.; Hall, B.; Borg, J.P.; Schwartz, M.A. Regulation of LKB1/STRAD localization and function by E-cadherin. Curr. Biol. 2009, 19, 37–42. [Google Scholar] [CrossRef]
- Matoba, R.; Morizane, Y.; Shiode, Y.; Hirano, M.; Doi, S.; Toshima, S.; Araki, R.; Hosogi, M.; Yonezawa, T.; Shiraga, F. Suppressive effect of AMP-activated protein kinase on the epithelial-mesenchymal transition in retinal pigment epithelial cells. PLoS ONE 2017, 12, e0181481. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Surendran, H.; Chowdhury, D.R.; Prabhakar, K.; Pal, R. Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J. Mol. Med. 2016, 94, 1397–1409. [Google Scholar] [CrossRef]
- Chou, C.C.; Lee, K.H.; Lai, I.L.; Wang, D.; Mo, X.; Kulp, S.K.; Shapiro, C.L.; Chen, C.S. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014, 74, 4783–4795. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Lu, M.; Yan, Q.; Li, S.; Feng, Y. Inhibition of Proliferation, Migration, and Invasion by Knockdown of Pyruvate Kinase-M2 (PKM2) in Ovarian Cancer SKOV3 and OVCAR3 Cells. Oncol. Res. 2016, 24, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.; Albergaria, A.; Oliveira, J.T.; Jeronimo, C.; Milanezi, F.; Schmitt, F.C. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin. Cancer Res. 2005, 11, 5869–5877. [Google Scholar] [CrossRef]
- Gamallo, C.; Moreno-Bueno, G.; Sarrio, D.; Calero, F.; Hardisson, D.; Palacios, J. The prognostic significance of P-cadherin in infiltrating ductal breast carcinoma. Mod. Pathol. 2001, 14, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Peralta Soler, A.; Knudsen, K.A.; Salazar, H.; Han, A.C.; Keshgegian, A.A. P-cadherin expression in breast carcinoma indicates poor survival. Cancer 1999, 86, 1263–1272. [Google Scholar] [CrossRef]
- Turashvili, G.; McKinney, S.E.; Goktepe, O.; Leung, S.C.; Huntsman, D.G.; Gelmon, K.A.; Los, G.; Rejto, P.A.; Aparicio, S.A. P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod. Pathol. 2011, 24, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Sousa, B.; Paredes, J.; Milanezi, F.; Lopes, N.; Martins, D.; Dufloth, R.; Vieira, D.; Albergaria, A.; Veronese, L.; Carneiro, V.; et al. P-cadherin, vimentin and CK14 for identification of basal-like phenotype in breast carcinomas: An immunohistochemical study. Histol. Histopathol. 2010, 25, 963–974. [Google Scholar]
- Sousa, B.; Ribeiro, A.S.; Nobre, A.R.; Lopes, N.; Martins, D.; Pinheiro, C.; Vieira, A.F.; Albergaria, A.; Gerhard, R.; Schmitt, F.; et al. The basal epithelial marker P-cadherin associates with breast cancer cell populations harboring a glycolytic and acid-resistant phenotype. BMC Cancer 2014, 14, 734. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Paredes, J. P-Cadherin Linking Breast Cancer Stem Cells and Invasion: A Promising Marker to Identify an “Intermediate/Metastable” EMT State. Front. Oncol. 2014, 4, 371. [Google Scholar] [CrossRef] [PubMed]
- Albergaria, A.; Ribeiro, A.S.; Vieira, A.F.; Sousa, B.; Nobre, A.R.; Seruca, R.; Schmitt, F.; Paredes, J. P-cadherin role in normal breast development and cancer. Int. J. Dev. Biol. 2011, 55, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.; Correia, A.L.; Ribeiro, A.S.; Albergaria, A.; Milanezi, F.; Schmitt, F.C. P-cadherin expression in breast cancer: A review. Breast Cancer Res. 2007, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.S.; Albergaria, A.; Sousa, B.; Correia, A.L.; Bracke, M.; Seruca, R.; Schmitt, F.C.; Paredes, J. Extracellular cleavage and shedding of P-cadherin: A mechanism underlying the invasive behaviour of breast cancer cells. Oncogene 2010, 29, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.; Stove, C.; Stove, V.; Milanezi, F.; Van Marck, V.; Derycke, L.; Mareel, M.; Bracke, M.; Schmitt, F. P-cadherin is up-regulated by the antiestrogen ICI 182,780 and promotes invasion of human breast cancer cells. Cancer Res. 2004, 64, 8309–8317. [Google Scholar] [CrossRef]
- Rozhin, J.; Sameni, M.; Ziegler, G.; Sloane, B.F. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res. 1994, 54, 6517–6525. [Google Scholar] [PubMed]
- Montcourrier, P.; Silver, I.; Farnoud, R.; Bird, I.; Rochefort, H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin. Exp. Metastasis 1997, 15, 382–392. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Carvalho, F.A.; Figueiredo, J.; Carvalho, R.; Mestre, T.; Monteiro, J.; Guedes, A.F.; Fonseca, M.; Sanches, J.; Seruca, R.; et al. Atomic force microscopy and graph analysis to study the P-cadherin/SFK mechanotransduction signalling in breast cancer cells. Nanoscale 2016, 8, 19390–19401. [Google Scholar] [CrossRef]
- Vieira, A.F.; Ribeiro, A.S.; Dionisio, M.R.; Sousa, B.; Nobre, A.R.; Albergaria, A.; Santiago-Gomez, A.; Mendes, N.; Gerhard, R.; Schmitt, F.; et al. P-cadherin signals through the laminin receptor alpha6beta4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget 2014, 5, 679–692. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Sousa, B.; Carreto, L.; Mendes, N.; Nobre, A.R.; Ricardo, S.; Albergaria, A.; Cameselle-Teijeiro, J.F.; Gerhard, R.; Soderberg, O.; et al. P-cadherin functional role is dependent on E-cadherin cellular context: A proof of concept using the breast cancer model. J. Pathol. 2013, 229, 705–718. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Nobre, A.R.; Mendes, N.; Almeida, J.; Vieira, A.F.; Sousa, B.; Carvalho, F.A.; Monteiro, J.; Polonia, A.; Fonseca, M.; et al. SRC inhibition prevents P-cadherin mediated signaling and function in basal-like breast cancer cells. Cell Commun. Signal. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.; Correia, A.L.; Ribeiro, A.S.; Milanezi, F.; Cameselle-Teijeiro, J.; Schmitt, F.C. Breast carcinomas that co-express E- and P-cadherin are associated with p120-catenin cytoplasmic localisation and poor patient survival. J. Clin. Pathol. 2008, 61, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Bazellieres, E.; Conte, V.; Elosegui-Artola, A.; Serra-Picamal, X.; Bintanel-Morcillo, M.; Roca-Cusachs, P.; Munoz, J.J.; Sales-Pardo, M.; Guimera, R.; Trepat, X. Control of cell–cell forces and collective cell dynamics by the intercellular adhesome. Nat. Cell Biol. 2015, 17, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Yu, Q.; Liu, T.J.; Gebreamlak, E.P.; Wang, S.L.; Zhang, R.J.; Zhang, J.; Niu, Y. P-cadherin expression and basal-like subtype in breast cancers. Med. Oncol. 2012, 29, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.F.; Dionisio, M.R.; Gomes, M.; Cameselle-Teijeiro, J.F.; Lacerda, M.; Amendoeira, I.; Schmitt, F.; Paredes, J. P-cadherin: A useful biomarker for axillary-based breast cancer decisions in the clinical practice. Mod. Pathol. 2017, 30, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.F.; Ricardo, S.; Ablett, M.P.; Dionisio, M.R.; Mendes, N.; Albergaria, A.; Farnie, G.; Gerhard, R.; Cameselle-Teijeiro, J.F.; Seruca, R.; et al. P-cadherin is coexpressed with CD44 and CD49f and mediates stem cell properties in basal-like breast cancer. Stem Cells 2012, 30, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Louie, E.; Nik, S.; Chen, J.S.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010, 12, R94. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.; McNeill, H. Big roles for Fat cadherins. Curr. Opin. Cell Biol. 2018, 51, 73–80. [Google Scholar] [CrossRef]
- Thomas, C.; Strutt, D. The roles of the cadherins Fat and Dachsous in planar polarity specification in Drosophila. Dev. Dyn. 2012, 241, 27–39. [Google Scholar] [CrossRef]
- Staley, B.K.; Irvine, K.D. Hippo signaling in Drosophila: Recent advances and insights. Dev. Dyn 2012, 241, 3–15. [Google Scholar] [CrossRef]
- Castillejo-Lopez, C.; Arias, W.M.; Baumgartner, S. The fat-like gene of Drosophila is the true orthologue of vertebrate fat cadherins and is involved in the formation of tubular organs. J. Biol. Chem. 2004, 279, 24034–24043. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, S.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, K.; Yamagata, H.D.; Arita, N.; Nakashiro, K.I.; Nose, M.; Miki, T.; Hamakawa, H. Identification of homozygous deletions of tumor suppressor gene FAT in oral cancer using CGH-array. Oncogene 2007, 26, 5300–5308. [Google Scholar] [CrossRef] [PubMed]
- De Bock, C.E.; Ardjmand, A.; Molloy, T.J.; Bone, S.M.; Johnstone, D.; Campbell, D.M.; Shipman, K.L.; Yeadon, T.M.; Holst, J.; Spanevello, M.D.; et al. The Fat1 cadherin is overexpressed and an independent prognostic factor for survival in paired diagnosis–relapse samples of precursor B-cell acute lymphoblastic leukemia. Leukemia 2011, 26, 918. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Miyazaki, T.; Nakashiro, K.; Yamagata, H.; Isokane, M.; Goda, H.; Tanaka, H.; Oka, R.; Hamakawa, H. Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of beta-catenin. Oncol. Rep. 2011, 26, 587–592. [Google Scholar] [PubMed]
- Valletta, D.; Czech, B.; Spruss, T.; Ikenberg, K.; Wild, P.; Hartmann, A.; Weiss, T.S.; Oefner, P.J.; Muller, M.; Bosserhoff, A.K.; et al. Regulation and function of the atypical cadherin FAT1 in hepatocellular carcinoma. Carcinogenesis 2014, 35, 1407–1415. [Google Scholar] [CrossRef]
- Dikshit, B.; Irshad, K.; Madan, E.; Aggarwal, N.; Sarkar, C.; Chandra, P.S.; Gupta, D.K.; Chattopadhyay, P.; Sinha, S.; Chosdol, K. FAT1 acts as an upstream regulator of oncogenic and inflammatory pathways, via PDCD4, in glioma cells. Oncogene 2012, 32, 3798. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Feng, D.; Hu, L.; Chen, H.; Yang, G.; Cai, Q.; Gao, C.; Wei, D. FAT4 functions as a tumour suppressor in gastric cancer by modulating Wnt/beta-catenin signalling. Br. J. Cancer 2015, 113, 1720–1729. [Google Scholar] [CrossRef][Green Version]
- Sing, A.; Tsatskis, Y.; Fabian, L.; Hester, I.; Rosenfeld, R.; Serricchio, M.; Yau, N.; Bietenhader, M.; Shanbhag, R.; Jurisicova, A.; et al. The atypical cadherin fat directly regulates mitochondrial function and metabolic state. Cell 2014, 158, 1293–1308. [Google Scholar] [CrossRef]
- Cao, L.L.; Riascos-Bernal, D.F.; Chinnasamy, P.; Dunaway, C.M.; Hou, R.; Pujato, M.A.; O’Rourke, B.P.; Miskolci, V.; Guo, L.; Hodgson, L.; et al. Control of mitochondrial function and cell growth by the atypical cadherin Fat1. Nature 2016, 539, 575–578. [Google Scholar] [CrossRef]
- Madan, E.; Dikshit, B.; Gowda, S.H.; Srivastava, C.; Sarkar, C.; Chattopadhyay, P.; Sinha, S.; Chosdol, K. FAT1 is a novel upstream regulator of HIF1α and invasion of high grade glioma. Int. J. Cancer 2016, 139, 2570–2582. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, B.; Pereira, J.; Paredes, J. The Crosstalk Between Cell Adhesion and Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 1933. https://doi.org/10.3390/ijms20081933
Sousa B, Pereira J, Paredes J. The Crosstalk Between Cell Adhesion and Cancer Metabolism. International Journal of Molecular Sciences. 2019; 20(8):1933. https://doi.org/10.3390/ijms20081933
Chicago/Turabian StyleSousa, Bárbara, Joana Pereira, and Joana Paredes. 2019. "The Crosstalk Between Cell Adhesion and Cancer Metabolism" International Journal of Molecular Sciences 20, no. 8: 1933. https://doi.org/10.3390/ijms20081933
APA StyleSousa, B., Pereira, J., & Paredes, J. (2019). The Crosstalk Between Cell Adhesion and Cancer Metabolism. International Journal of Molecular Sciences, 20(8), 1933. https://doi.org/10.3390/ijms20081933