Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Lactate Dehydrogenases and Lactate
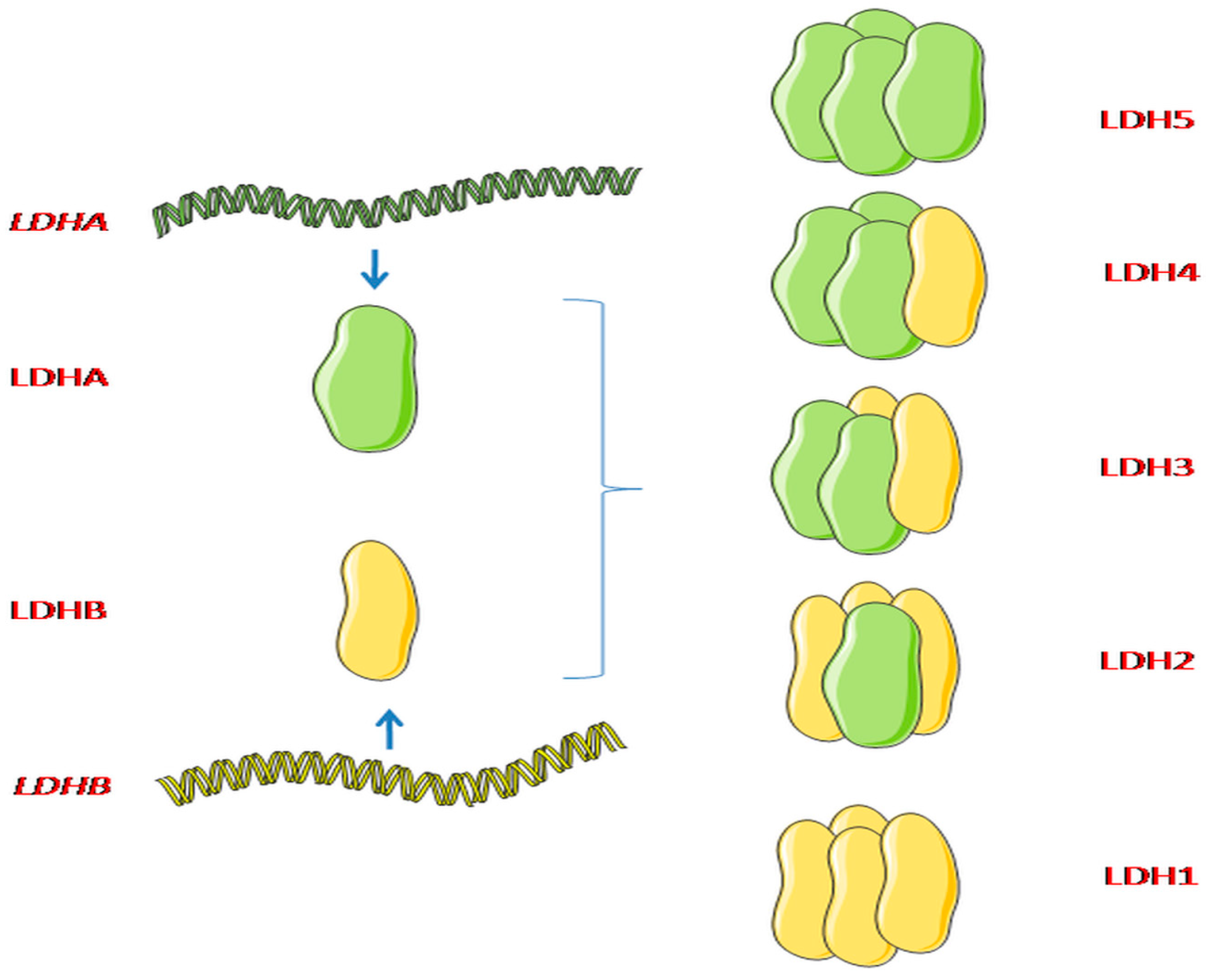
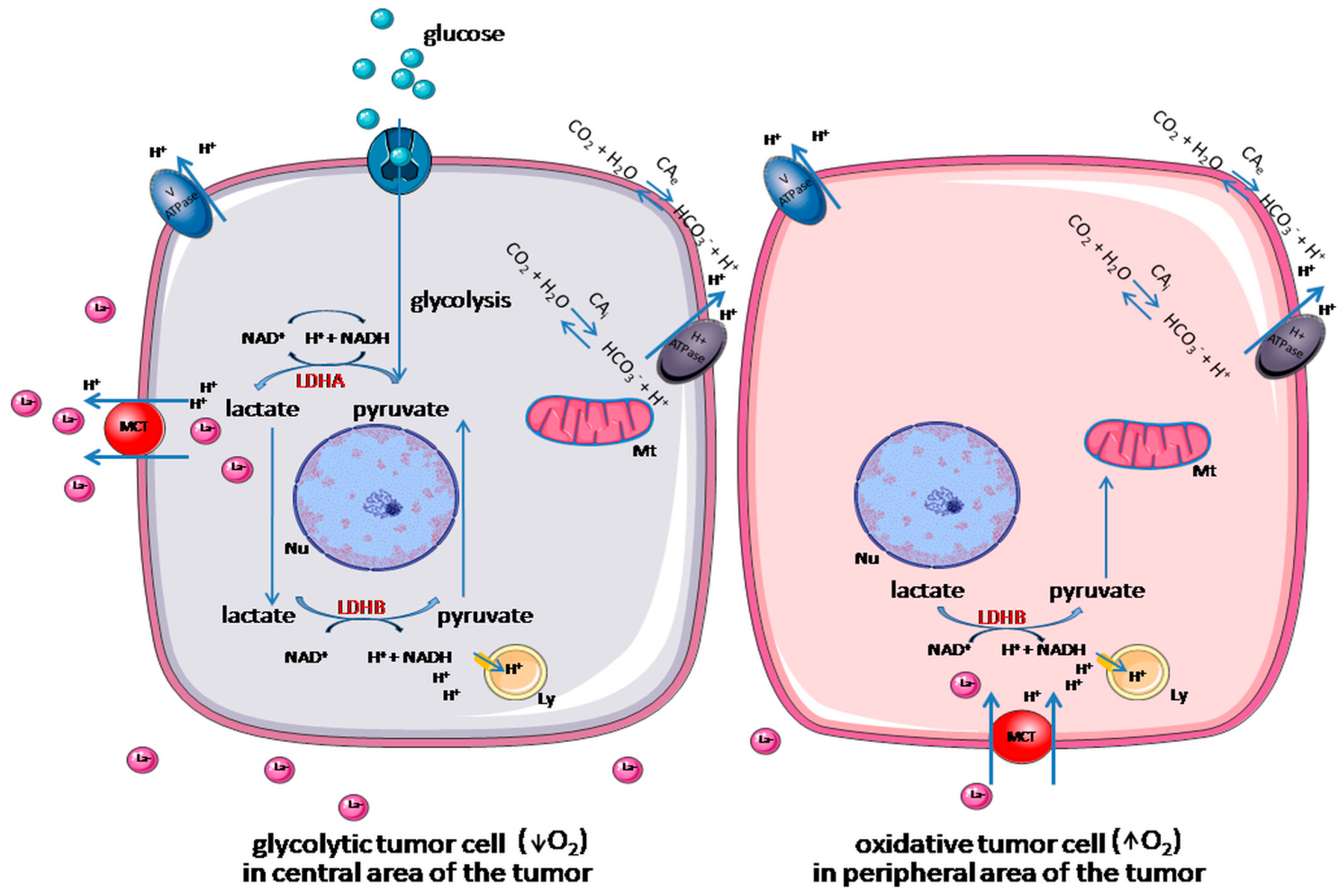
1.1. LDHA and LDHB in Tumors
1.2. LDHA and LDHB Regulation
2. LDHA, LDHB and Lactic Acid in the Cell Death of Tumor Cells
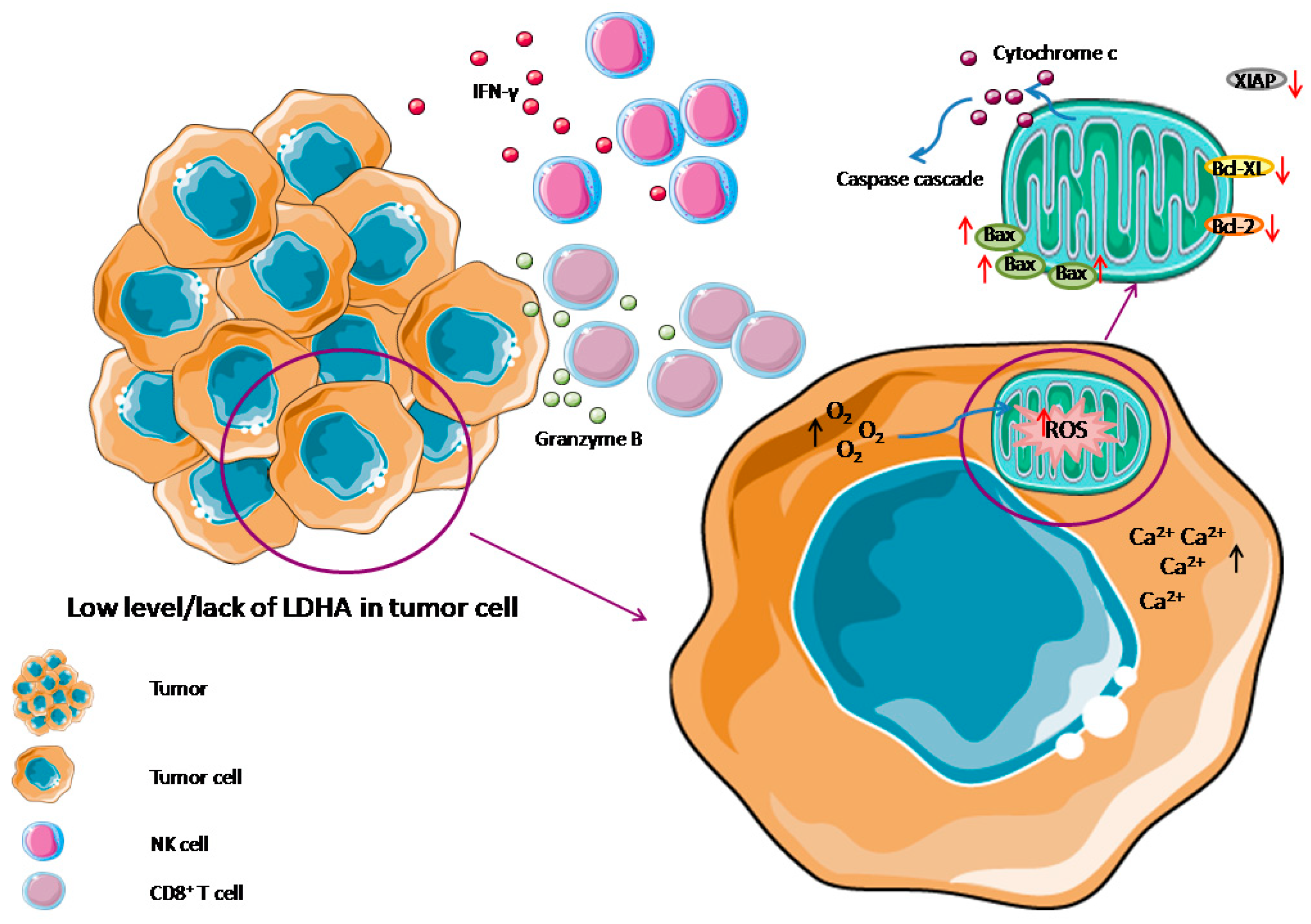
2.1. Apoptosis
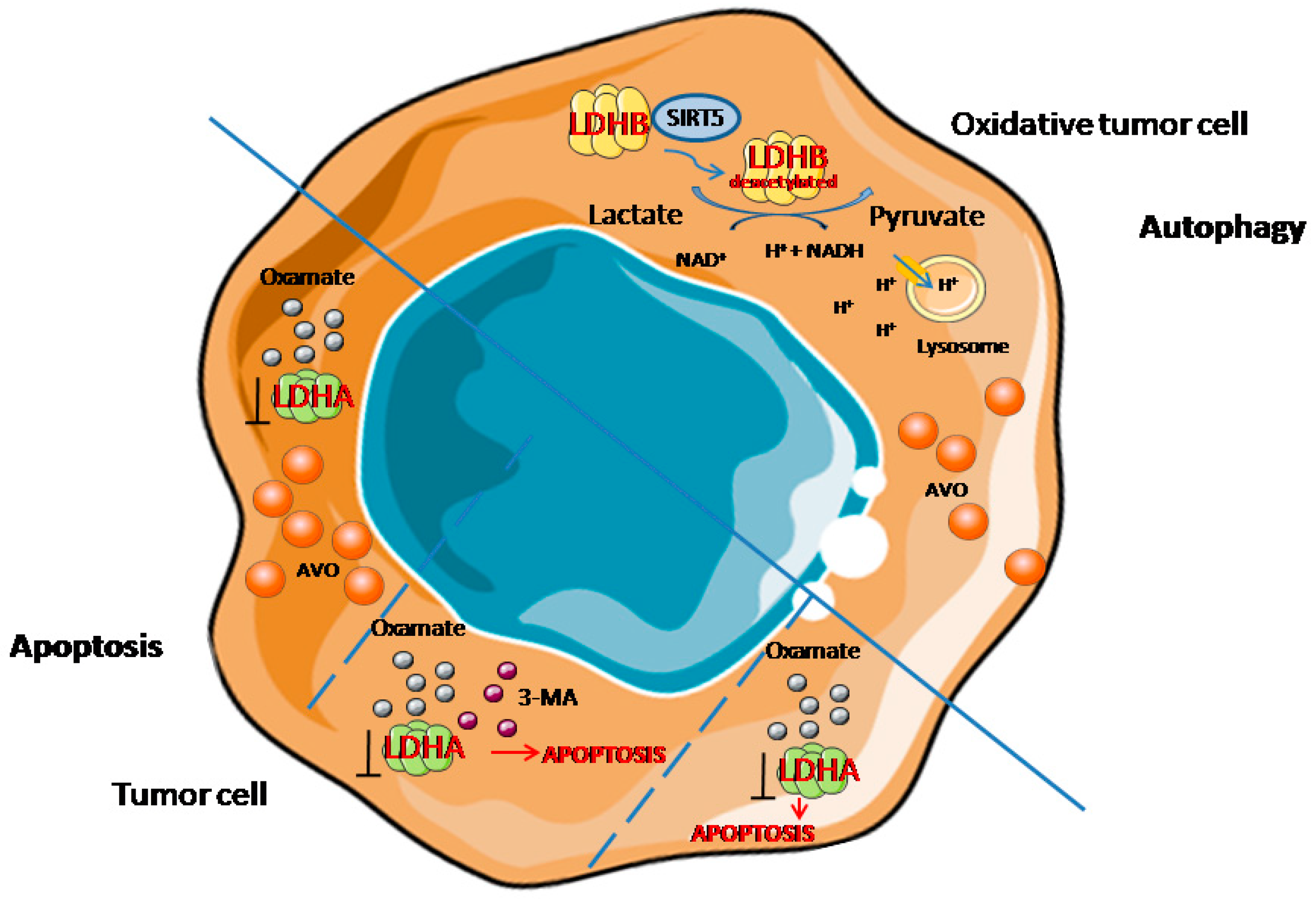
2.2. Autophagy
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenine |
| AIF | Apoptosis-inducing factor |
| AKT | AKT Serine/Threonine Kinase |
| AMP | Adenosine monophosphate |
| ATP | Adenosine triphosphate |
| ATPase | Adenosine triphosphatase |
| AVO | Acidic Vacuolar Organelles |
| BAX | BCL2 associated X protein |
| BCL-2 | B-cell lymphoma 2 |
| BH-3 | Interacting-domain death agonist |
| Ca2+ | Calcium ion |
| EGFR | Epidermal Growth Factor Receptor |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FOXM1 | Forkhead Box Protein M1 |
| H+ | Hydrogen ion |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HIF | Hypoxia-inducible factor |
| HMGB2 | High-mobility group box 2 |
| IFN-γ | Interferon gamma |
| JMJD2A | Jumonji C Domain 2A |
| KRAS | KRAS proto-oncogene |
| La− | Lactate |
| LC3-II | Microtubule-associated protein 1A/1B-light chain 3 (LC3) conjugated to phosphatidylethanolamine |
| LDH | Lactate dehydrogenase |
| LDHA | Lactate dehydrogenase A |
| LDHB | Lactate dehydrogenase B |
| LDHC | Lactate dehydrogenase C |
| LDHD | Lactate dehydrogenase D |
| MCC | Merkel cell carcinoma |
| MCPyV | Merkel cell polyomavirus |
| MCT | Monocarboxylate Transporter |
| MDR | Multidrug resistance |
| miR | microRNA |
| MPT | Mitochondrial permeability transition |
| mTOR | Mammalian Target of Rapamycin |
| NAD+ | Nicotinamide adenine dinucleotide (oxidized form) |
| NADH | Nicotinamide adenine dinucleotide (reduced form) |
| NCCD | Nomenclature Committee on Cell Death |
| NOXA | Phorbol-12-myristate-13-acetate-induced protein 1 |
| OXPHOS | Oxidative phosphorylation |
| PGC1β | Peroxisome proliferator-activated receptor-gamma coactivator 1 – beta |
| PI3K | Phosphoinositide-3-kinase |
| PPAR-γ | Peroxisome proliferator-activated receptor-gamma |
| PUMA | p53 upregulated modulator of apoptosis |
| ROS | Reactive oxygen species |
| SIRT | Sirtuin |
| SIRT5 | Sirtuin 5 |
| Sp1 | Specificity protein 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
References
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double Genetic Disruption of Lactate Dehydrogenases A and B Is Required to Ablate the “Warburg Effect” Restricting Tumor Growth to Oxidative Metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Inomata, M.; Sasaki, M.S.; Kaneko, A.; Ushiama, M.; Sugano, K.; Takayama, J.; Kanno, T. Electrophoretic Variant of a Lactate Dehydrogenase Isoenzyme and Selective Promoter Methylation of the LDHA Gene in a Human Retinoblastoma Cell Line. Clin. Chem. 2002, 48, 1938–1945. [Google Scholar]
- Li, S.S.; Luedemann, M.; Sharief, F.S.; Takano, T.; Deaven, L.L. Mapping of Human Lactate Dehydrogenase-A, -B, and -C Genes and Their Related Sequences: The Gene for LDHC Is Located with That for LDHA on Chromosome 11. Cytogenet. Cell Genet. 1988, 48, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting Lactate Metabolism for Cancer Therapeutics. J. Clin. Invest. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Yang, J.; Thomas, K. Molecular and Functional Characterization of the Promoter Region of the Mouse LDH/C Gene: Enhancer-Assisted, Sp1-Mediated Transcriptional Activation. Nucleic. Acids Res. 1997, 25, 2213–2220. [Google Scholar] [CrossRef][Green Version]
- Flick, M.J.; Konieczny, S.F. Identification of Putative Mammalian d-Lactate Dehydrogenase Enzymes. Biochem. Biophys. Res. Commun. 2002, 295, 910–916. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, E. Functional and Physical Interaction between Human Lactate Dehydrogenase B and Na + /H + Exchanger Isoform 1. Anim. Cells Syst. 2009, 13, 283–288. [Google Scholar] [CrossRef]
- Hussien, R.; Brooks, G.A. Mitochondrial and Plasma Membrane Lactate Transporter and Lactate Dehydrogenase Isoform Expression in Breast Cancer Cell Lines. Physiol. Genom. 2011, 43, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Mack, N.; Mazzio, E.A.; Bauer, D.; Flores-Rozas, H.; Soliman, K.F.A. Stable ShRNA Silencing of Lactate Dehydrogenase A (LDHA) in Human MDA-MB-231 Breast Cancer Cells Fails to Alter Lactic Acid Production, Glycolytic Activity, ATP or Survival. Anticancer Res. 2017, 37, 1205–1212. [Google Scholar] [CrossRef]
- Sola-Penna, M. Metabolic Regulation by Lactate. IUBMB Life 2008, 60, 605–608. [Google Scholar] [CrossRef]
- De Groot, R.; Sprenger, R.A.; Imholz, A.L.T.; Gerding, M.N. Type B Lactic Acidosis in Solid Malignancies. Neth. J. Med. 2011, 69, 120–123. [Google Scholar]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate Is Always the End Product of Glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef]
- Hu, X.; Chao, M.; Wu, H. Central Role of Lactate and Proton in Cancer Cell Resistance to Glucose Deprivation and Its Clinical Translation. Signal Transduct. Target. 2017, 2, 16047. [Google Scholar] [CrossRef]
- Huang, R.; Zong, X. Aberrant Cancer Metabolism in Epithelial-Mesenchymal Transition and Cancer Metastasis: Mechanisms in Cancer Progression. Crit. Rev. Oncol. Hematol. 2017, 115, 13–22. [Google Scholar] [CrossRef]
- San-Millán, I.; Brooks, G.A. Reexamining Cancer Metabolism: Lactate Production for Carcinogenesis Could Be the Purpose and Explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.-J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Dumas, J.-F.; Brisson, L.; Chevalier, S.; Mahéo, K.; Fromont, G.; Moussata, D.; Besson, P.; Roger, S. Metabolic Reprogramming in Cancer Cells, Consequences on PH and Tumour Progression: Integrated Therapeutic Perspectives with Dietary Lipids as Adjuvant to Anticancer Treatment. Semin. Cancer Biol. 2017, 43, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and Chemotherapy Resistance: A Promising Therapeutic Target for Cancer Treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. PH Sensing and Regulation in Cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.H.; Olde Damink, S.W.M.; Malago, M.; Dhar, D.K.; Pereira, S.P. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS ONE 2016, 11, e0151635. [Google Scholar] [CrossRef]
- Hu, S.; Jiang, Q.; Luo, D.; Zhao, L.; Fu, X.; Chen, Y.; Song, X.; Li, L.; Zhao, H.; He, Y.; et al. MiR-200b Is a Key Regulator of Tumor Progression and Metabolism Targeting Lactate Dehydrogenase A in Human Malignant Glioma. Oncotarget 2016, 7, 48423–48431. [Google Scholar] [CrossRef]
- Rong, Y.; Wu, W.; Ni, X.; Kuang, T.; Jin, D.; Wang, D.; Lou, W. Lactate Dehydrogenase A Is Overexpressed in Pancreatic Cancer and Promotes the Growth of Pancreatic Cancer Cells. Tumour Biol. 2013, 34, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Xian, Z.-Y.; Liu, J.-M.; Chen, Q.-K.; Chen, H.-Z.; Ye, C.-J.; Xue, J.; Yang, H.-Q.; Li, J.-L.; Liu, X.-F.; Kuang, S.-J. Inhibition of LDHA Suppresses Tumor Progression in Prostate Cancer. Tumour Biol. 2015, 36, 8093–8100. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Zhu, W.; Ma, L.; Qian, J.; Xu, J.; Xu, T.; Pang, L.; Zhou, H.; Shu, Y.; Zhou, J. The Molecular Mechanism and Clinical Significance of LDHA in HER2-Mediated Progression of Gastric Cancer. Am. J. Transl. Res. 2018, 10, 2055–2067. [Google Scholar]
- Su, Y.; Yu, Q.-H.; Wang, X.-Y.; Yu, L.-P.; Wang, Z.-F.; Cao, Y.-C.; Li, J.-D. JMJD2A Promotes the Warburg Effect and Nasopharyngeal Carcinoma Progression by Transactivating LDHA Expression. BMC Cancer 2017, 17, 477. [Google Scholar] [CrossRef]
- Held-Warmkessel, J.; Dell, D.D. Lactic Acidosis in Patients with Cancer. Clin. J. Oncol. Nurs. 2014, 18, 592–594. [Google Scholar] [CrossRef]
- Munoz, J.; Khushman, M.; Hanbali, A.; Stoltenberg, M. Severe Lactic Acidosis in a Patient with Metastatic Prostate Cancer. J. Cancer Res. 2011, 7, 201–202. [Google Scholar] [CrossRef]
- Claudino, W.M.; Dias, A.; Tse, W.; Sharma, V.R. Type B Lactic Acidosis: A Rare but Life Threatening Hematologic Emergency. A Case Illustration and Brief Review. Am. J. Blood Res. 2015, 5, 25–29. [Google Scholar] [PubMed]
- Oh, D.J.; Dinerman, E.; Matthews, A.H.; Aron, A.W.; Berg, K.M. Refractory Lactic Acidosis in Small Cell Carcinoma of the Lung. Case Rep. Crit. Care 2017, 6148350. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Shang, Y.; Hunsaker, T.; Truong, T.; Peterson, D.; Torres, E.; Li, L.; Haley, B.; Stephan, J.-P.; et al. An Integrated Genomic Screen Identifies LDHB as an Essential Gene for Triple-Negative Breast Cancer. Cancer Res. 2012, 72, 5812–5823. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate Dehydrogenase B Is Required for the Growth of KRAS-Dependent Lung Adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef]
- Kumar, S.; Xie, H.; Scicluna, P.; Lee, L.; Björnhagen, V.; Höög, A.; Larsson, C.; Lui, W.-O. MiR-375 Regulation of LDHB Plays Distinct Roles in Polyomavirus-Positive and -Negative Merkel Cell Carcinoma. Cancers 2018, 10, 443. [Google Scholar] [CrossRef]
- Chen, R.; Zhou, X.; Yu, Z.; Liu, J.; Huang, G. Low Expression of LDHB Correlates With Unfavorable Survival in Hepatocellular Carcinoma: Strobe-Compliant Article. Medicine 2015, 94, e1583. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, X.; Ding, X.; Li, H.; Geng, M.; Xie, Z.; Wu, H.; Huang, M. Lactate Dehydrogenase B Is Associated with the Response to Neoadjuvant Chemotherapy in Oral Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0125976. [Google Scholar] [CrossRef]
- Cui, J.; Quan, M.; Jiang, W.; Hu, H.; Jiao, F.; Li, N.; Jin, Z.; Wang, L.; Wang, Y.; Wang, L. Suppressed Expression of LDHB Promotes Pancreatic Cancer Progression via Inducing Glycolytic Phenotype. Med. Oncol. 2015, 32, 143. [Google Scholar] [CrossRef]
- Hidalgo, M. New Insights into Pancreatic Cancer Biology. Ann. Oncol. 2012, 23, x135–x138. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Thompson, C.B. The Roles of Therapy-Induced Autophagy and Necrosis in Cancer Treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Jungmann, R.A. Transcriptional Regulation of the Lactate Dehydrogenase A Subunit Gene by the Phorbol Ester 12-O-Tetradecanoylphorbol-13-Acetate. Mol. Cell. Endocrinol. 1995, 108, 87–94. [Google Scholar]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 Promotes the Warburg Effect and Pancreatic Cancer Progression via Transactivation of LDHA Expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhou, F.; Li, N.; Li, Q.; Wang, L. FOXM1-LDHA Signaling Promoted Gastric Cancer Glycolytic Phenotype and Progression. Int. J. Clin. Exp. Pathol. 2015, 8, 6756–6763. [Google Scholar]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. C-Myc Transactivation of LDH-A: Implications for Tumor Metabolism and Growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar]
- Cui, X.-G.; Han, Z.-T.; He, S.-H.; Wu, X.; Chen, T.-R.; Shao, C.-H.; Chen, D.-L.; Su, N.; Chen, Y.-M.; Wang, T.; et al. HIF1/2α Mediates Hypoxia-Induced LDHA Expression in Human Pancreatic Cancer Cells. Oncotarget 2017, 8, 24840–24852. [Google Scholar] [CrossRef]
- Challapalli, A.; Carroll, L.; Aboagye, E.O. Molecular Mechanisms of Hypoxia in Cancer. Clin. Transl Imaging 2017, 5, 225–253. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Lara, A.R.; Thannickal, V.J. Lactate, a Novel Trigger of Transforming Growth Factor-β Activation in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; Harris, S.C.; McGuire, W.L. Lactate Dehydrogenase in Estrogen-Responsive Human Breast Cancer Cells. Cancer Res. 1978, 38, 2773–2776. [Google Scholar]
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC1β Regulates Multiple Myeloma Tumor Growth through LDHA-Mediated Glycolytic Metabolism. Mol Oncol 2018, 12, 1579–1595. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Liu, A.; Fang, C.; Hao, J.; Wang, Z. Lactate Dehydrogenase A Negatively Regulated by MiRNAs Promotes Aerobic Glycolysis and Is Increased in Colorectal Cancer. Oncotarget 2015, 6, 19456–19468. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, X.; Ye, F.; Chen, B.; Song, C.; Wen, J.; Zhang, Z.; Zheng, G.; Tang, H.; Xie, X. The MiR-34a-LDHA Axis Regulates Glucose Metabolism and Tumor Growth in Breast Cancer. Sci. Rep. 2016, 6, 21735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Min, Z.; Sun, K.; Qu, S.; Zhou, J.; Duan, H.; Liu, H.; Liu, X.; Gong, Z.; Li, D. MiR-199a-3p/Sp1/LDHA Axis Controls Aerobic Glycolysis in Testicular Tumor Cells. Int. J. Mol. Med. 2018, 42, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Xiong, Y.; Lei, Q.-Y.; Guan, K.-L. LDH-A Acetylation: Implication in Cancer. Oncotarget 2013, 4, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-Mediated Activation of LDHA Promotes Cancer Cell Invasion and Tumour Metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hitosugi, T.; Chung, T.-W.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine Phosphorylation of Lactate Dehydrogenase A Is Important for NADH/NAD(+) Redox Homeostasis in Cancer Cells. Mol. Cell. Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, G.; Liu, Z.; Liu, S.; Cai, Z.; You, P.; Ke, Y.; Lai, L.; Huang, Y.; Gao, H.; et al. Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res. 2018, 78, 4459–4470. [Google Scholar] [CrossRef]
- Zha, X.; Wang, F.; Wang, Y.; He, S.; Jing, Y.; Wu, X.; Zhang, H. Lactate Dehydrogenase B Is Critical for Hyperactive MTOR-Mediated Tumorigenesis. Cancer Res. 2011, 71, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Li, J.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. HMGB2 Is Associated with Malignancy and Regulates Warburg Effect by Targeting LDHB and FBP1 in Breast Cancer. Cell Commun. Signal 2018, 16, 8. [Google Scholar] [CrossRef]
- Maekawa, M.; Taniguchi, T.; Ishikawa, J.; Sugimura, H.; Sugano, K.; Kanno, T. Promoter Hypermethylation in Cancer Silences LDHB, Eliminating Lactate Dehydrogenase Isoenzymes 1-4. Clin. Chem. 2003, 49, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, Y.; Hoshino, I.; Nohata, N.; Kinoshita, T.; Akutsu, Y.; Hanari, N.; Mori, M.; Yoneyama, Y.; Akanuma, N.; Takeshita, N.; et al. Identification of Novel Molecular Targets Regulated by Tumor Suppressive MiR-375 Induced by Histone Acetylation in Esophageal Squamous Cell Carcinoma. Int. J. Oncol. 2012, 41, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Billard, C. Apoptosis Inducers in Chronic Lymphocytic Leukemia. Oncotarget 2014, 5, 309–325. [Google Scholar] [CrossRef]
- Letai, A. Apoptosis and Cancer. Annu. Rev. Cancer Biol. 2017, 1, 275–294. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Gamero, A.M. Interferons as Inducers of Apoptosis in Malignant Cells. J. Interferon Cytokine Res. 2013, 33, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis-the P53 Network. J. Cell. Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. The Role of P53 in Apoptosis. Discov. Med. 2010, 9, 145–152. [Google Scholar] [PubMed]
- Allison, S.J.; Knight, J.R.P.; Granchi, C.; Rani, R.; Minutolo, F.; Milner, J.; Phillips, R.M. Identification of LDH-A as a Therapeutic Target for Cancer Cell Killing via (i) P53/NAD(H)-Dependent and (Ii) P53-Independent Pathways. Oncogenesis 2014, 3, e102. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Wang, Z.; Wang, Q.; Han, J.; Zhao, C.; Hong, Y.; Zeng, L.; Tang, L.; Ying, W. Roles of NAD(+) / NADH and NADP(+) / NADPH in Cell Death. Curr. Pharm. Des. 2009, 15, 12–19. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Loo, T.Y.; Shen, J.-G.; Wang, N.; Wang, D.-M.; Yang, D.-P.; Mo, S.-L.; Guan, X.-Y.; Chen, J.-P. LDH-A Silencing Suppresses Breast Cancer Tumorigenicity through Induction of Oxidative Stress Mediated Mitochondrial Pathway Apoptosis. Breast Cancer Res. Treat. 2012, 131, 791–800. [Google Scholar] [CrossRef]
- Yu, Y.; Liao, M.; Liu, R.; Chen, J.; Feng, H.; Fu, Z. Overexpression of Lactate Dehydrogenase-A in Human Intrahepatic Cholangiocarcinoma: Its Implication for Treatment. World J. Surg. Oncol. 2014, 12, 78. [Google Scholar] [CrossRef]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of Lactate Dehydrogenase A Suppresses Tumor Growth and Metastasis of Human Hepatocellular Carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef]
- McConkey, D.J.; Orrenius, S. The Role of Calcium in the Regulation of Apoptosis. Biochem. Biophys. Res. Commun. 1997, 239, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by Oxamate Induces G2/M Arrest, Apoptosis and Increases Radiosensitivity in Nasopharyngeal Carcinoma Cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial Control of Apoptosis: The Role of Cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar] [CrossRef]
- Kuida, K. Caspase-9. Int. J. Biochem. Cell Biol. 2000, 32, 121–124. [Google Scholar] [CrossRef]
- Pawlowski, J.; Kraft, A.S. Bax-Induced Apoptotic Cell Death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef]
- Vogler, M.; Hamali, H.A.; Sun, X.-M.; Bampton, E.T.W.; Dinsdale, D.; Snowden, R.T.; Dyer, M.J.S.; Goodall, A.H.; Cohen, G.M. BCL2/BCL-X(L) Inhibition Induces Apoptosis, Disrupts Cellular Calcium Homeostasis, and Prevents Platelet Activation. Blood 2011, 117, 7145–7154. [Google Scholar] [CrossRef]
- Flanagan, L.; Kehoe, J.; Fay, J.; Bacon, O.; Lindner, A.U.; Kay, E.W.; Deasy, J.; McNamara, D.A.; Prehn, J.H.M. High Levels of X-Linked Inhibitor-of-Apoptosis Protein (XIAP) Are Indicative of Radio Chemotherapy Resistance in Rectal Cancer. Radiat. Oncol. 2015, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of Autophagy in Cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Kania, E.; Pająk, B.; O’Prey, J.; Sierra Gonzalez, P.; Litwiniuk, A.; Urbańska, K.; Ryan, K.M.; Orzechowski, A. Verapamil Treatment Induces Cytoprotective Autophagy by Modulating Cellular Metabolism. FEBS J. 2017, 284, 1370–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Han, F.; Yang, S.; Wu, J.; Zhan, W. Oxamate-Mediated Inhibition of Lactate Dehydrogenase Induces Protective Autophagy in Gastric Cancer Cells: Involvement of the Akt-MTOR Signaling Pathway. Cancer Lett. 2015, 358, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and Multidrug Resistance in Cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Han, M.W.; Lee, J.C.; Choi, J.-Y.; Kim, G.C.; Chang, H.W.; Nam, H.Y.; Kim, S.W.; Kim, S.Y. Autophagy Inhibition Can Overcome Radioresistance in Breast Cancer Cells through Suppression of TAK1 Activation. Anticancer Res. 2014, 34, 1449–1455. [Google Scholar] [PubMed]
- Gewirtz, D.A. The Four Faces of Autophagy: Implications for Cancer Therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-A.; Chao, Y.; Shiah, S.-G.; Lin, W.-W. Nutrient Deprivation Induces the Warburg Effect through ROS/AMPK-Dependent Activation of Pyruvate Dehydrogenase Kinase. Biochim. Biophys. Acta 2013, 1833, 1147–1156. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 Regulates Energy Metabolism, Differentiation and Survival of Philadelphia-Chromosome-Positive Cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef]
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different Effects of LDH-A Inhibition by Oxamate in Non-Small Cell Lung Cancer Cells. Oncotarget 2014, 5, 11886–11896. [Google Scholar] [CrossRef]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT5-Mediated Deacetylation of LDHB Promotes Autophagy and Tumorigenesis in Colorectal Cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbańska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 2085. https://doi.org/10.3390/ijms20092085
Urbańska K, Orzechowski A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. International Journal of Molecular Sciences. 2019; 20(9):2085. https://doi.org/10.3390/ijms20092085
Chicago/Turabian StyleUrbańska, Kaja, and Arkadiusz Orzechowski. 2019. "Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells" International Journal of Molecular Sciences 20, no. 9: 2085. https://doi.org/10.3390/ijms20092085
APA StyleUrbańska, K., & Orzechowski, A. (2019). Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. International Journal of Molecular Sciences, 20(9), 2085. https://doi.org/10.3390/ijms20092085