Vascular Diseases and Gangliosides


Abstract
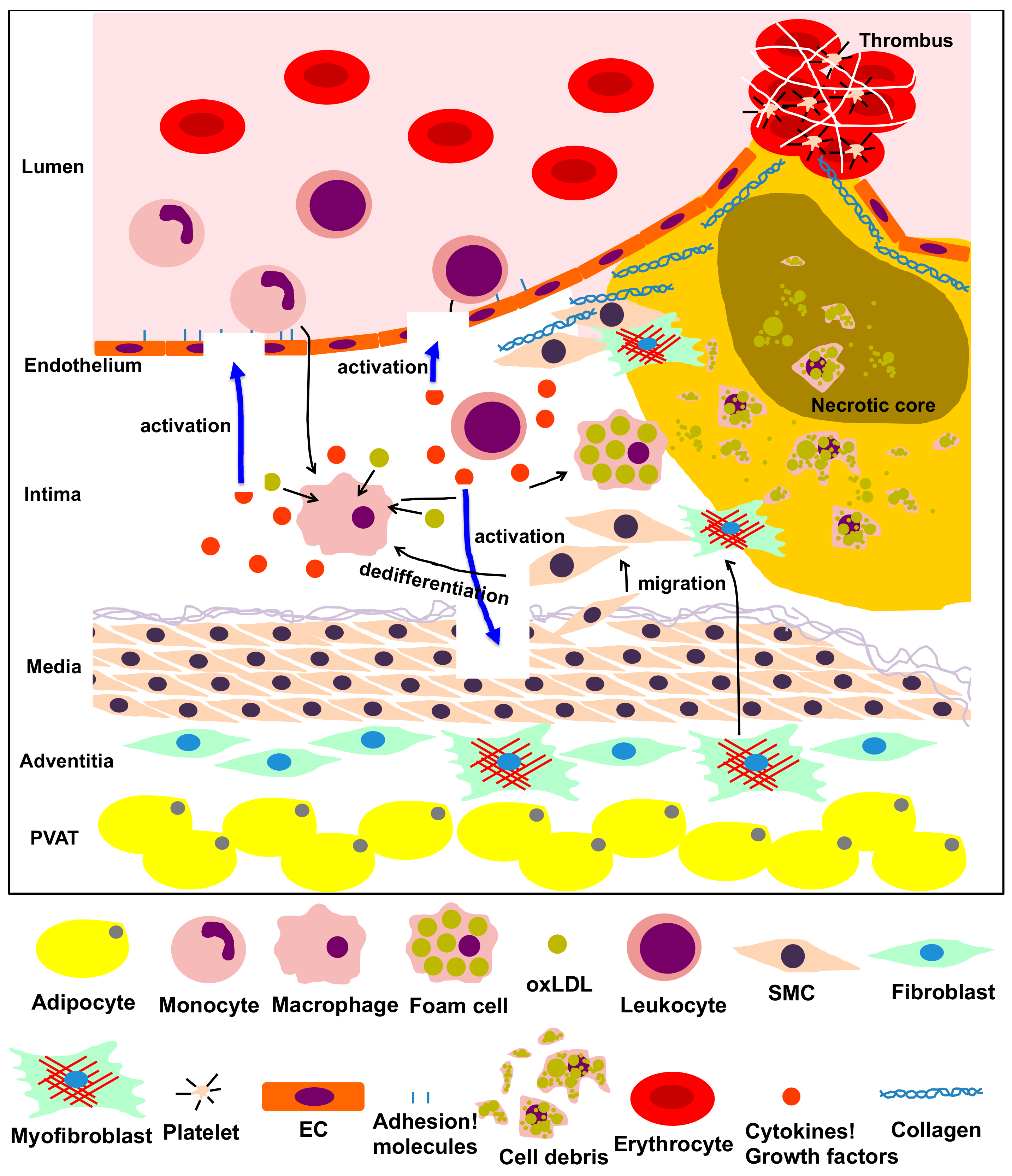
1. Introduction
2. Gangliosides in Vascular and Vascular Associated-Cells (Table.1)
2.1. ECs and Gangliosides
2.2. VSMCs and Gangliosides
2.3. Fibroblasts and Gangliosides
2.4. Inflammatory Cells and Gangliosides
2.5. Other Types of Cells and Gangliosides
3. Relevance to Aging
Author Contributions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Torzewski, M. Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis. Biomolecules 2019, 9, 472. [Google Scholar] [CrossRef] [PubMed]
- Bot, I.; Shi, G.P.; Kovanen, P.T. Mast cells as effectors in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Qi, X.Y.; Qu, S.L.; Xiong, W.H.; Rom, O.; Chang, L.; Jiang, Z.S. Perivascular adipose tissue (PVAT) in atherosclerosis: A double-edged sword. Cardiovasc. Diabetol. 2018, 17, 134. [Google Scholar] [CrossRef]
- Jackson, A.O.; Zhang, J.; Jiang, Z.; Yin, K. Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases. Trends. Cardiovasc. Med. 2017, 27, 383–393. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Lagraauw, H.M.; Wezel, A.; van der Velden, D.; Kuiper, J.; Bot, I. Stress-induced mast cell activation contributes to atherosclerotic plaque destabilization. Sci. Rep. 2019, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Neupane, R.; Jin, X.; Sasaki, T.; Li, X.; Murohara, T.; Cheng, X.W. Immune Disorder in Atherosclerotic Cardiovascular Disease- Clinical Implications of Using Circulating T-Cell Subsets as Biomarkers. Circ. J. 2019, 83, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, J.E.; Kavousi, M.; Leebeek, F.W.; Felix, J.F.; Hofman, A.; Witteman, J.C.; de Maat, M.P. von Willebrand factor plasma levels, genetic variations and coronary heart disease in an older population. J. Thromb. Haemost. 2012, 10, 1262–1269. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Schmitt, M.M. Platelets and their chemokines in atherosclerosis-clinical applications. Front. Physiol. 2014, 5, 294. [Google Scholar] [CrossRef]
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front. Immunol. 2017, 8, 928. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Sonnino, S.; Chiricozzi, E.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Gangliosides in Membrane Organization. Prog. Mol. Biol. Transl. Sci. 2018, 156, 83–120. [Google Scholar] [PubMed]
- Zhuo, D.; Guan, F. Ganglioside GM1 promotes contact inhibition of growth by regulating the localization of epidermal growth factor receptor from glycosphingolipid-enriched microdomain to caveolae. Cell Prolif. 2019, 52, e12639. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Gangliosides Contribute to Vascular Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1819. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L. Ganglioside designation. Adv. Exp. Med. Biol. 1980, 125, 11. [Google Scholar]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef]
- Rusnati, M.; Urbinati, C.; Tanghetti, E.; Dell’Era, P.; Lortat-Jacob, H.; Presta, M. Cell membrane GM1 ganglioside is a functional coreceptor for fibroblast growth factor 2. Proc. Natl. Acad. Sci. USA 2002, 99, 4367–4372. [Google Scholar] [CrossRef]
- Slevin, M.; Kumar, S.; He, X.; Gaffney, J. Physiological concentrations of gangliosides GM1, GM2 and GM3 differentially modify basic-fibroblast-growth-factor-induced mitogenesis and the associated signalling pathway in endothelial cells. Int. J. Cancer 1999, 82, 412–423. [Google Scholar] [CrossRef]
- Liu, Y.; McCarthy, J.; Ladisch, S. Membrane ganglioside enrichment lowers the threshold for vascular endothelial cell angiogenic signaling. Cancer Res. 2006, 66, 10408–10414. [Google Scholar] [CrossRef]
- Chung, T.W.; Kim, S.J.; Choi, H.J.; Kim, K.J.; Kim, M.J.; Kim, S.H.; Lee, H.J.; Ko, J.H.; Lee, Y.C.; Suzuki, A.; et al. Ganglioside GM3 inhibits VEGF/VEGFR-2-mediated angiogenesis: Direct interaction of GM3 with VEGFR-2. Glycobiology 2009, 19, 229–239. [Google Scholar] [CrossRef]
- Kim, S. J1.; Chung, T.W.; Choi, H.J.; Jin, U.H.; Ha, K.T.; Lee, Y.C.; Kim, C.H. Monosialic ganglioside GM3 specifically suppresses the monocyte adhesion to endothelial cells for inflammation. Int. J. Biochem. Cell Biol. 2014, 46, 32–38. [Google Scholar] [CrossRef]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Ganglioside GM1 Contributes to the State of Insulin Resistance in Senescent Human Arterial Endothelial Cells. J. Biol. Chem. 2015, 290, 25475–25486. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Ganglioside GM1 contributes to extracellular/intracellular regulation of insulin resistance, impairment of insulin signaling and down-stream eNOS activation, in human aortic endothelial cells after short- or long-term exposure to TNFα. Oncotarget 2017, 9, 5562–5577. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bhunia, A.K.; Schwarzmann, G.; Chatterjee, S. GD3 recruits reactive oxygen species to induce cell proliferation and apoptosis in human aortic smooth muscle cells. J. Biol. Chem. 2002, 277, 16396–16402. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.K.; Kim, H.M.; Lee, Y.C.; Kim, C.H. Disialoganglioside (GD3) synthase gene expression suppresses vascular smooth muscle cell responses via the inhibition of ERK1/2 phosphorylation, cell cycle progression, and matrix metalloproteinase-9 expression. J. Biol. Chem. 2004, 279, 33063–33070. [Google Scholar] [CrossRef] [PubMed]
- Gouni-Berthold, I.; Seul, C.; Ko, Y.; Hescheler, J.; Sachinidis, A. Gangliosides GM1 and GM2 induce vascular smooth muscle cell proliferation via extracellular signal-regulated kinase 1/2 pathway. Hypertension 2001, 38, 1030–1037. [Google Scholar] [CrossRef]
- Li, R.; Manela, J.; Kong, Y.; Ladisch, S. Cellular gangliosides promote growth factor-induced proliferation of fibroblasts. J. Biol. Chem. 2000, 275, 34213–34223. [Google Scholar] [CrossRef]
- Liu, Y.; Su, Y.; Wiznitzer, M.; Epifano, O.; Ladisch, S. Ganglioside depletion and EGF responses of human GM3 synthase-deficient fibroblasts. Glycobiology 2008, 18, 593–601. [Google Scholar] [CrossRef]
- Matarrese, P.; Garofalo, T.; Manganelli, V.; Gambardella, L.; Marconi, M.; Grasso, M.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Evidence for the involvement of GD3 ganglioside in autophagosome formation and maturation. Autophagy 2014, 10, 750–765. [Google Scholar] [CrossRef]
- Hashiramoto, A.; Mizukami, H.; Yamashita, T. Ganglioside GM3 promotes cell migration by regulating MAPK and c-Fos/AP-1. Oncogene 2006, 25, 3948–3955. [Google Scholar] [CrossRef]
- Cavallini, L.; Venerando, R.; Miotto, G.; Alexandre, A. Ganglioside GM1 protection from apoptosis of rat heart fibroblasts. Arch. Biochem. Biophys. 1999, 370, 156–162. [Google Scholar] [CrossRef]
- Stroud, M.R.; Handa, K.; Salyan, M.E.; Ito, K.; Levery, S.B.; Hakomori, S.; Reinhold, B.B.; Reinhold, W.N. Monosialogangliosides of human myelogenous leukemia HL60 cells and normal human leukocytes. 1. Separation of E-selectin binding from nonbinding gangliosides, and absence of sialosyl-Le(x) having tetraosyl to octaosyl core. Biochemistry 1996, 35, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Macher, B.A.; Klock, J.C.; Fukuda, M.N.; Fukuda, M. Isolation and structural characterization of human lymphocyte and neutrophil gangliosides. J. Biol. Chem. 1981, 256, 1968–1974. [Google Scholar] [PubMed]
- Sheriff, A.; Gaipl, U.S.; Franz, S.; Heyder, P.; Voll, R.E.; Kalden, J.R.; Herrmann, M. Loss of GM1 surface expression precedes annexin V-phycoerythrin binding of neutrophils undergoing spontaneous apoptosis during in vitro aging. Cytom. A 2004, 62, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zuberbier, T.; Guhl, S.; Hantke, T.; Hantke, C.; Welker, P.; Grabbe, J.; Henz, B.M. Alterations in ganglioside expression during the differentiation of human mast cells. Exp. Dermatol. 1999, 8, 380–387. [Google Scholar] [CrossRef]
- Fujimaki, H.; Nohara, O.; Katayama, N.; Abe, T.; Nohara, K. Ganglioside GM3 inhibits interleukin-3-dependent bone marrow-derived mast cell proliferation. Int. Arch. Allergy Immunol. 1995, 107, 527–532. [Google Scholar] [CrossRef]
- Filho, E.G.; da Silva, E.Z.; Zanotto, C.Z.; Oliver, C.; Jamur, M.C. Cross-Linking Mast Cell Specific Gangliosides Stimulates the Release of Newly Formed Lipid Mediators and Newly Synthesized Cytokines. Mediators Inflamm. 2016, 2016, 9160540. [Google Scholar] [CrossRef]
- Nojiri, H.; Takaku, F.; Terui, Y.; Miura, Y.; Saito, M. Ganglioside GM3: An acidic membrane component that increases during macrophage-like cell differentiation can induce monocytic differentiation of human myeloid and monocytoid leukemic cell lines HL-60 and U937. Proc. Natl. Acad. Sci. USA 1986, 83, 782–786. [Google Scholar] [CrossRef]
- Chung, T.W.; Choi, H.J.; Park, M.J.; Choi, H.J.; Lee, S.O.; Kim, K.J.; Kim, C.H.; Hong, C.; Kim, K.H.; Joo, M. The function of cancer-shed gangliosides in macrophage phenotype: Involvement with angiogenesis. Oncotarget. 2017, 8, 4436–4448. [Google Scholar] [CrossRef]
- Rosenfelder, G.; Ziegler, A.; Wernet, P.; Braun, D.G. Ganglioside patterns: New biochemical markers for human hematopoietic cell lines. J. Natl. Cancer Inst. 1982, 68, 203–209. [Google Scholar]
- Cho, J.H.; Kim, H.O.; Surh, C.D.; Sprent, J. T cell receptor-dependent regulation of lipid rafts controls naive CD8+ T cell homeostasis. Immunity 2010, 32, 214–226. [Google Scholar] [CrossRef]
- Barbat, C.; Trucy, M.; Sorice, M.; Garofalo, T.; Manganelli, V.; Fischer, A.; Mazerolles, F. p56lck, LFA-1 and PI3K but not SHP-2 interact with GM1- or GM3-enriched microdomains in a CD4-p56lck association-dependent manner. Biochem. J. 2007, 402, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, P.; Lenti, L.; Martini, F.; Ciatti, F.; Pontieri, G.M.; Gazzaniga, P.P. Ganglioside content of human platelets--differences in resting and activated platelets. Thromb. Haemost. 1997, 77, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, A.V.; Prokazova, N.V.; Mikhailenko, I.A.; Mukjin, D.N.; Repin, V.S.; Bergelson, L.D. Stimulation of platelet adhesion and activation by ganglioside GD3 adsorbed to plastic. Biochim. Biophys. Acta 1988, 968, 167–171. [Google Scholar] [CrossRef]
- Yatomi, Y.; Igarashi, Y.; Hakomori, S. Effects of exogenous gangliosides on intracellular Ca2+ mobilization and functional responses in human platelets. Glycobiology 1996, 6, 347–353. [Google Scholar] [CrossRef]
- Nguyen, K.A.; Hamzeh-Cognasse, H.; Palle, S.; Anselme-Bertrand, I.; Arthaud, C.A.; Chavarin, P.; Pozzetto, B.; Garraud, O.; Cognasse, F. Role of Siglec-7 in apoptosis in human platelets. PLoS ONE 2014, 9, e106239. [Google Scholar] [CrossRef]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 13678–13683. [Google Scholar] [CrossRef]
- Duvar, S.; Peter-Katalinić, J.; Hanisch, F.G.; Müthing, J. Isolation and structural characterization of glycosphingolipids of in vitro propagated bovine aortic endothelial cells. Glycobiology 1997, 7, 1099–1109. [Google Scholar] [CrossRef][Green Version]
- Minciullo, P.L.; Catalano, A.; Mandraffino, G.; Casciaro, M.; Crucitti, A.; Maltese, G.; Morabito, N.; Lasco, A.; Gangemi, S.; Basile, G. Inflammaging and Anti-Inflammaging: The Role of Cytokines in Extreme Longevity. Arch. Immunol. Ther. Exp. (Warsz) 2016, 64, 111–126. [Google Scholar] [CrossRef]
- Kim, S.J.; Chung, T.W.; Choi, H.J.; Kwak, C.H.; Song, K.H.; Suh, S.J.; Kwon, K.M.; Chang, Y.C.; Park, Y.G.; Chang, H.W.; et al. Ganglioside GM3 participates in the TGF-β1-induced epithelial-mesenchymal transition of human lens epithelial cells. Biochem. J. 2013, 449, 241–251. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef]
- Chatterjee, S.B.; Dey, S.; Shi, W.Y.; Thomas, K.; Hutchins, G.M. Accumulation of glycosphingolipids in human atherosclerotic plaque and unaffected aorta tissues. Glycobiology 1997, 7, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Vacek, T.P.; Rehman, S.; Neamtu, D.; Yu, S.; Givimani, S.; Tyagi, S.C. Matrix metalloproteinases in atherosclerosis: Role of nitric oxide, hydrogen sulfide, homocysteine, and polymorphisms. Vasc. Health Risk Manag. 2015, 11, 173–183. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Golovanova, N.K.; Tran, D.; Samovilova, N.N.; Gracheva, E.V.; Efremov, E.E.; Sobolev, A.Y.; Yurchenko, Y.V.; Lord, R.S.; Cao, W.; et al. Expression of GM3 synthase in human atherosclerotic lesions. Atherosclerosis 2006, 184, 63–71. [Google Scholar] [CrossRef]
- Park, S.S.; Kim, W.J.; Moon, S.K. Suppression of vascular smooth muscle cell responses induced by TNF-α in GM3 synthase gene transfected cells. Int. J. Mol. Med. 2011, 27, 147–154. [Google Scholar] [PubMed]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef]
- Deaton, R.A.; Gan, Q.; Owens, G.K. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1027–H1037. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Hynds, D.L.; Summers, M.; Van Brocklyn, J.; O’Dorisio, M.S.; Yates, A.J. Gangliosides inhibit platelet-derived growth factor-stimulated growth, receptor phosphorylation, and dimerization in neuroblastoma SH-SY5Y cells. J. Neurochem. 1995, 65, 2251–2258. [Google Scholar] [CrossRef]
- Mitsuda, T.; Furukawa, K.; Fukumoto, S.; Miyazaki, H.; Urano, T.; Furukawa, K. Overexpression of ganglioside GM1 results in the dispersion of platelet-derived growth factor receptor from glycolipid-enriched microdomains and in the suppression of cell growth signals. J. Biol. Chem. 2002, 277, 11239–11246. [Google Scholar] [CrossRef]
- Bergante, S.; Creo, P.; Piccoli, M.; Ghiroldi, A.; Menon, A.; Cirillo, F.; Rota, P.; Monasky, M.M.; Ciconte, G.; Pappone, C.; et al. GM1 Ganglioside Promotes Osteogenic Differentiation of Human Tendon Stem Cells. Stem Cells Int. 2018, 2018, 4706943. [Google Scholar] [CrossRef]
- Ohsawa, T.; Nagai, Y. Ganglioside changes during cell aging in human diploid fibroblast TIG-1. Exp. Gerontol. 1982, 17, 287–293. [Google Scholar] [CrossRef]
- Meran, S.; Luo, D.D.; Simpson, R.; Martin, J.; Wells, A.; Steadman, R.; Phillips, A.O. Hyaluronan facilitates transforming growth factor-β1-dependent proliferation via CD44 and epidermal growth factor receptor interaction. J. Biol. Chem. 2011, 286, 17618–17630. [Google Scholar] [CrossRef] [PubMed]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Sialylation regulates myofibroblast differentiation of human skin fibroblasts. Stem Cell Res. Ther. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Kambe, N.; Du, Z.; Li, Y.; Xia, H.Z.; Kambe, M.; Bieberich, E.; Pozez, A.; Grimes, M.; Yu, R.K.; et al. Disialoganglioside GD3 is selectively expressed by developing and mature human mast cells. J. Allergy Clin. Immunol. 2001, 107, 322–330. [Google Scholar] [CrossRef]
- Hakomori, S. Structure, organization, and function of glycosphingolipids in membrane. Curr. Opin. Hematol. 2003, 10, 16–24. [Google Scholar] [CrossRef]
- Gracheva, E.V.; Samovilova, N.N.; Golovanova, N.K.; Andreeva, E.R.; Andrianova, I.V.; Tararak, E.M.; Prokazova, N.V. Activation of ganglioside GM3 biosynthesis in human monocyte/macrophages during culturing in vitro. Biochemistry (Mosc) 2007, 72, 772–777. [Google Scholar] [CrossRef]
- Gracheva, E.V.; Samovilova, N.N.; Golovanova, N.K.; Kashirina, S.V.; Shevelev, A.; Rybalkin, I.; Gurskaya, T.; Vlasik, T.N.; Andreeva, E.R.; Prokazova, N.V. Enhancing of GM3 synthase expression during differentiation of human blood monocytes into macrophages as in vitro model of GM3 accumulation in atherosclerotic lesion. Mol. Cell Biochem. 2009, 330, 121–129. [Google Scholar] [CrossRef]
- Sorice, M.; Parolini, I.; Sansolini, T.; Garofalo, T.; Dolo, V.; Sargiacomo, M.; Tai, T.; Peschle, C.; Torrisi, M.R.; Pavan, A. Evidence for the existence of ganglioside-enriched plasma membrane domains in human peripheral lymphocytes. J. Lipid Res. 1997, 38, 969–980. [Google Scholar]
- Garofalo, T.; Sorice, M.; Misasi, R.; Cinque, B.; Giammatteo, M.; Pontieri, G.M.; Cifone, M.G.; Pavan, A. A novel mechanism of CD4 down-modulation induced by monosialoganglioside GM3. Involvement of serine phosphorylation and protein kinase c delta translocation. J. Biol. Chem. 1998, 273, 35153–35160. [Google Scholar] [CrossRef]
- Zhang, T.; de Waard, A.A.; Wuhrer, M.; Spaapen, R.M. The Role of Glycosphingolipids in Immune Cell Functions. Front. Immunol. 2019, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Nagafuku, M.; Sato, T.; Sato, S.; Shimizu, K.; Taira, T.; Inokuchi, J. Control of homeostatic and pathogenic balance in adipose tissue by ganglioside GM3. Glycobiology 2015, 25, 303–318. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Invest. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef]
- Cudejko, C.; Wouters, K.; Fuentes, L.; Hannou, S.A.; Paquet, C.; Bantubungi, K.; Bouchaert, E.; Vanhoutte, J.; Fleury, S.; Remy, P.; et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood 2011, 118, 2556–2566. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Sources | Types of Gangliosides | Functional Roles | References |
|---|---|---|---|---|
| GM 7373 cells (ECs) | Bovine | GM1 | Coreceptor of bFGF | [26] |
| BAECs | Bovine | GM2, GM1 | Inhibition of proliferation | [27] |
| GM3 | Promotion of proliferation | [27] | ||
| HUVECs | Human | GD1a | Enhancement of VEGF-induced signaling, proliferation and migration | [28] |
| GM3 | Inhibition of VEGF signaling, angiogenesis and adhesion molecules | [29,30] | ||
| HAECs | Human | GM1 | Association with aging and Inhibition of insulin signaling | [31,32] |
| VSMCs | Human | GD3 | Modulation of proliferation and apoptosis | [33] |
| VSMCs | Mouse | GD3 | Inhibition of PDGF-induced ERK pathway and proliferation | [34] |
| GD3 | Inhibition of TNFα-induced MMP9 expression | [34] | ||
| VSMCs | Rat | GM2, GM1 | Activation of ERK pathway and promotion of proliferation | [35] |
| Fibroblasts (dermal) | Human | GM3, GD1a | Promotion of EGF or bFGF stimulated proliferation | [36,37] |
| GD3 | Activation of autophagic process | [38] | ||
| Fibroblasts (embryonic) | Mouse | GM3 | Attenuation of FBS stimulated MAPK pathway | [39] |
| Fibroblasts (heart) | Rat | GM1 | Protection from apoptosis caused from protein kinase C inhibition | [40] |
| Neutrophils | Human | GM1 | Association with maturation | [41,42] |
| GM1 | Decrease at early stage of apoptosis | [43] | ||
| HMC-1 (mast cell line) | Human | GM3, GM2, GM1, GD1a | Association with maturation | [44] |
| Mast cells | Mouse | GM3 | Inhibition of IL-3 stimulated proliferation | [45] |
| RBL-2H3 (mast cell line) | Rat | GD1b | Activation and induction of inflammatory cytokines | [46] |
| HL-60, U937 (monocyte) | Human | GM3 | Induction of cell differentiation | [47] |
| Raw264.7 (macrophage) | Mouse | GM1 | Induction of arginase-1 and MCP-1 | [48] |
| T cells | Human | GM3, GM1 | Association with activation | [49] |
| CD8+ T cells | Human | GM1 | Increase with IL-2 stimulation | [50] |
| CD4+ T cells | Human | GM3, GM1 | Downregulation of CD4 expression | [51] |
| Platelets | Human | GD3 | Association with activation | [52,53] |
| GM3, GM1 | Induction of activation with Ca2+ mobilization and shape change | [54] | ||
| GD2 | Induction of apoptosis | [55] | ||
| 3T3-L1 (adipocyte) | Mouse | GM3 | Inhibition of insulin signaling | [56] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasaki, N.; Toyoda, M. Vascular Diseases and Gangliosides. Int. J. Mol. Sci. 2019, 20, 6362. https://doi.org/10.3390/ijms20246362
Sasaki N, Toyoda M. Vascular Diseases and Gangliosides. International Journal of Molecular Sciences. 2019; 20(24):6362. https://doi.org/10.3390/ijms20246362
Chicago/Turabian StyleSasaki, Norihiko, and Masashi Toyoda. 2019. "Vascular Diseases and Gangliosides" International Journal of Molecular Sciences 20, no. 24: 6362. https://doi.org/10.3390/ijms20246362
APA StyleSasaki, N., & Toyoda, M. (2019). Vascular Diseases and Gangliosides. International Journal of Molecular Sciences, 20(24), 6362. https://doi.org/10.3390/ijms20246362