Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics

 ,
,  ,
,  ,
, 
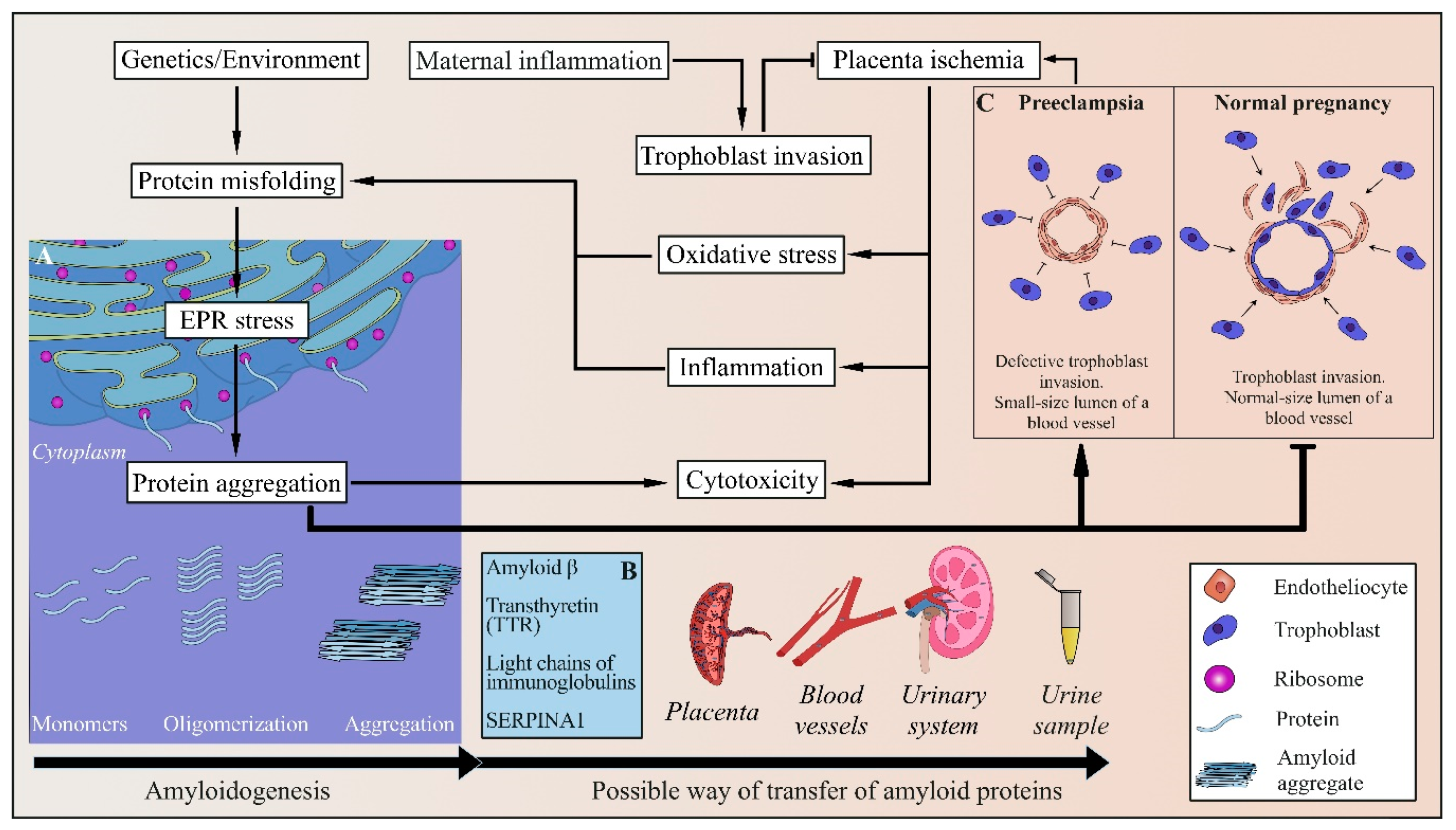{kind=link}
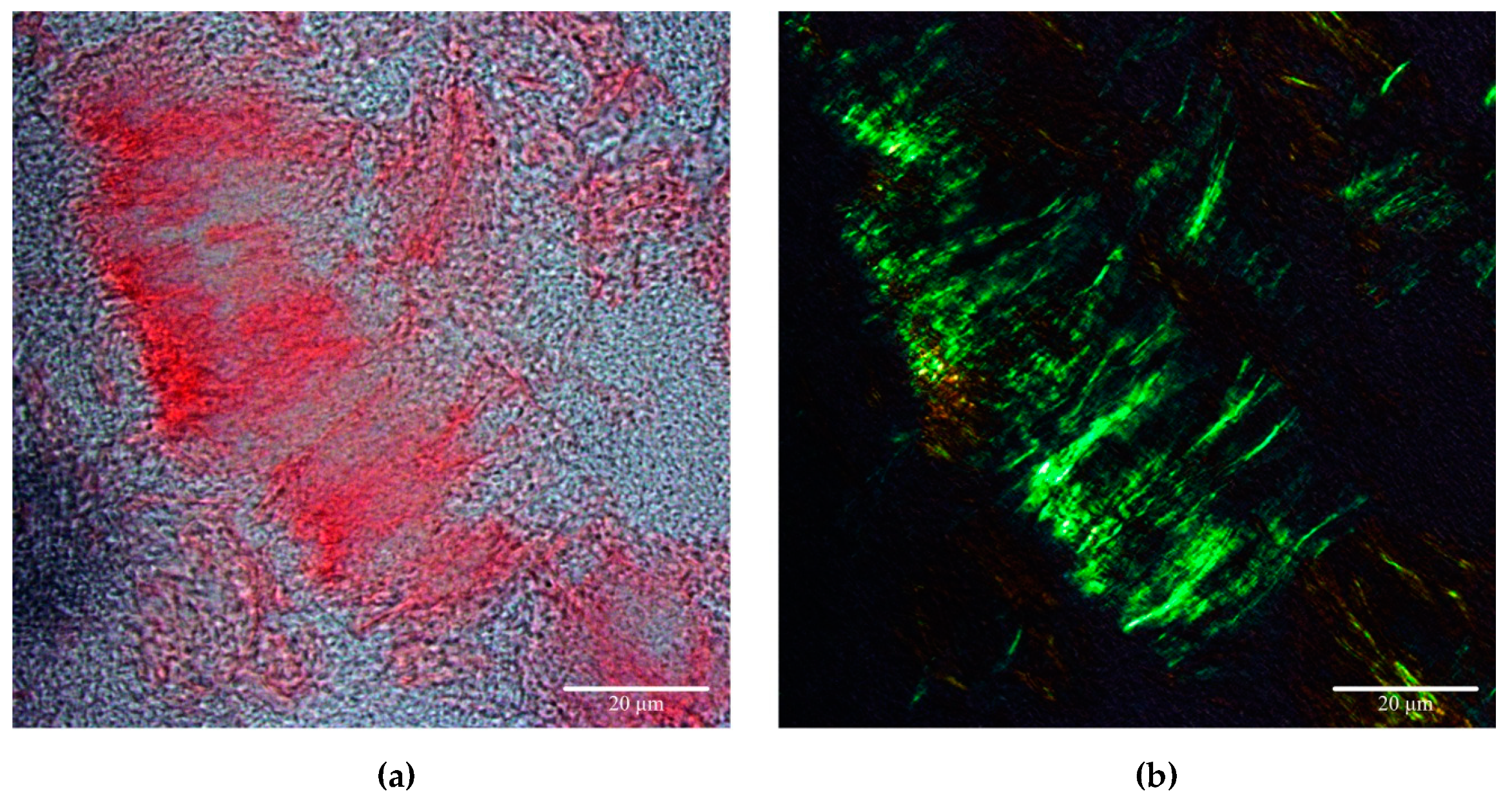{kind=link}
{kind=link}
Abstract
1. Introduction
2. Diagnosis of PE
3. Etiology and Pathogenesis of PE
4. Protein Misfolding and Amyloid Aggregation in PE
4.1. Amyloids and Amyloidogenic Diseases
4.2. Amyloids in PE
4.3. Alpha-1 Antitrypsin in PE
4.4. Light Chains of Immunoglobulins in PE
4.5. Amyloid β in PE
4.6. Transthyretin in PE
4.7. Possible Role of the Human Pregnancy Zone Protein
5. New Approaches to PE Diagnostics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PE | Preeclampsia |
| CR | Congo Red |
| CRD | Congo Red |
| BP | Blood Pressure |
| sFlt-1 | Soluble Fms-like tyrosine kinase-1 |
| sEng | soluble Endoglin |
| PLGF | Placental Growth Factor |
| sVEGFR | Vascular Endothelial Growth Factor |
| VEGF | Vascular Endothelial Growth Factor |
| ROS | Reactive Oxygen Species |
| HO | Heme Oxygenase |
| mRNA | messenger Ribonucleic Acid |
| NkB | Neurokinin B |
| AT1-AA | Autoantibodies to Angiotensin II receptor 1 |
| Apo E | Apolipoprotein E |
| TSEs | Transmissible Spongiform Encephalopathies |
| Aβ | Amyloid β peptide |
| EM | Electron Microscopy |
| ER | Endoplasmic Reticulum |
| TTR | Transthyretin |
| MS | Mass Spectrometry |
| igG | immunoglobulins |
| IFI-6 | Interferon-inducible protein 6-16 |
| APP | Amyloid Precursor Protein |
| sAPPa | soluble N-terminal fragment of APP |
| α2M | alpha-2-macroglobulin |
| PZP | Pregnancy Zone Protein |
| ThT | Thioflavin-T |
References
- Duley, L. The Global Impact of Pre-eclampsia and Eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians; Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2014, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Sircar, M.; Thadhani, R.; Karumanchi, S.A. Pathogenesis of preeclampsia. Curr. Opin. Nephrol. Hypertens. 2015, 24, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.; Sibai, B. Maternal Mortality from Preeclampsia/Eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular Mechanisms of Preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5, a023473. [Google Scholar] [CrossRef]
- Wu, P.; van den Berg, C.; Alfirevic, Z.; O’Brien, S.; Röthlisberger, M.; Baker, P.; Kenny, L.; Kublickiene, K.; Duvekot, J. Early Pregnancy Biomarkers in Pre-Eclampsia: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2015, 16, 23035–23056. [Google Scholar] [CrossRef]
- Wright, D.; Tan, M.Y.; O’Gorman, N.; Poon, L.C.; Syngelaki, A.; Wright, A.; Nicolaides, K.H. Predictive performance of the competing risk model in screening for preeclampsia. Am. J. Obstet. Gynecol. 2019, 220, e1–e199. [Google Scholar] [CrossRef]
- Schiettecatte, J.; Russcher, H.; Anckaert, E.; Mees, M.; Leeser, B.; Tirelli, A.S.; Fiedler, G.M.; Luthe, H.; Denk, B.; Smitz, J. Multicenter evaluation of the first automated Elecsys sFlt-1 and PlGF assays in normal pregnancies and preeclampsia. Clin. Biochem. 2010, 43, 768–770. [Google Scholar] [CrossRef]
- Salahuddin, S.; Lee, Y.; Vadnais, M.; Sachs, B.P.; Karumanchi, S.A.; Lim, K.-H. Diagnostic utility of soluble fms-like tyrosine kinase 1 and soluble endoglin in hypertensive diseases of pregnancy. Am. J. Obstet. Gynecol. 2007, 197, e1–e28. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.-H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Sovio, U.; Gaccioli, F.; Cook, E.; Hund, M.; Charnock-Jones, D.S.; Smith, G.C.S. Prediction of Preeclampsia Using the Soluble fms-Like Tyrosine Kinase 1 to Placental Growth Factor Ratio: A Prospective Cohort Study of Unselected Nulliparous Women. Hypertension 2017, 69, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Wortelboer, E.; Koster, M.; Cuckle, H.; Stoutenbeek, P.; Schielen, P.; Visser, G. First-trimester placental protein 13 and placental growth factor: Markers for identification of women destined to develop early-onset pre-eclampsia. Int. J. Obstet. Gynaecol. 2010, 117, 1384–1389. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Bindra, R.; Turan, O.M.; Chefetz, I.; Sammar, M.; Meiri, H.; Tal, J.; Cuckle, H.S. A novel approach to first-trimester screening for early pre-eclampsia combining serum PP-13 and Doppler ultrasound. Ultrasound Obstet. Gynecol. 2005, 27, 13–17. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.-H.; Yuan, H.-T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Foidart, J.-M.; Munaut, C.; Chantraine, F.; Akolekar, R.; Nicolaides, K.H. Maternal plasma soluble endoglin at 11–13 weeks’ gestation in pre-eclampsia. Ultrasound Obstet. Gynecol. 2010, 35, 680–687. [Google Scholar] [CrossRef]
- Spencer, K.; Cowans, N.J.; Nicolaides, K.H. Low levels of maternal serum PAPP-A in the first trimester and the risk of pre-eclampsia. Prenat. Diagn. 2008, 28, 7–10. [Google Scholar] [CrossRef]
- Poon, L.C.; Nicolaides, K.H. Early Prediction of Preeclampsia. Obstet. Gynecol. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Diagnosis and management of preeclampsia and eclampsia. Int. J. Gynecol. Obstet. 2002, 77, 67–75. [CrossRef]
- Lowe, S.A.; Brown, M.A.; Dekker, G.A.; Gatt, S.; McLintock, C.K.; McMahon, L.P.; Mangos, G.; Moore, M.P.; Muller, P.; Paech, M.; et al. Guidelines for the management of hypertensive disorders of pregnancy 2008. Aust. N. Z. J. Obstet. Gynaecol. 2009, 49, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Thurnau, G.R. Pregnancy-induced hypertension without proteinuria: Is it true preeclampsia? South. Med. J. 1988, 81, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.L.; Mercer, B.M.; Friedman, S.A.; Sibai, B.M. Urinary dipstick protein: A poor predictor of absent or severe proteinuria. Am. J. Obstet. Gynecol. 1994, 170, 137–141. [Google Scholar] [CrossRef]
- Lindheimer, M.D.; Kanter, D. Interpreting abnormal proteinuria in pregnancy: The need for a more pathophysiological approach. Obstet. Gynecol. 2010, 115, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Buddle, M.L. Inadequacy of dipstick proteinuria in hypertensive pregnancy. Aust. N. Z. J. Obstet. Gynaecol. 1995, 35, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Grauer, G.F. Proteinuria: Measurement and interpretation. Top. Companion Anim. Med. 2011, 26, 121–127. [Google Scholar] [CrossRef]
- Shamshirsaz, A.A.; Paidas, M.; Krikun, G. Preeclampsia, Hypoxia, Thrombosis, and Inflammation. J. Pregnancy 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Roberts, J.M.; Gammill, H.S. Preeclampsia: Recent insights. Hypertension 2005, 46, 1243–1249. [Google Scholar] [CrossRef]
- Von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of Preeclampsia. Hypertens. Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef]
- Ødegård, R.A.; Vatten, L.J.; Nilsen, S.T.; Salvesen, K.; Austgulen, R. Risk factors and clinical manifestations of pre-eclampsia. Br. J. Obstet. Gynaecol. 2000, 107, 1410–1416. [Google Scholar] [CrossRef]
- Masuyama, H.; Segawa, T.; Sumida, Y.; Masumoto, A.; Inoue, S.; Akahori, Y.; Hiramatsu, Y. Different profiles of circulating angiogenic factors and adipocytokines between early- and late-onset pre-eclampsia. Int. J. Obstet. Gynaecol. 2010, 117, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in pathogenesis, definitions, and guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.M. Evaluation and management of severe preeclampsia before 34 weeks’ gestation. Am. J. Obstet. Gynecol. 2011, 205, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Leavey, K.; Bainbridge, S.A. Cox Large scale aggregate microarray analysis reveals three distinct molecular subclasses of human preeclampsia. PLoS ONE 2015, 10, e0116508. [Google Scholar] [CrossRef]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension Produced by Reduced Uterine Perfusion in Pregnant Rats Is Associated With Increased Soluble Fms-Like Tyrosine Kinase-1 Expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef]
- Brosens, I.; Renaer, M. On the pathogenesis of placental infarcts in pre-eclampsia. Int. J. Obstet. Gynaecol. 1972, 79, 794–799. [Google Scholar] [CrossRef]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble Endoglin and Other Circulating Antiangiogenic Factors in Preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Harihana, N.; Shoemaker, A.; Wagner, S. Pathophysiology of hypertension in preeclampsia. Clin. Pr. 2016, 13, 33–37. [Google Scholar]
- Than, N.G.; Romero, R.; Tarca, A.L.; Kekesi, K.A.; Xu, Y.; Xu, Z.; Juhasz, K.; Bhatti, G.; Leavitt, R.J.; Gelencser, Z.; et al. Integrated Systems Biology Approach Identifies Novel Maternal and Placental Pathways of Preeclampsia. Front. Immunol. 2018, 9, 1661. [Google Scholar] [CrossRef]
- El-Sayed, A.A.F. Preeclampsia: A review of the pathogenesis and possible management strategies based on its pathophysiological derangements. Taiwan. J. Obstet. Gynecol. 2017, 56, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Ohkuchi, A.; Hirashima, C.; Suzuki, H.; Takahashi, K.; Yoshida, M.; Matsubara, S.; Suzuki, M. Evaluation of a new and automated electrochemiluminescence immunoassay for plasma sFlt-1 and PlGF levels in women with preeclampsia. Hypertens. Res. 2010, 33, 422–427. [Google Scholar] [CrossRef]
- Knudsen, U.B.; Kronborg, C.S.; von Dadelszen, P.; Kupfer, K.; Lee, S.-W.; Vittinghus, E.; Allen, J.G.; Redman, C.W. A single rapid point-of-care placental growth factor determination as an aid in the diagnosis of preeclampsia. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2012, 2, 8–15. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef]
- Myatt, L.; Cui, X. Oxidative stress in the placenta. Histochem. Cell Biol. 2004, 122, 369–382. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Spasic-Boskovic, O.; Jauniaux, E.; Charnock-Jones, D.S.; Burton, G.J. Nuclear Factor-κB, p38, and Stress-Activated Protein Kinase Mitogen-Activated Protein Kinase Signaling Pathways Regulate Proinflammatory Cytokines and Apoptosis in Human Placental Explants in Response to Oxidative Stress. Am. J. Pathol. 2007, 170, 1511–1520. [Google Scholar] [CrossRef]
- Zenclussen, A.C.; Lim, E.; Knoeller, S.; Knackstedt, M.; Hertwig, K.; Hagen, E.; Klapp, B.F.; Arck, P.C. Heme Oxygenases in Pregnancy II: HO-2 is Downregulated in Human Pathologic Pregnancies. Am. J. Reprod. Immunol. 2003, 50, 66–76. [Google Scholar] [CrossRef]
- Lyall, F.; Barber, A.; Myatt, L.; Bulmer, J.N.; Robson, S.C. Hemeoxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J. 2000, 14, 208–219. [Google Scholar] [CrossRef] [PubMed]
- George, E.M.; Granger, J.P. Heme oxygenase in pregnancy and preeclampsia. Curr. Opin. Nephrol. Hypertens. 2013, 22, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, S.; Farley, A.; McLaughlin, B.; Graham, C.; Marks, G.; Nakatsu, K.; Brien, J.; Smith, G. Carbon Monoxide Decreases Perfusion Pressure in Isolated Human Placenta. Placenta 2002, 23, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative Regulation of Soluble Flt-1 and Soluble Endoglin Release by Heme Oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- McCaig, D.; Lyall, F. Inhibitors of Heme Oxygenase Reduce Invasion of Human Primary Cytotrophoblast Cells In vitro. Placenta 2009, 30, 536–538. [Google Scholar] [CrossRef]
- Costantine, M.M.; Tamayo, E.; Lu, F.; Bytautiene, E.; Longo, M.; Hankins, G.D.V.; Saade, G.R. Using Pravastatin to Improve the Vascular Reactivity in a Mouse Model of Soluble Fms-Like Tyrosine Kinase-1–Induced Preeclampsia. Obstet. Gynecol. 2010, 116, 114–120. [Google Scholar] [CrossRef]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Kim, Y.M.; Bujold, E.; Chaiworapongsa, T.; Gomez, R.; Yoon, B.H.; Thaler, H.T.; Rotmensch, S.; Romero, R. Failure of physiologic transformation of the spiral arteries in patients with preterm labor and intact membranes. Am. J. Obstet. Gynecol. 2003, 189, 1063–1069. [Google Scholar] [CrossRef]
- Dommisse, J.; Tiltman, A.J. Placental bed biopsies in placental abruption. Int. J. Obstet. Gynaecol. 1992, 99, 651–654. [Google Scholar] [CrossRef]
- Page, N.; Butlin, D.; Manyonda, I.; Lowry, P. The development of a genetic profile of placental gene expression during the first trimester of pregnancy: A potential tool for identifying novel secreted markers. Fetal Diagn. Ther. 2000, 15, 237–245. [Google Scholar] [CrossRef]
- Zulfikaroglu, E.; Ugur, M.; Taflan, S.; Ugurlu, N.; Atalay, A.; Kalyoncu, S. Neurokinin B levels in maternal and umbilical cord blood in preeclamptic and normal pregnancies. J. Perinat. Med. 2007, 35, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Page, N.M.; Kemp, C.F.; Butlin, D.J.; Lowry, P.J. Placental peptides as markers of gestational disease. Reproduction 2002, 123, 487–495. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laliberte, C.; DiMarzo, L.; Morrish, D.W.; Kaufman, S. Neurokinin B causes concentration-dependent relaxation of isolated human placental resistance vessels. Regul. Pept. 2004, 117, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Wu, J.; Murray, J.K.; Gellman, S.H.; Wozniak, M.A.; Keely, P.J.; Boyer, M.E.; Gomez, T.M.; Hasso, S.M.; Fallon, J.F.; et al. An antiangiogenic neurokinin-B/thromboxane A2 regulatory axis. J. Cell Biol. 2006, 174, 1047–1058. [Google Scholar] [CrossRef]
- Page, N.M. Neurokinin B and pre-eclampsia: A decade of discovery. Reprod. Biol. Endocrinol. 2010, 8, 4. [Google Scholar] [CrossRef]
- Hoang, V.M.; Foulk, R.; Clauser, K.; Burlingame, A.; Gibson, B.W.; Fisher, S.J. Functional Proteomics: Examining the Effects of Hypoxia on the Cytotrophoblast Protein Repertoire. Biochemistry 2001, 40, 4077–4086. [Google Scholar] [CrossRef]
- Sawicki, G.; Dakour, J.; Morrish, D.W. Functional proteomics of neurokinin B in the placenta indicates a novel role in regulating cytotrophoblast antioxidant defences. Proteomics 2003, 3, 2044–2051. [Google Scholar] [CrossRef]
- Xie, F.; von Dadelszen, P.; Nadeau, J. CMV infection, TLR-2 and -4 expression, and cytokine profiles in early-onset preeclampsia with HELLP syndrome. Am. J. Reprod. Immunol. 2014, 71, 379–386. [Google Scholar] [CrossRef]
- Molvarec, A.; Szarka, A.; Walentin, S.; Bekő, G.; Karádi, I.; Prohászka, Z.; Rigó, J., Jr. Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod. Biol. Endocrinol. 2011, 9, 124. [Google Scholar] [CrossRef]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jüpner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Investig. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Romero, R.; Yoshimatsu, J.; Espinoza, J.; Kim, Y.M.; Park, K.; Kalache, K.; Edwin, S.; Bujold, E.; Gomez, R. Soluble adhesion molecule profile in normal pregnancy and pre-eclampsia. J. Matern. Neonatal Med. 2002, 12, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Bretelle, F.; Sabatier, F.; Blann, A.; D’Ercole, C.; Boutière, B.; Mutin, M.; Boubli, L.; Sampol, J.; Dignat-George, F. Maternal endothelial soluble cell adhesion molecules with isolated small for gestational age fetuses: Comparison with pre-eclampsia. Br. J. Obstet. Gynaecol. 2001, 108, 1277–1282. [Google Scholar]
- Francoual, J.; Audibert, F.; Trioche, P.; Chalas, J.; Capel, L.; Lindenbaum, A.; Labrune, P.; Frydman, R. Erratum: Is a Polymorphism of the Apolipoprotein E Gene Associated with Preeclampsia? Hypertens. Pregnancy 2003, 21, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Belo, L.; Gaffney, D.; Caslake, M.; Santos-Silva, A.; Pereira-Leite, L.; Quintanilha, A.; Rebelo, I. Apolipoprotein E and cholesteryl ester transfer protein polymorphisms in normal and preeclamptic pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 112, 9–15. [Google Scholar] [CrossRef]
- Aguzzi, A.; Haass, C. Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science 2003, 302, 814–818. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Wiggins, R.C. Prions and the Transmissible Spongiform Encephalopathies. In Metabolic Encephalopathy; McCandless, D., Ed.; Springer: New York, NY, USA, 2009; pp. 531–550. [Google Scholar]
- Belay, E.D. Transmissible Spongiform Encephalopathies in Humans. Annu. Rev. Microbiol. 1999, 53, 283–314. [Google Scholar] [CrossRef]
- Geschwind, M.D.; Legname, G. Transmissible spongiform encephalopathies. In Protein Misfolding in Neurodegenerative Diseases: Mechanisms and Therapeutic Strategies; CRC Press; Smith, H.J., Simons, C., Sewell, R.D.E., Eds.; Taylor and Francis Group: Abingdon, UK, 2007; pp. 221–248. [Google Scholar]
- Iwasaki, Y. Creutzfeldt-Jakob disease. Neuropathology. 2017, 2, 174–188. [Google Scholar] [CrossRef]
- Brown, K.; Mastrianni, J.A. The Prion Diseases. J. Geriatr. Psychiatry Neurol. 2010, 23, 277–298. [Google Scholar] [CrossRef]
- Bhadbhade, A.; Cheng, D.W. Amyloid Precursor Protein Processing in Alzheimer’s Disease. Iran. J. Child Neurol. 2012, 34, 185–204. [Google Scholar]
- Iqbal, K.; Alonso, A.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. α-Synuclein and neuronal cell death. Mol. Neurobiol. 2013, 47, 466–483. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar] [CrossRef]
- Wu, C.; Wang, Z.; Lei, H.; Zhang, W.; Duan, Y. Dual binding modes of Congo red to amyloid protofibril surface observed in molecular dynamics simulations. J. Am. Chem. Soc. 2007, 129, 1225–1232. [Google Scholar] [CrossRef]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- Ivancic, V.A.; Ekanayake, O.; Lazo, N.D. Binding Modes of Thioflavin T on the Surface of Amyloid Fibrils Studied by NMR. ChemPhysChem 2016, 17, 2461–2464. [Google Scholar] [CrossRef]
- Levine, H. Thioflavine t interaction with amyloid βsheet structures. Amyloid 1995, 2, 1–6. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β (1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and x-ray diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar] [PubMed]
- Chandramowlishwaran, P.; Sun, M.; Casey, K.L.; Romanyuk, A.V.; Grizel, A.V.; Sopova, J.V.; Rubel, A.A.; Nussbaum-Krammer, C.; Vorberg, I.M.; Chernoff, Y.O. Mammalian amyloidogenic proteins promote prion nucleation in yeast. J. Biol. Chem. 2018, 293, 3436–3450. [Google Scholar] [CrossRef] [PubMed]
- Rubel, A.A.; Ryzhova, T.A.; Antonets, K.S.; Chernoff, Y.O.; Galkin, A.P. Identification of PrP sequences essential for the interaction between the PrP polymers and Aβ peptide in a yeast-based assay. Prion 2013, 7, 469–476. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Zhao, G.; Funai, E.F.; Harris, N.; Sasson, I.E.; Bernstein, I.M.; Saade, G.R.; Buhimschi, C.S. Proteomic profiling of urine identifies specific fragments of SERPINA1 and albumin as biomarkers of preeclampsia. Am. J. Obstet. Gynecol. 2008, 199, e1–e551. [Google Scholar] [CrossRef]
- Millen, K.R.; Buhimschi, C.S.; Zhao, G.; Rood, K.M.; Tabbah, S.; Buhimschi, I.A. Serum and Urine Thioflavin-T-Enhanced Fluorescence in Severe Preeclampsia. Hypertension 2018, 71, 1185–1192. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra92. [Google Scholar] [CrossRef]
- Tong, M.; Cheng, S.; Chen, Q.; DeSousa, J.; Stone, P.R.; James, J.L.; Chamley, L.W.; Sharma, S. Aggregated transthyretin is specifically packaged into placental nano-vesicles in preeclampsia. Sci. Rep. 2017, 7, 6694. [Google Scholar] [CrossRef]
- Cater, J.H.; Kumita, J.R.; Zeineddine Abdallah, R.; Zhao, G.; Bernardo-Gancedo, A.; Henry, A.; Winata, W.; Chi, M.; Grenyer, B.S.F.; Townsend, M.L.; et al. Human pregnancy zone protein stabilizes misfolded proteins including preeclampsia- and Alzheimer’s-associated amyloid beta peptide. Proc. Natl. Acad. Sci. USA 2019, 116, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-B.; Nakashima, A.; Sharma, S. Understanding Pre-Eclampsia Using Alzheimer’s Etiology: An Intriguing Viewpoint. Am. J. Reprod. Immunol. 2016, 75, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.R.; Crowder, C.M. Protein Misfolding Induces Hypoxic Preconditioning via a Subset of the Unfolded Protein Response Machinery. Mol. Cell. Biol. 2010, 30, 5033–5042. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W.; Mengesdorf, T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 2005, 38, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Jian, B.; Hsieh, C.H.; Chen, J.; Choudhry, M.; Bland, K.; Chaudry, I.; Raju, R. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 621–626. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Yoshiike, Y.; Kayed, R.; Milton, S.C.; Takashima, A.; Glabe, C.G. Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromol. Med. 2007, 9, 270–275. [Google Scholar] [CrossRef]
- Shirahama, T. High-resolution electron microscopic analysis of the amyloid fibril. J. Cell Biol. 1967, 33, 679–708. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Katzmann, J.A.; Larson, D.R.; Colby, C.L.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Leung, N.; Zeldenrust, S.R.; et al. Serum immunoglobulin free light-chain measurement in primary amyloidosis: Prognostic value and correlations with clinical features. Blood 2010, 116, 5126–5129. [Google Scholar] [CrossRef]
- Huntington, J.A. Serpin structure, function and dysfunction. J. Thromb. Haemost. 2011, 9, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Engström, G.; Janzon, L.; Berglund, G.; Lind, P.; Stavenow, L.; Hedblad, B.; Lindgärde, F. Blood Pressure Increase and Incidence of Hypertension in Relation to Inflammation-Sensitive Plasma Proteins. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2054–2058. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matheson, N.R.; Wong, P.S.; Travis, J. Enzymatic inactivation of human alpha-1-proteinase inhibitor by neutrophil myeloperoxidase. Biochem. Biophys. Res. Commun. 1979, 88, 402–409. [Google Scholar] [CrossRef]
- Lomas, D.A. Polymerisation underlies alpha1-antitrypsin deficiency, dementia and other serpinopathies. Front. Biosci. 2004, 9, 2873. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Alvarado, M. Amyloid Formation in Light Chain Amyloidosis. Curr. Top. Med. Chem. 2013, 12, 2523–2533. [Google Scholar] [CrossRef]
- Bellotti, V.; Mangione, P.; Merlini, G. Review: Immunoglobulin Light Chain Amyloidosis—The Archetype of Structural and Pathogenic Variability. J. Struct. Biol. 2000, 130, 280–289. [Google Scholar] [CrossRef]
- Benson, M.D.; Liepnieks, J.J.; Kluve-Beckerman, B. Hereditary systemic immunoglobulin light-chain amyloidosis. Blood 2015, 125, 3281–3286. [Google Scholar] [CrossRef]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Buxbaum, J.N. The systemic amyloidoses. Curr. Opin. Rheumatol. 2004, 16, 67–75. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid β peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993, 75, 1039–1042. [Google Scholar] [CrossRef]
- Clarris, H.J.; Key, B.; Beyreuther, K.; Masters, C.L.; Small, D.H. Expression of the amyloid protein precursor of Alzheimer’s disease in the developing rat olfactory system. Brain Res. Dev. Brain Res. 1995, 88, 87–95. [Google Scholar] [CrossRef]
- Muresan, V.; Varvel, N.H.; Lamb, B.T.; Muresan, Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J. Neurosci. 2009, 29, 3565–3578. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease Beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanetj. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef]
- Kalkunte, S.S.; Neubeck, S.; Norris, W.E.; Cheng, S.-B.; Kostadinov, S.; Vu Hoang, D.; Ahmed, A.; von Eggeling, F.; Shaikh, Z.; Padbury, J.; et al. Transthyretin Is Dysregulated in Preeclampsia, and Its Native Form Prevents the Onset of Disease in a Preclinical Mouse Model. Am. J. Pathol. 2013, 183, 1425–1436. [Google Scholar] [CrossRef]
- Kalkunte, S.; Boij, R.; Norris, W.; Friedman, J.; Lai, Z.; Kurtis, J.; Lim, K.-H.; Padbury, J.F.; Matthiesen, L.; Sharma, S. Sera from Preeclampsia Patients Elicit Symptoms of Human Disease in Mice and Provide a Basis for an in Vitro Predictive Assay. Am. J. Pathol. 2010, 177, 2387–2398. [Google Scholar] [CrossRef]
- Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular Chaperones and Proteostasis. Annu. Rev. Biochem. 2013, 82, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin Has Chaperone-like Activity Similar to That of Small Heat Shock Proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Kumita, J.R.; Meehan, S.; Dobson, C.M.; Wilson, M.R. α2-Macroglobulin and Haptoglobin Suppress Amyloid Formation by Interacting with Prefibrillar Protein Species. J. Biol. Chem. 2009, 284, 4246–4254. [Google Scholar] [CrossRef] [PubMed]
- Blacker, D.; Wilcox, M.A.; Laird, N.M.; Rodes, L.; Horvath, S.M.; Go, R.C.P.; Perry, R.; Watson, B.; Bassett, S.S.; McInnis, M.G.; et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat. Genet. 1998, 19, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Ekelund, L.; Laurell, C.B. The pregnancy zone protein response during gestation: A metabolic challenge. Scand. J. Clin. Lab. Investig. 1994, 54, 623–629. [Google Scholar] [CrossRef]
- Tatarinov, I.S.; Mesniankina, N.V.; Nikulina, D.M. Immunochemical indentification of beta globulin of the “pregnancy zone” in the blood serum of patients with hydatid mole and chorioepithelioma]. Akush. Ginekol. 1974, 5, 67–68. [Google Scholar]
- Tatarinov, I.S.; Masiukevich, V.N.; Mesniankina, N.V.; Parfenova, L.F. Immunochemical identification of a new alpha 2 globulin in the blood serum of pregnant women]. Akush. Ginekol. 1970, 46, 25–28. [Google Scholar]
- Perni, U.; Sison, C.; Sharma, V.; Helseth, G.; Hawfield, A.; Suthanthiran, M.; August, P. Angiogenic Factors in Superimposed Preeclampsia. Hypertension 2012, 59, 740–746. [Google Scholar] [CrossRef]
- Rana, S.; Cerdeira, A.S.; Wenger, J.; Salahuddin, S.; Lim, K.-H.; Ralston, S.J.; Thadhani, R.I.; Karumanchi, S.A. Plasma Concentrations of Soluble Endoglin versus Standard Evaluation in Patients with Suspected Preeclampsia. PLoS ONE 2012, 7, e48259. [Google Scholar] [CrossRef]
- Akolekar, R.; Syngelaki, A.; Poon, L.; Wright, D.; Nicolaides, K.H. Competing risks model in early screening for preeclampsia by biophysical and biochemical markers. Fetal Diagn. Ther. 2013, 33, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, V.A.; Zakharova, N.V.; Bugrova, A.E.; Starodubtseva, N.L.; Indeykina, M.I.; Kononikhin, A.S.; Frankevich, V.E.; Nikolaev, E.N. The high-resolution mass spectrometry study of the protein composition of amyloid-like urine aggregates associated with preeclampsia. Eur. J. Mass Spectrom. 2019, 146906671986007. [Google Scholar] [CrossRef] [PubMed]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39, BSR20181415. [Google Scholar] [CrossRef] [PubMed]
- Halimi, M.; Dayan-Amouyal, Y.; Kariv-Inbal, Z.; Friedman-Levi, Y.; Mayer-Sonnenfeld, T.; Gabizon, R. Prion urine comprises a glycosaminoglycan-light chain IgG complex that can be stained by Congo red. J. Virol. Methods 2006, 133, 205–210. [Google Scholar] [CrossRef]
- Rood, K.M.; Buhimschi, C.S.; Dible, T.; Webster, S.; Zhao, G.; Samuels, P.; Buhimschi, I.A. Congo Red Dot Paper Test for Antenatal Triage and Rapid Identification of Preeclampsia. EClinicalMedicine 2019, 8, 47–56. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerasimova, E.M.; Fedotov, S.A.; Kachkin, D.V.; Vashukova, E.S.; Glotov, A.S.; Chernoff, Y.O.; Rubel, A.A. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. Int. J. Mol. Sci. 2019, 20, 6183. https://doi.org/10.3390/ijms20246183
Gerasimova EM, Fedotov SA, Kachkin DV, Vashukova ES, Glotov AS, Chernoff YO, Rubel AA. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. International Journal of Molecular Sciences. 2019; 20(24):6183. https://doi.org/10.3390/ijms20246183
Chicago/Turabian StyleGerasimova, Elizaveta M., Sergey A. Fedotov, Daniel V. Kachkin, Elena S. Vashukova, Andrey S. Glotov, Yury O. Chernoff, and Aleksandr A. Rubel. 2019. "Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics" International Journal of Molecular Sciences 20, no. 24: 6183. https://doi.org/10.3390/ijms20246183
APA StyleGerasimova, E. M., Fedotov, S. A., Kachkin, D. V., Vashukova, E. S., Glotov, A. S., Chernoff, Y. O., & Rubel, A. A. (2019). Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. International Journal of Molecular Sciences, 20(24), 6183. https://doi.org/10.3390/ijms20246183