Ischemic Postconditioning Reduces Reperfusion Arrhythmias by Adenosine Receptors and Protein Kinase C Activation but Is Independent of KATP Channels or Connexin 43

 ,
, 
Abstract
1. Introduction
2. Results
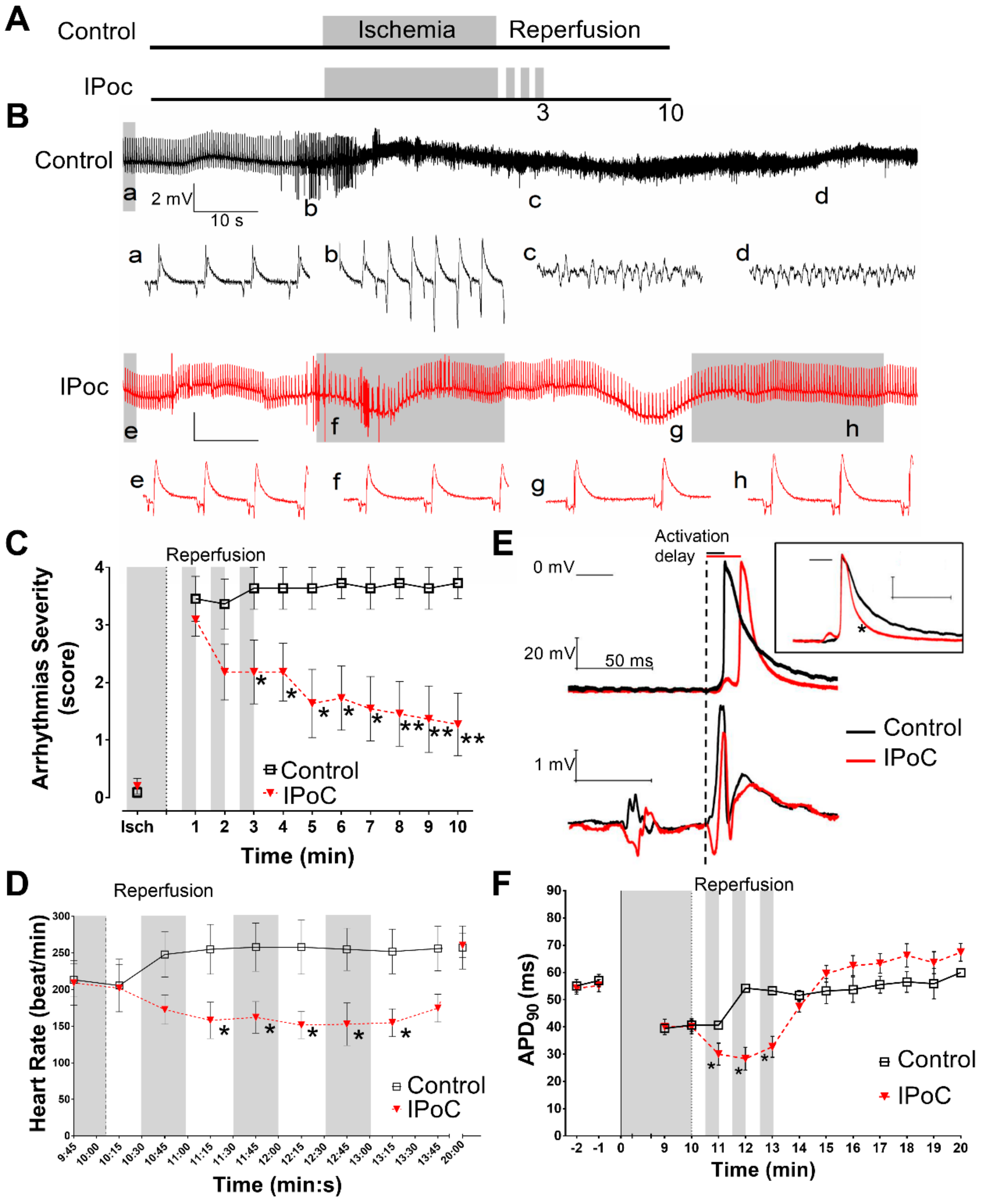
2.1. Electrophysiological Effects of IPoC in Isolated Rat Hearts Submitted to Regional Ischemia
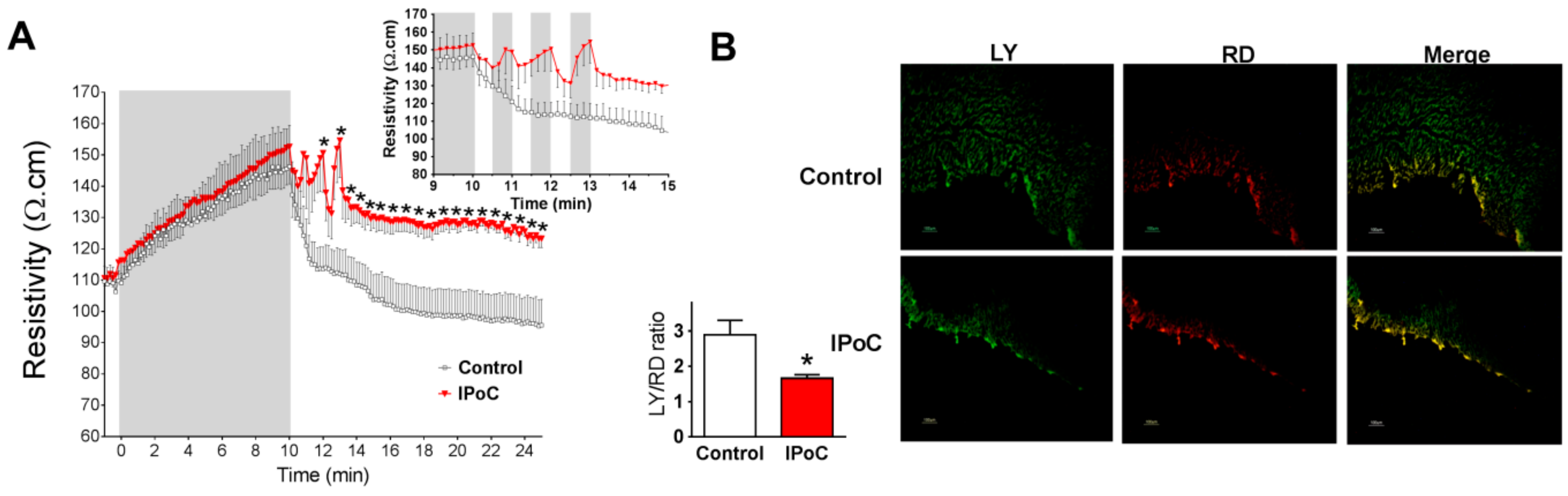
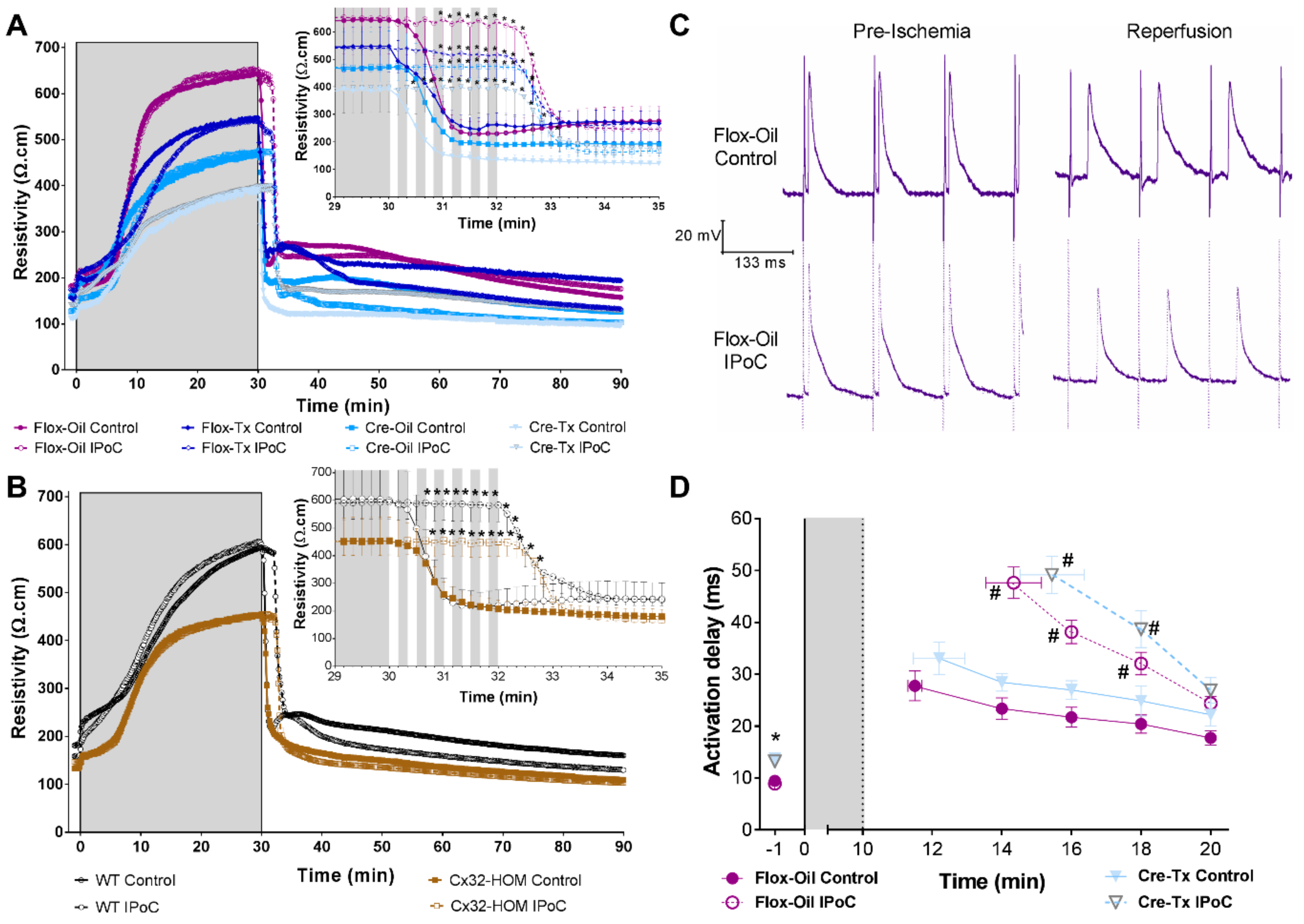
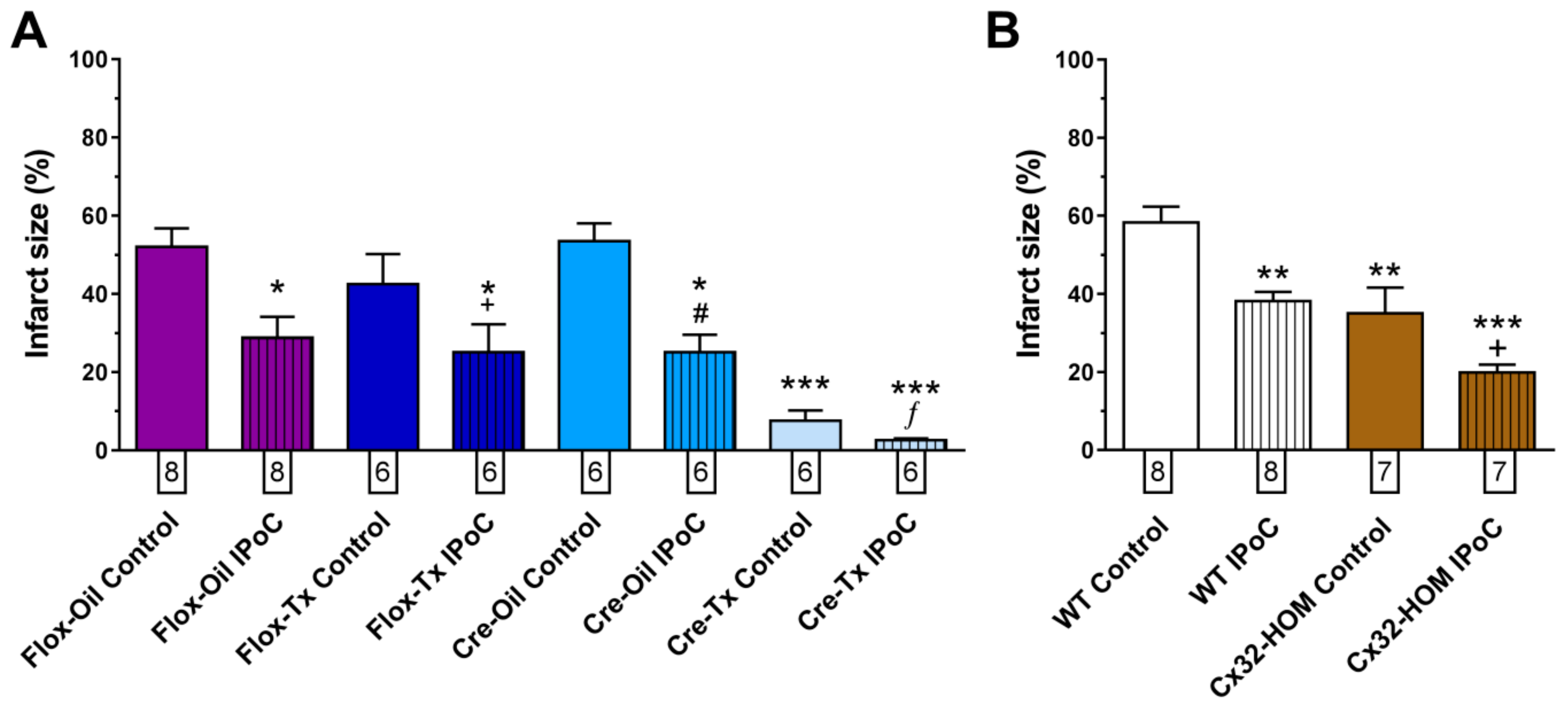
2.2. Connexin 43 Is Not Essential for IPoC Effects in Isolated Mice Hearts Submitted to Global Myocardial Ischemia
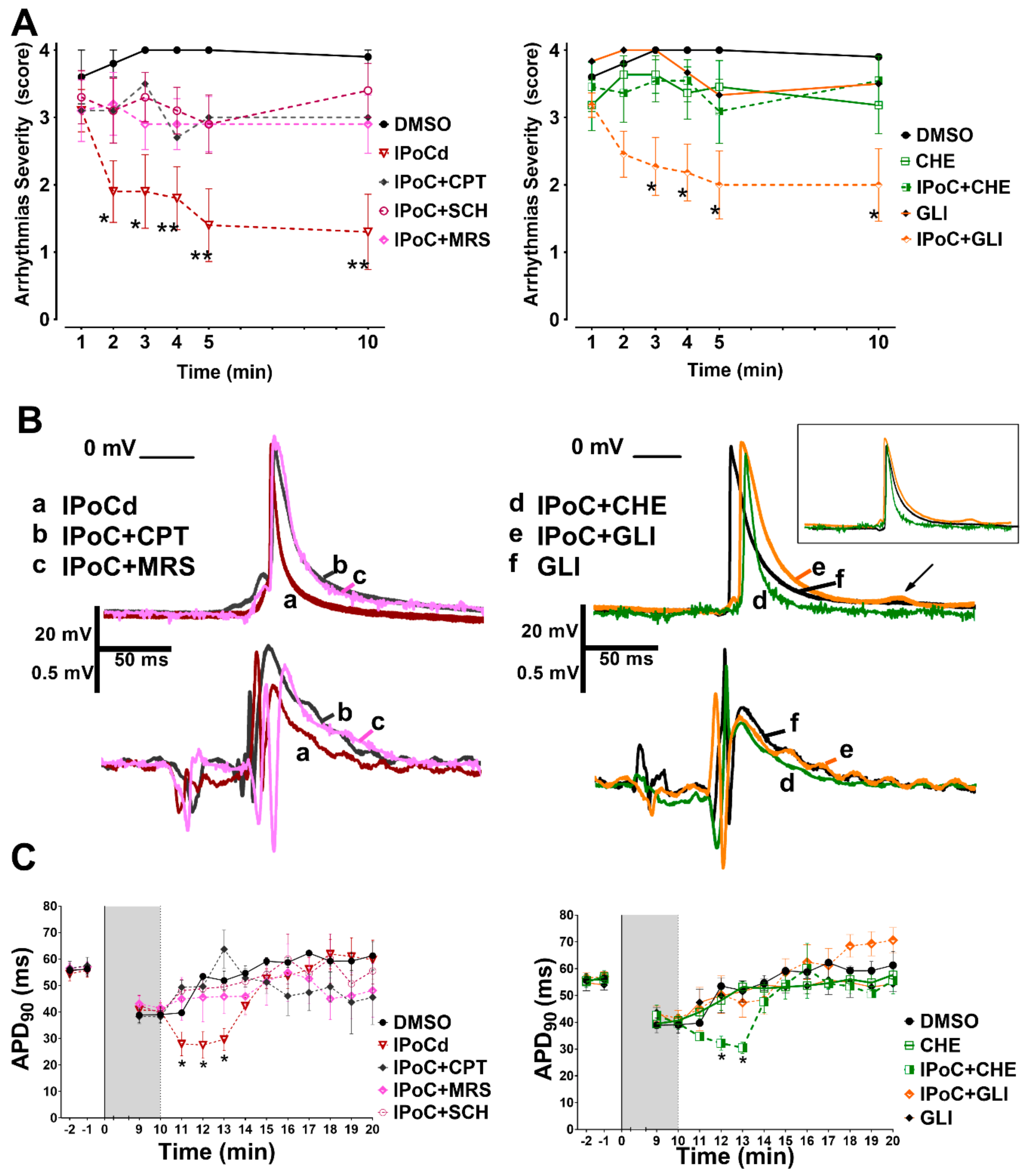
2.3. Adenosine Receptors and PKC Activations, but Not KATP Channels, Are Needed for IPoC Antiarrhythmic Protection
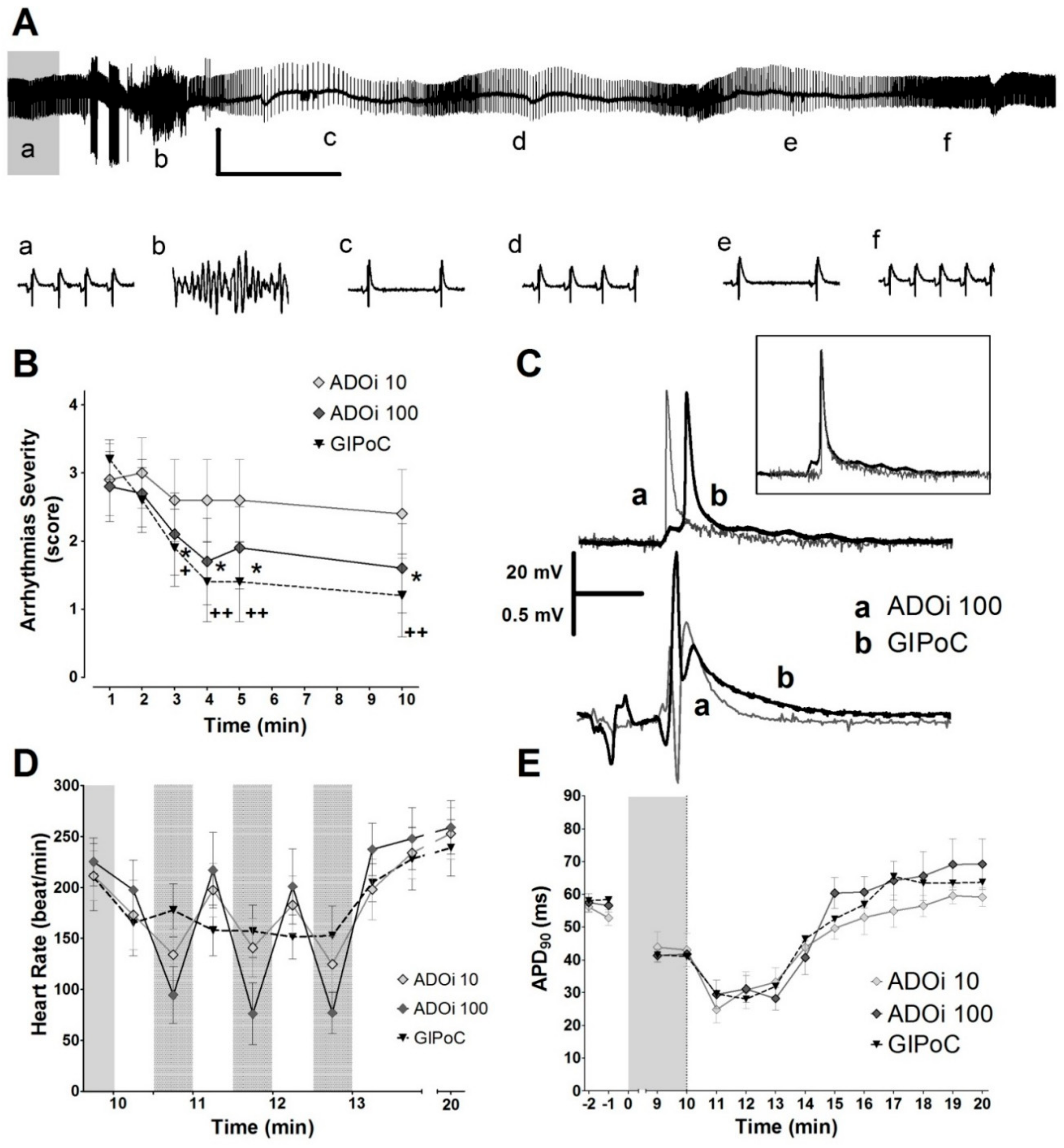
2.4. Adenosine Intermitet (ADOi) Administration during Reperfusion Reproduced Some of the Electrophysiological Effects of IPoC
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Experimental Protocols
4.3. Electrophysiological Studies
4.3.1. Arrhythmias
4.3.2. Electrograms and Action Potentials
4.3.3. Impedance
4.4. Intercellular Chemical Communication
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APD | action potential duration |
| IPoC | ischemic postconditioning |
| PKC | protein kinase C |
| KATP | ATP-dependent potassium channels |
| Cx32 | connexin 32 |
| Cx43 | connexin 43 |
| CPT | adenosine A1 receptor antagonist, cyclopentyl theophylline |
| SCH | adenosine A2A receptor antagonist, SCH 58261 |
| MRS | adenosine A3 receptor antagonist, MRS-1523 |
| GLI | non selective KATP blocker, glibenclamide |
| LY | Licifer Yellow |
| RD | Rhodamine |
References
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Kalarus, Z.; Svendsen, J.H.; Capodanno, D.; Dan, G.-A.; De Maria, E.; Gorenek, B.; Jędrzejczyk-Patej, E.; Mazurek, M.; Podolecki, T.; Sticherling, C.; et al. Cardiac arrhythmias in the emergency settings of acute coronary syndrome and revascularization: An European Heart Rhythm Association (EHRA) consensus document, endorsed by the European Association of Percutaneous Cardiovascular Interventions (EAPCI), and European Acute Cardiovascular Care Association (ACCA). EP Eur. 2019, 21, 1603–1604. [Google Scholar]
- Caccioppo, A.; Franchin, L.; Grosso, A.; Angelini, F.; D’Ascenzo, F.; Brizzi, M.F. Ischemia Reperfusion Injury: Mechanisms of Damage/Protection and Novel Strategies for Cardiac Recovery/Regeneration. Int. J. Mol. Sci. 2019, 20, 5024. [Google Scholar] [CrossRef] [PubMed]
- Spannbauer, A.; Traxler, D.; Lukovic, D.; Zlabinger, K.; Winkler, J.; Gugerell, A.; Ferdinandy, P.; Hausenloy, D.J.; Pavo, N.; Emmert, M.Y.; et al. Effect of Ischemic Preconditioning and Postconditioning on Exosome-Rich Fraction microRNA Levels, in Relation with Electrophysiological Parameters and Ventricular Arrhythmia in Experimental Closed-Chest Reperfused Myocardial Infarction. Int. J. Mol. Sci. 2019, 20, 2140. [Google Scholar] [CrossRef] [PubMed]
- Van der Weg, K.; Prinzen, F.W.; Gorgels, A.P. Editor’s Choice- Reperfusion cardiac arrhythmias and their relation to reperfusion-induced cell death. Eur. Hear. J. Acute Cardiovasc. Care 2019, 8, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Avezum, Á.; Piegas, L.S.; Goldberg, R.J.; Brieger, D.; Stiles, M.K.; Paolini, R.; Huang, W.; Gore, J.M. GRACE Investigators Magnitude and Prognosis Associated With Ventricular Arrhythmias in Patients Hospitalized With Acute Coronary Syndromes (from the GRACE Registry). Am. J. Cardiol. 2008, 102, 1577–1582. [Google Scholar] [CrossRef]
- Mehta, R.H.; Yu, J.; Piccini, J.P.; Tcheng, J.E.; Farkouh, M.E.; Reiffel, J.; Fahy, M.; Mehran, R.; Stone, G.W. Prognostic significance of postprocedural sustained ventricular tachycardia or fibrillation in patients undergoing primary percutaneous coronary intervention (from the HORIZONS-AMI Trial). Am. J. Cardiol. 2012, 109, 805–812. [Google Scholar] [CrossRef]
- Jabbari, R.; Risgaard, B.; Fosbøl, E.L.; Scheike, T.; Philbert, B.T.; Winkel, B.G.; Albert, C.M.; Glinge, C.; Ahtarovski, K.A.; Haunsø, S.; et al. Factors Associated with and Outcomes after Ventricular Fibrillation before and during Primary Angioplasty in Patients with ST-Segment Elevation Myocardial Infarction. Am. J. Cardiol. 2015, 116, 678–685. [Google Scholar] [CrossRef]
- Galagudza, M.; Kurapeev, D.; Minasian, S.; Valen, G.; Vaage, J. Ischemic postconditioning: Brief ischemia during reperfusion converts persistent ventricular fibrillation into regular rhythm. Eur. J. Cardiothorac. Surg. 2004, 25, 1006–1010. [Google Scholar] [CrossRef]
- Kloner, R.A.; Dow, J.; Bhandari, A. Postconditioning markedly attenuates ventricular arrhythmias after ischemia-reperfusion. J. Cardiovasc. Pharmacol. 2006, 11, 55–63. [Google Scholar] [CrossRef]
- Dow, J.; Bhandari, A.; Kloner, R.A. Ischemic postconditioning’s benefit on reperfusion ventricular arrhythmias is maintained in the senescent heart. J. Cardiovasc. Pharmacol. 2008, 13, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Diez, E.R.; Prado, N.J.; Ponce Zumino, A.Z.; Miatello, R.M. Efecto del poscondicionamiento isquémico sobre las arritmias de reperfusión en un modelo de hipertrofia miocárdica. Rev. Urug. Cardiol. 2011, 26, 101–107. [Google Scholar]
- Okishige, K.; Kanda, S.; Shimura, T.; Kurabayashi, M.; Ueshima, D.; Miwa, N.; Sugiyama, K.; Aoyagi, H.; Yoshimura, K.; Yanagi, H.; et al. Clinical study of the electrophysiological effects of ischemic post-conditioning in patients with acute myocardial infarctions. J. Cardiol. 2011, 58, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Araszkiewicz, A.; Grygier, M.; Pyda, M.; Rajewska, J.; Lesiak, M.; Grajek, S. Postconditioning attenuates early ventricular arrhythmias in patients with high-risk ST-segment elevation myocardial infarction. J. Cardiol. 2015, 65, 459–465. [Google Scholar] [CrossRef][Green Version]
- Mykytenko, J.; Reeves, J.G.; Kin, H.; Wang, N.-P.; Zatta, A.J.; Jiang, R.; Guyton, R.A.; Vinten-Johansen, J.; Zhao, Z.-Q. Persistent beneficial effect of postconditioning against infarct size: Role of mitochondrial KATP channels during reperfusion. Basic Res. Cardiol. 2008, 103, 472–484. [Google Scholar] [CrossRef]
- Dow, J.; Bhandari, A.; Kloner, R.A. The Mechanism by Which Ischemic Postconditioning Reduces Reperfusion Arrhythmias in Rats Remains Elusive. J. Cardiovasc. Pharmacol. 2009, 14, 99–103. [Google Scholar] [CrossRef]
- Kolettis, T.M.; Vilaeti, A.D.; Tsalikakis, D.G.; Zoga, A.; Valenti, M.; Tzallas, A.T.; Papalois, A.; Iliodromitis, E.K. Effects of Pre- and Postconditioning on Arrhythmogenesis in the In Vivo Rat Model. J. Cardiovasc. Pharmacol. 2013, 18, 376–385. [Google Scholar] [CrossRef]
- Wainwright, C.L.; Kang, L.; Ross, S. Studies on the mechanism underlying the antifibrillatory effect of the A1-adenosine agonist, R-PIA, in rat isolated hearts. Cardiovasc. Drugs 1997, 11, 669–678. [Google Scholar] [CrossRef]
- Schreieck, J.; Richardt, G. Endogenous adenosine reduces the occurrence of ischemia-induced ventricular fibrillation in rat heart. J. Mol. Cell. Cardiol. 1999, 31, 123–134. [Google Scholar] [CrossRef]
- Kin, H.; Zatta, A.J.; Lofye, M.T.; Amerson, B.S.; Halkos, M.E.; Kerendi, F.; Zhao, Z.-Q.; Guyton, R.A.; Headrick, J.P.; Vinten-Johansen, J. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc. Res. 2005, 67, 124–133. [Google Scholar] [CrossRef][Green Version]
- Xi, J.; McIntosh, R.; Shen, X.; Lee, S.; Chanoit, G.; Criswell, H.; Zvara, D.A.; Xu, Z. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J. Mol. Cell. Cardiol. 2009, 47, 684–690. [Google Scholar] [CrossRef]
- Xi, L.; Das, A.; Zhao, Z.-Q.; Merino, V.F.; Bader, M.; Kukreja, R.C. Loss of myocardial ischemic postconditioning in adenosine A1 and bradykinin B2 receptors gene knockout mice. Circulation 2008, 118, S32–S37. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Rodriguez-Sinovas, A.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Effects of a reduction in the number of gap junction channels or in their conductance on ischemia-reperfusion arrhythmias in isolated mouse hearts. Ajp Hear. Circ. Physiol. 2011, 301, H2442–H2453. [Google Scholar] [CrossRef][Green Version]
- Stables, C.L.; Curtis, M.J. Development and characterization of a mouse in vitro model of ischaemia-induced ventricular fibrillation. Cardiovasc. Res. 2009, 83, 397–404. [Google Scholar] [CrossRef]
- Cole, W.C.; McPherson, C.D.; Sontag, D. ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ. Res. 1991, 69, 571–581. [Google Scholar] [CrossRef]
- Spinelli, W.; Sorota, S.; Siegal, M.; Hoffman, B.F. Antiarrhythmic actions of the ATP-regulated K+ current activated by pinacidil. Circ. Res. 1991, 68, 1127–1137. [Google Scholar] [CrossRef]
- Fish, F.A.; Prakash, C.; Roden, D.M. Suppression of repolarization-related arrhythmias in vitro and in vivo by low-dose potassium channel activators. Circulation 1990, 82, 1362–1369. [Google Scholar] [CrossRef]
- Munch-Ellingsen, J.; Løkebø, J.E.; Bugge, E.; Jonassen, A.K.; Ravingerová, T.; Ytrehus, K. 5-HD abolishes ischemic preconditioning independently of monophasic action potential duration in the heart. Basic Res. Cardiol. 2000, 95, 228–234. [Google Scholar] [CrossRef]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res. Cardiol. 2006, 101, 180–189. [Google Scholar] [CrossRef]
- Zatta, A.J.; Kin, H.; Lee, G.; Wang, N.; Jiang, R.; Lust, R.; Reeves, J.G.; Mykytenko, J.; Guyton, R.A.; Zhao, Z.-Q.; et al. Infarct-sparing effect of myocardial postconditioning is dependent on protein kinase C signalling. Cardiovasc. Res. 2006, 70, 315–324. [Google Scholar] [CrossRef]
- Restivo, M.; Kozhevnikov, D.O.; Qu, Y.S.; Yue, Y.; Mochly-Rosen, D.; El-Sherif, N.; Boutjdir, M. Activation of εPKC reduces reperfusion arrhythmias and improves recovery from ischemia: Optical mapping of activation patterns in the isolated guinea-pig heart. Biochem. Biophys. Res. Commun. 2012, 426, 237–241. [Google Scholar] [CrossRef][Green Version]
- Carmeliet, E. Cardiac ionic currents and acute ischemia: From channels to arrhythmias. Physiol. Rev. 1999, 79, 917–1017. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Szilvássy, Z.; Droy-Lefaix, M.T.; Tarrade, T.; Koltai, M. KATP channel modulation in working rat hearts with coronary occlusion: Effects of cromakalim, cicletanine, and glibenclamide. Cardiovasc. Res. 1995, 30, 781–787. [Google Scholar] [CrossRef]
- Cohen, M.V.; Downey, J.M. Adenosine: Trigger and mediator of cardioprotection. Basic Res. Cardiol. 2008, 103, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Downey, J.M.; Davis, A.M.; Cohen, M.V. Signaling pathways in ischemic preconditioning. Heart Fail. Rev. 2007, 12, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Simkhovich, B.Z.; Przyklenk, K.; Kloner, R.A. Role of Protein Kinase C in Ischemic “Conditioning”. J. Cardiovasc. Pharmacol. 2013, 18, 525–532. [Google Scholar] [CrossRef]
- Workman, A.J.; MacKenzie, I.; Northover, B.J. A K(ATP) channel opener inhibited myocardial reperfusion action potential shortening and arrhythmias. Eur. J. Pharmacol. 2001, 419, 73–83. [Google Scholar] [CrossRef]
- Bril, A.; Laville, M.-P.; Gout, B. Effects of glibenclamide on ventricular arrhythmias and cardiac function in ischaemia and reperfusion in isolated rat heart. Cardiovasc. Res. 1992, 26, 1069–1076. [Google Scholar] [CrossRef]
- Bernauer, W. Concerning the effect of the K+ channel blocking agent glibenclamide on ischaemic and reperfusion arrhythmias. Eur. J. Pharmacol. 1997, 326, 147–156. [Google Scholar] [CrossRef]
- Smallwood, J.K.; Ertel, P.J.; Steinberg, M.I. Modification by glibenclamide of the electrophysiological consequences of myocardial ischaemia in dogs and rabbits. Naunyn Schmiedebergs Arch. Pharmacol. 1990, 342, 214–220. [Google Scholar] [CrossRef]
- Del Valle, H.F.; Lascano, E.C.; Negroni, J.A.; Crottogini, A.J. Glibenclamide effects on reperfusion-induced malignant arrhythmias and left ventricular mechanical recovery from stunning in conscious sheep. Cardiovasc. Res. 2001, 50, 474–485. [Google Scholar] [CrossRef][Green Version]
- Maruyama, I.; Tomiyama, Y.; Maruyama, K.; Ojima, K.; Kobayashi, K.; Kobayashi, M.; Yamazaki, Y.; Kojima, M.; Shibata, N. Effects of mitiglinide and sulfonylureas in isolated canine coronary arteries and perfused rat hearts. Eur. J. Pharmacol. 2006, 531, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Waldo, A.L.; Camm, A.J.; DeRuyter, H.; Friedman, P.L.; MacNeil, D.J.; Pauls, J.F.; Pitt, B.; Pratt, C.M.; Schwartz, P.J.; Veltri, E.P. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet (Lond. Engl.) 1996, 348, 7–12. [Google Scholar] [CrossRef]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L.; et al. Mortality and Morbidity in Patients Receiving Encainide, Flecainide, or Placebo. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, J.G.; Moreno, J.; Archondo, T.; Chin, A.; Pérez-Castellano, N.; Usandizaga, E.; García-Torrent, M.J.; Molina-Morúa, R.; González, P.; Rodríguez-Bobada, C.; et al. KATP channel opening accelerates and stabilizes rotors in a swine heart model of ventricular fibrillation. Cardiovasc. Res. 2013, 99, 576–585. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Andres-Villarreal, M.; Ruiz-Meana, M.; Inserte, J.; Barba, I. Myocardial edema: A translational view. J. Mol. Cell. Cardiol. 2012, 52, 931–939. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.-P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Changes in anisotropic conduction caused by remodeling cell size and the cellular distribution of gap junctions and Na (+) channels. J. Electrocardiol. 2001, 34, 69–76. [Google Scholar] [CrossRef]
- Heusch, G.; Büchert, A.; Feldhaus, S.; Schulz, R. No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res. Cardiol. 2006, 101, 354–356. [Google Scholar] [CrossRef]
- Tu, R.H.; Li, Q.J.; Huang, Z.; He, Y.; Meng, J.J.; Zheng, H.L.; Zeng, Z.Y.; Zhong, G.Q. Novel functional role of heat shock protein 90 in mitochondrial connexin 43-mediated hypoxic postconditioning. Cell. Physiol. Biochem. 2017, 44, 982–997. [Google Scholar] [CrossRef]
- Penna, C.; Mancardi, D.; Tullio, F.; Pagliaro, P. Intermittent Adenosine at the Beginning of Reperfusion Does Not Trigger Cardioprotection. J. Surg. Res. 2009, 153, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Diez, E.R.; Renna, N.F.; Prado, N.J.; Lembo, C.; Ponce Zumino, A.Z.; Vazquez-Prieto, M.; Miatello, R.M. Melatonin, given at the time of reperfusion, prevents ventricular arrhythmias in isolated hearts from fructose-fed rats and spontaneously hypertensive rats. J. Pineal Res. 2013, 55, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sinovas, A.; Sánchez, J.A.; González-Loyola, A.; Barba, I.; Morente, M.; Aguilar, R.; Agulló, E.; Miró-Casas, E.; Esquerda, N.; Ruiz-Meana, M.; et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischaemia and preconditioning protection. J. Physiol. 2010, 588, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.A.; Rodríguez-Sinovas, A.; Barba, I.; Miró-Casas, E.; Fernández-Sanz, C.; Ruiz-Meana, M.; Alburquerque-Béjar, J.J.; García-Dorado, D. Activation of RISK and SAFE pathways is not involved in the effects of Cx43 deficiency on tolerance to ischemia–reperfusion injury and preconditioning protection. Basic Res. Cardiol. 2013, 108, 351. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M.; Augereau, J.M.; Gleye, J.; Maffrand, J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Curtis, M.J.; Hancox, J.C.; Farkas, A.; Wainwright, C.L.; Stables, C.L.; Saint, D.A.; Clements-Jewery, H.; Lambiase, P.D.; Billman, G.E.; Janse, M.J.; et al. The Lambeth Conventions (II): Guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacology 2013, 139, 213–248. [Google Scholar] [CrossRef]
- Curtis, M.J.; Walker, M.J. Quantification of arrhythmias using scoring systems: An examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc. Res. 1988, 22, 656–665. [Google Scholar] [CrossRef]
- Rodríguez-Sinovas, A.; García-Dorado, D.; Padilla, F.; Inserte, J.; Barrabés, J.A.; Ruiz-Meana, M.; Agulló, L.; Soler-Soler, J. Pre-treatment with the Na+/H+ exchange inhibitor cariporide delays cell-to-cell electrical uncoupling during myocardial ischemia. Cardiovasc. Res. 2003, 58, 109–117. [Google Scholar] [CrossRef][Green Version]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Lane, S.; Pina, P.; Inserte, J.; Mirabet, M.; Soler-Soler, J. Persistence of gap junction communication during myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2563–H2571. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Groups | Ventricular Tachycardia | Ventricular Fibrillation |
|---|---|---|
| Control | 45.0 (10–142) | 476.0 (198–555) |
| IPoC | 17.0 (3–146) | 5.0 (0–115) ** |
| CPT | 11.0 (0–41) | 459.0 (228–555) |
| IPoC + CPT | 68.0 (28–235) | 250.5 (121–291) |
| DMSO | 45.0 (24–123) | 529.0 (205–582) |
| IPoCd | 27.5 (3–146) | 10.5 (0–141) ** |
| SCH | 50.5 (33–110) | 398.5 (124–460) |
| IPoC + SCH | 64.0 (25–128) | 282.0 (112–505) |
| MRS | 35.0 (8–99) | 520.0 (233–563) |
| IPoC + MRS | 82.5 (20–186) | 263.0 (0–415) |
| CHE | 41.0 (20–51) | 467.5 (295–545) |
| IPoC + CHE | 71.5 (38–133) | 360.0 (224–460) |
| GLI | 401.0 (260–542) ** | 61.5 (18–75) * |
| IPoC + GLI | 33.5 (12–63) | 66.0 (12–90) * |
| ADOi10 | 141.0 (60–242) | 91.5 (30–135) * |
| ADOi100 | 120.5 (22–195) | 66.5 (0–106) * |
| GIPoC | 39.5 (18–87) | 48.0 (0–78) ** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diez, E.R.; Sánchez, J.A.; Prado, N.J.; Ponce Zumino, A.Z.; García-Dorado, D.; Miatello, R.M.; Rodríguez-Sinovas, A. Ischemic Postconditioning Reduces Reperfusion Arrhythmias by Adenosine Receptors and Protein Kinase C Activation but Is Independent of KATP Channels or Connexin 43. Int. J. Mol. Sci. 2019, 20, 5927. https://doi.org/10.3390/ijms20235927
Diez ER, Sánchez JA, Prado NJ, Ponce Zumino AZ, García-Dorado D, Miatello RM, Rodríguez-Sinovas A. Ischemic Postconditioning Reduces Reperfusion Arrhythmias by Adenosine Receptors and Protein Kinase C Activation but Is Independent of KATP Channels or Connexin 43. International Journal of Molecular Sciences. 2019; 20(23):5927. https://doi.org/10.3390/ijms20235927
Chicago/Turabian StyleDiez, Emiliano Raúl, Jose Antonio Sánchez, Natalia Jorgelina Prado, Amira Zulma Ponce Zumino, David García-Dorado, Roberto Miguel Miatello, and Antonio Rodríguez-Sinovas. 2019. "Ischemic Postconditioning Reduces Reperfusion Arrhythmias by Adenosine Receptors and Protein Kinase C Activation but Is Independent of KATP Channels or Connexin 43" International Journal of Molecular Sciences 20, no. 23: 5927. https://doi.org/10.3390/ijms20235927
APA StyleDiez, E. R., Sánchez, J. A., Prado, N. J., Ponce Zumino, A. Z., García-Dorado, D., Miatello, R. M., & Rodríguez-Sinovas, A. (2019). Ischemic Postconditioning Reduces Reperfusion Arrhythmias by Adenosine Receptors and Protein Kinase C Activation but Is Independent of KATP Channels or Connexin 43. International Journal of Molecular Sciences, 20(23), 5927. https://doi.org/10.3390/ijms20235927