The Burden of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: Screening Issue and Future Perspectives

 ,
,
Abstract
1. Introduction
2. The Burden of NAFLD-Related HCC
3. The Impact of Metabolic and Genetic Risk Factors
3.1. Obesity
3.2. Diabetes
3.3. Genetics Findings
3.3.1. PNPLA3
3.3.2. TM6SF2
3.3.3. MBOAT7
3.3.4. Alpha-fetoprotein
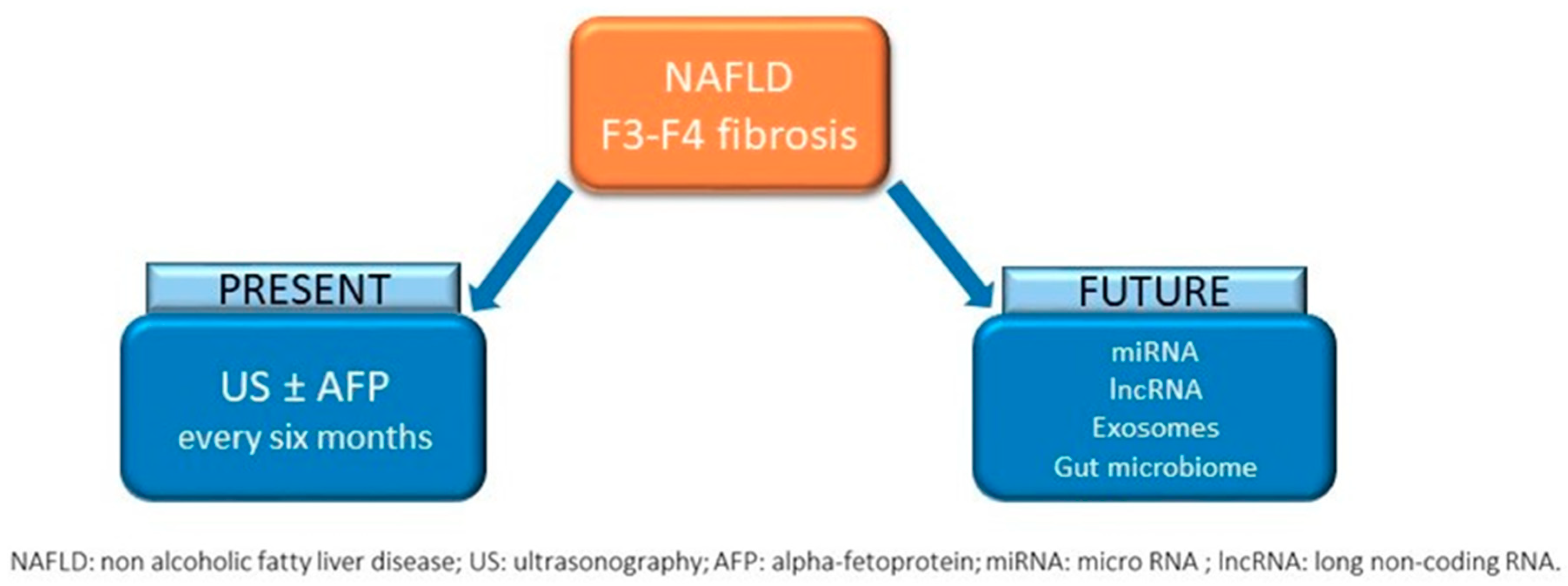
4. The Issue of Screening
4.1. Who Screen
4.2. How to Screen
5. Prevention Strategies
5.1. Lifestyle Interventions
5.2. Statins and Metformin
5.3. Anti-fibrotic Therapies
6. Future perspectives
6.1. miRNA
6.2. lncRNA
6.3. Exosomes
6.4. Epigenetic
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B. Global epidemiology of non-alcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagström, H. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Akinyemiju, T.; Abera, S. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [PubMed]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- Pais, R.; Fartoux, L.; Goumard, C.; Scatton, O.; Wendum, D.; Rosmorduc, O.; Ratziu, V. Temporal trends, clinical patterns and outcomes of NAFLD-related HCC in patients undergoing liver resection over a 20-year period. Aliment. Pharm. Ther. 2017, 46, 856–863. [Google Scholar] [CrossRef]
- Goldberg, D.; Ditah, I.C.; Saeian, K.; Lalehzari, M.; Aronsohn, A.; Gorospe, E.C.; Charlton, M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients with Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology 2017, 152, 1090–1099. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, F.J. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J. Hepatol. 2016, 70, 674–683. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755. [Google Scholar] [CrossRef]
- Belli, L.S.; Perricone, G.; Adam, R.; Cortesi, P.A.; Strazzabosco, M.; Facchetti, R.; Karam, V.; Salizzoni, M.; Andújar, R.L.; Fondevila, C.; et al. Impact of DAAs on liver transplantation: Major effects on the evolution of indications and results. An ELITA study based on the ELTR registry. J. Hepatol. 2018, 69, 810–817. [Google Scholar] [CrossRef]
- Kim, D.; Li, A.A.; Gadiparthi, C.; Khan, M.A.; Cholankeril, G.; Glenn, J.S.; Ahmed, A. Changing Trends in Etiology-Based Annual Mortality from Chronic Liver Disease. Gastroenterology 2018, 155, 1154–1163. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Basen-Engquist, K.; Chang, M. Obesity and Cancer Risk: Recent Review and Evidence. Curr. Oncol. Rep. 2011, 13, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Tahergorabi, Z.; Khazaei, M.; Moodi, M.; Chamani, E. From obesity to cancer: A review on proposed mechanisms. Cell Biochem. Funct. 2016, 34, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 2012, 1271, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Walker-Thurmond, K.; Thun, M.J.; Calle, E.E.; Rodriguez, C. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar]
- Yao, K.F.; Ma, M. Meta-analysis reveals gender difference in the association of liver cancer incidence and excess BMI. Oncotarget 2017, 8, 72959–72971. [Google Scholar] [CrossRef] [PubMed]
- Villa, E. Role of estrogen in liver cancer. Womens Health 2008, 4, 41–50. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef]
- Dávila, J.; Morgan, R.; Shaib, Y.; McGlynn, K.; El-Serag, H.B. Diabetes increases the risk of hepatocellular carcinoma in the United States: A population based case control study. Gut 2005, 54, 533–539. [Google Scholar] [CrossRef]
- Kawamura, Y.; Arase, Y.; Ikeda, K.; Seko, Y.; Imai, N.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Sezaki, H.; Akuta, N.; et al. Large-Scale Long-Term Follow-Up Study of Japanese Patients with Non-Alcoholic Fatty Liver Disease for the Onset of Hepatocellular Carcinoma. Am. J. Gastroenterol. 2012, 107, 253–261. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; HCC-NAFLD Italian Study Group. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Diabetes is Associated with Increased Risk of Hepatocellular Carcinoma in Cirrhosis Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2019, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Edefonti, V.C.; Talamini, R.; Ferraroni, M.; Malvezzi, M.; Bravi, F.; Franceschi, S.; Montella, M.; Polesel, J.; Zucchetto, A.; et al. Family history of liver cancer and hepatocellular carcinoma. Hepatology 2012, 55, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef]
- Trépo, E.; Nahon, P.; Bontempi, G.; Valenti, L.; Falleti, E.; Nischalke, H.-D.; Hamza, S.; Corradini, S.G.; Burza, M.A.; Guyot, E.; et al. Association between thePNPLA3 (rs738409 C > G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59, 2170–2177. [Google Scholar] [CrossRef]
- Valenti, L.; Alisi, A.; Galmozzi, E.; Bartuli, A.; Del Menico, B.; Alterio, A. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1274–1280. [Google Scholar] [CrossRef]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.C.; Burt, A.D.; Dufour, J.F. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Grimaudo, S.; Pipitone, R.M.; Pennisi, G.; Celsa, C.; Cammà, C.; Di Marco, V.; Barcellona, M.R.; Boemi, R.; Enea, M.; Giannetti, A.; et al. Association Between PNPLA3 rs738409 C > G Variant and Liver-Related Outcomes in Patients with Non-alcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, in press. [Google Scholar] [CrossRef]
- Valenti, L.; Motta, B.M.; Soardo, G.; Iavarone, M.; Donati, B.; Sangiovanni, A. PNPLA3 I148M polymorphism, clinical presentation, and survival in patients with hepatocellular carcinoma. PLoS ONE 2013, 8, e75982. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-Wide Association Analysis Identifies Variants Associated with Nonalcoholic Fatty Liver Disease That Have Distinct Effects on Metabolic Traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Miele, L.; Bugianesi, E.; Cammà, C.; Rosso, C.; Boccia, S. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e87523. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef]
- Falleti, E.; Cussigh, A.; Cmet, S.; Fabris, C.; Toniutto, P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. 2016, 48, 69–75. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef]
- Björnson, E.; Mukhopadhyay, B.; Asplund, A.; Pristovšek, N.; Cinar, R.; Romeo, S.; Uhlén, M.; Kunos, G.; Nielsen, J.; Mardinoglu, A. Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell Rep. 2015, 13, 2014–2026. [Google Scholar] [CrossRef]
- Li, L.; Che, L.; Tharp, K.M.; Park, H.-M.; Pilo, M.G.; Cao, D.; Cigliano, A.; Latte, G.; Xu, Z.; Ribback, S.; et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 2016, 63, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.J. The role of serum alpha-fetoprotein estimation in the diagnosis and management of hepatocellular carcinoma. Clin. Liver Dis. 2001, 5, 145–159. [Google Scholar] [CrossRef]
- Colombo, M. Screening for cancer in viral hepatitis. Clin. Liver Dis. 2001, 5, 109–122. [Google Scholar] [CrossRef]
- Tsukuma, H.; Hiyama, T.; Tanaka, S.; Nakao, M.; Yabuuchi, T.; Kitamura, T.; Nakanishi, K.; Fujimoto, I.; Inoue, A.; Yamazaki, H.; et al. Risk Factors for Hepatocellular Carcinoma among Patients with Chronic Liver Disease. N. Engl. J. Med. 1993, 328, 1797–1801. [Google Scholar] [CrossRef]
- Chen, J.-G.; Parkin, D.M.; Chen, Q.-G.; Lu, J.-H.; Shen, Q.-J.; Zhang, B.-C.; Zhu, Y.-R. Screening for liver cancer: Results of a randomised controlled trial in Qidong, China. J. Med. Screen. 2003, 10, 204–209. [Google Scholar] [CrossRef]
- Trevisani, F.; D’Intino, P.; Morselli-Labate, A.M.; Mazzella, G.; Accogli, E.; Caraceni, P.; Domenicali, M.; De Notariis, S.; Roda, E.; Bernardi, M. Serum alpha-fetoprotein for diagnosis of hepatocellular carcinoma in patients with chronic liver disease: Influence of HBsAg and anti-HCV status. J. Hepatol. 2001, 34, 570–575. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Sposito, C.; Zhou, J.; Pinna, A.D.; De Carlis, L.; Fan, J. Metroticket 2.0 model for analysis of competing risks of death following liver transplantation for hepatocellular carcinoma. Gastroenterology 2018, 154, 128–139. [Google Scholar] [CrossRef]
- Taketa, K.; Endo, Y.; Sekiya, C.; Tanikawa, K.; Koji, T.; Taga, H. A collaborative study for the evaluation of lectin-reactive alpha-fetoproteins in early detection of hepatocellular carcinoma. Cancer Res. 1993, 53, 5419–5423. [Google Scholar]
- Sterling, R.K.; Jeffers, L.; Gordon, F.; Sherman, M.; Venook, A.P.; Reddy, K.R.; Satomura, S.; Schwartz, M.E. Clinical Utility of AFP-L3% Measurement in North American Patients with HCV-Related Cirrhosis. Am. J. Gastroenterol. 2007, 102, 2196–2205. [Google Scholar] [CrossRef]
- Johnson, P.; Leung, N.; Cheng, P.; Welby, C.; Leung, W.; Lau, W.; Yu, S.; Ho, S. ‘Hepatoma-specific’ alphafetoprotein may permit preclinical diagnosis of malignant change in patients with chronic liver disease. Br. J. Cancer 1997, 75, 236–240. [Google Scholar] [CrossRef]
- Mittal, S.; Sada, Y.H.; El-Serag, H.B.; Kanwal, F.; Duan, Z.; Temple, S. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin. Gastroenterol. Hepatol. 2015, 13, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysi. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharm. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A. Risk of Hepatocellular Cancer in Patients with Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.-R.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Sharma, S.A.; Kowgier, M.; Hansen, B.E.; Brouwer, W.P.; Maan, R.; Wong, D. Toronto HCC risk index: A validated scoring system to predict 10-year risk of HCC in patients with cirrhosis. J. Hepatol. 2017, 86, 92–96. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Green, P.; Kerr, K.F.; Berry, K. Models estimating risk of hepatocellular carcinoma in patients with alcohol or NAFLD-related cirrhosis for risk stratification. J. Hepatol. 2019, 71, 523–533. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef]
- Korean Liver Cancer Association; National Cancer Center. 2018 Korean Liver Cancer Association-National Cancer Center Korea Practice Guidelines for the Management of Hepatocellular Carcinoma. Gut Liver 2019, 13, 227–299. [Google Scholar] [CrossRef]
- Tzartzeva, K.; Obi, J.; Rich, N.E.; Parikh, N.D.; Marrero, J.A.; Yopp, A. Surveillance Imaging and Alpha Fetoprotein for Early Detection of Hepatocellular Carcinoma in Patients with Cirrhosis: A Meta-analysis. Gastroenterology 2018, 154, 1706–1718. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Jin, M.; Le, R.H.; Le, M.H.; Chen, V.L.; Jin, M. Poor adherence to hepatocellular carcinoma surveillance: A systematic review and meta-analysis of a complex issue. Liver Int. 2018, 38, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Bucci, L.; Garuti, F.; Lenzi, B.; Pecorelli, A.; Farinati, F.; Giannini, E.G. The evolutionary scenario of hepatocellular carcinoma in Italy: An update. Liver Int. 2017, 37, 259–270. [Google Scholar] [CrossRef]
- Singal, A.G.; Tiro, J.A.; Murphy, C.C.; Marrero, J.A.; McCallister, K.; Fullington, H. Mailed Outreach Invitations Significantly Improve HCC Surveillance Rates in Patients with Cirrhosis: A Randomized Clinical Trial. Hepatology 2019, 69, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Conjeevaram, H.S.; Volk, M.L.; Fu, S.; Fontana, R.J.; Askari, F.; Su, G.L.; Lok, A.S.; Marrero, J.A. Effectiveness of hepatocellular carcinoma surveillance in patients with cirrhosis. Cancer Epidemiol. Biomark. Prev. 2012, 21, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Simmons, O.; Fetzer, D.T.; Yokoo, T.; Marrero, J.A.; Yopp, A.; Kono, Y. Predictors of adequate ultrasound quality for hepatocellular carcinoma surveillance in patients with cirrhosis. Aliment. Pharm. Ther. 2017, 45, 169–177. [Google Scholar] [CrossRef]
- Samoylova, M.L.; Mehta, N.; Roberts, J.P.; Yao, F.Y. Predictors of Ultrasound Failure to Detect Hepatocellular Carcinoma. Liver Transplant. 2018, 24, 1171–1177. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hampel, H.; Javadi, F. The Association Between Diabetes and Hepatocellular Carcinoma: A Systematic Review of Epidemiologic Evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5·24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, D.; Feng, N.; Chen, G.; Liu, J.; Chen, G.; Zhu, Y. Increased intake of vegetables, but not fruit, reduces risk for hepatocellular carcinoma: A meta-analysis. Gastroenterology 2014, 147, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.-P.; Lin, J.; Yang, Y.C.; Tsai, M.K.; Tsao, C.K.; Etzel, C.; Huang, M.; Hsu, C.Y.; Ye, Y.; Mishra, L.; et al. Hepatocellular Carcinoma Risk Prediction Model for the General Population: The Predictive Power of Transaminases. J. Natl. Cancer Inst. 2012, 104, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Trichopoulos, D.; Polesel, J.; Bravi, F.; Rossi, M.; Talamini, R.; Franceschi, S.; Montella, M.; Trichopoulou, A.; La Vecchia, C.; et al. Mediterranean diet and hepatocellular carcinoma. J. Hepatol. 2014, 60, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Fan-Minogue, H.; Bellovin, D.I.; Yevtodiyenko, A.; Arzeno, J.; Yang, Q. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011, 71, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tokoro, T.; Higa, S.; Kitajima, I. Anti-inflammatory effect of pitavastatin on NF-kappaB activated by TNF-alpha in hepatocellular carcinoma cells. Boil. Pharm. Bull. 2006, 29, 634–639. [Google Scholar] [CrossRef]
- Higashi, T.; Hayashi, H.; Kitano, Y.; Yamamura, K.; Kaida, T.; Arima, K. Statin attenuates cell proliferative ability via TAZ (WWTR1) in hepatocellular carcinoma. Med. Oncol. 2016, 33, 123. [Google Scholar] [CrossRef]
- Yang, P.M.; Liu, Y.L.; Lin, Y.C.; Shun, C.T.; Wu, M.S.; Chen, C.C. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 2010, 70, 7699–7709. [Google Scholar] [CrossRef]
- Sutter, A.P.; Maaser, K.; Höpfner, M.; Huether, A.; Schuppan, D.; Scherübl, H. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J. Hepatol. 2005, 43, 808–816. [Google Scholar] [CrossRef]
- Kah, J.; Wüstenberg, A.; Keller, A.D.; Sirma, H.; Montalbano, R.; Ocker, M. Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol. Rep. 2012, 28, 1077–1083. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, C.; Zhou, J.; Zhen, Z.; Wang, Y.; Shen, C. Simvastatin Ameliorates Liver Fibrosis via Mediating Nitric Oxide Synthase in Rats with Non-Alcoholic Steatohepatitis-Related Liver Fibrosis. PLoS ONE 2013, 8, e76538. [Google Scholar] [CrossRef]
- Chong, L.W.; Hsu, Y.C.; Lee, T.F.; Lin, Y.; Chiu, Y.T.; Yang, K.C. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Russo, L.; Rosado, E.; Hide, D.; García-Cardeña, G. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J. Hepatol. 2013, 58, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Marinho, T.D.S.; Kawasaki, A.; Bryntesson, M.; Souza-Mello, V.; Barbosa-da-Silva, S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Rosuvastatin limits the activation of hepatic stellate cells in diet-induced obese mice. Hepatol. Res. 2017, 47, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Tsan, Y.T.; Lee, C.H.; Wang, J.D.; Chen, P.C. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J. Clin. Oncol. 2012, 30, 623–630. [Google Scholar] [CrossRef]
- Tsan, Y.T.; Lee, C.H.; Ho, W.C.; Lin, M.H.; Wang, J.D.; Chen, P.C. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J. Clin. Oncol. 2013, 31, 1514–1521. [Google Scholar] [CrossRef]
- Zhou, Y.-Y.; Zhu, G.-Q.; Wang, Y.; Zheng, J.-N.; Ruan, L.-Y.; Cheng, Z.; Hu, B.; Fu, S.-W.; Zheng, M.-H. Systematic review with network meta-analysis: Statins and risk of hepatocellular carcinoma. Oncotarget 2016, 7, 21753–21762. [Google Scholar] [CrossRef]
- Simon, T.G.; Bonilla, H.; Yan, P.; Chung, R.T.; Butt, A.A. Atorvastatin and fluvastatin are associated with dose-dependent reductions in cirrhosis and hepatocellular carcinoma, among patients with hepatitis C virus: Results from ERCHIVES. Hepatology 2016, 64, 47–57. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic Significance of AMPK Activation and Therapeutic Effects of Metformin in Hepatocellular Carcinoma. Clin. Cancer Res. 2013, 19, 5372–5380. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic Treatment with the Antidiabetic Drug Metformin Selectively Impairs p53-Deficient Tumor Cell Growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef]
- Deperalta, D.K.; Wei, L.; Ghoshal, S.; Schmidt, B.; Lauwers, G.Y.; Lanuti, M.; Chung, R.T.; Tanabe, K.K.; Fuchs, B.C. Metformin prevents hepatocellular carcinoma development by suppressing hepatic progenitor cell activation in a rat model of cirrhosis. Cancer 2016, 122, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zheng, Y.; Xiao, Y.; Zhou, P.; Tan, H. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients. Medicine 2017, 96, e6888. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA signature in NAFLD: A turning point for a non-invasive diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; El Andaloussi, S.; Wood, M.J. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, 125–134. [Google Scholar] [CrossRef]
- Enache, L.S.; Enache, E.L.; Ramière, C.; Diaz, O.; Bancu, L.; Sin, A.; André, P. Circulating RNA molecules as biomarkers in liver disease. Int. J. Mol. Sci. 2017, 15, 17644–17666. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating MicroRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Halász, T.; Horváth, G.; Pár, G.; Werling, K.; Kiss, A.; Schaff, Z.; Lendvai, G. miR-122 negatively correlates with liver fibrosis as detected by histology and FibroScan. World J. Gastroenterol. 2015, 21, 7814–7823. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiong, Y.; Sheng, Q.; Zhao, S.; Wattacheril, J.; Flynn, C.R. A micro-RNA expression signature for human NAFLD progression. J. Gastroenterol. 2016, 51, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Castaño, G.O.; Mallardi, P.; San Martino, J. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNA sto liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Moshiri, F.; Salvi, A.; Gramantieri, L.; SanGiovanni, A.; Guerriero, P.; De Petro, G.; Bassi, C.; Lupini, L.; Sattari, A.; Cheung, D.; et al. Circulating miR-106b-3p, miR-101-3p and miR-1246 as diagnostic biomarkers of hepatocellular carcinoma. Oncotarget 2018, 9, 15350–15364. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Ohno, M.; Takata, A.; Shibata, C.; Koike, K. The role of microRNAs in hepatocarcinogenesis: Current knowledge and future prospects. J. Gastroenterol. 2014, 49, 173–184. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hwang, S.; Cai, Y.; Kim, S.J.; Xu, M.; Yang, D. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenis genes in hepatocytes. Hepatology 2019, 70, 1150–1167. [Google Scholar] [CrossRef] [PubMed]
- Gori, M.; Arciello, M.; Balsano, C. MicroRNAs in Nonalcoholic Fatty Liver Disease: Novel Biomarkers and Prognostic Tools during the Transition from Steatosis to Hepatocarcinoma. BioMed Res. Int. 2014, 2014, 741465. [Google Scholar] [CrossRef]
- Peng, L.; Yuan, X.Q.; Zhang, C.Y.; Peng, J.Y.; Zhang, Y.Q.; Pan, X.; Li, G.C. The emergence of long non-coding RNAs in hepatocellular carcinoma: An update. J. Cancer 2018, 9, 2549–2558. [Google Scholar] [CrossRef]
- Xiong, D.; Sheng, Y.; Ding, S.; Chen, J.; Tan, X.; Zeng, T. LINC00052 regulates the expression of NTRK3 by miR-485-3p to strengthen HCC cells invasion and migration. Oncotarget 2016, 7, 41593–41608. [Google Scholar] [CrossRef]
- Li, T.; Xie, J.; Shen, C.; Cheng, D.; Shi, Y.; Wu, Z. Upregulation of long noncoding RNA ZEB1-AS1 promotes tumor metastasis and predicts poor prognosis in hepatocellular carcinoma. Oncogene 2016, 35, 1575–1584. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Tang, J.; Huang, R.; Li, J.; Xu, D.; Xie, Y.; Jiang, R.; Deng, L.; Zhang, X.; et al. LINC01225 promotes occurrence and metastasis of hepatocellular carcinoma in an epidermal growth factor receptor-dependent pathway. Cell Death Dis. 2016, 7, e2130. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jiang, B.; Yuan, X.; Qiu, Y.; Peng, J.; Huang, Y. Super-enhancer-associated long noncoding RNA HCCL5 is activated by ZEB1 and promotes the malignancy of hepatocellular carcinoma. Cancer Res. 2019, 79, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, J.; Ding, C.-M.; Jin, X.; Jia, Z.-M.; Peng, J. LncRNA NR2F1-AS1 regulates hepatocellular carcinoma oxaliplatin resistance by targeting ABCC1 via miR-. J. Cell. Mol. Med. 2018, 22, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Cheng, X.; Pan, X.; Li, J. Emerging role of exosomes in liver physiology and pathology. Hepatol. Res. 2017, 47, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, A.; Costa, V.; Dico, A.L.; Saieva, L.; Buccheri, S.; Dieli, F. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.A. Epigenetics in liver disease. Hepatology 2014, 60, 1418–1425. [Google Scholar] [CrossRef]
- Tian, W.; Xu, H.; Fang, F.; Chen, Q.; Xu, Y.; Shen, A. Brahma-related gene 1 bridges epigenetic regulation of proinflammatory cytokine production to steatohepatitis in mice. Hepatology 2013, 58, 576–588. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, K.; Deng, Q.; Li, W.; Zhang, X.; Liu, X. Identification of Key Hydroxymethylated Genes and Transcription Factors Associated with Alpha-Fetoprotein-Negative Hepatocellular Carcinoma DNA. Cell Biol. 2019, in press. [Google Scholar]



{kind=link}
{kind=link}
| SNP | Number of Patients | Study Design | Cirrhosis | Country | Covariate Adjustment | Reference |
|---|---|---|---|---|---|---|
| PNPLA3 I148M (rs738409, C > G) | 100 with HCC-related NAFLD 275 NAFLD controls without HCC | Case-control, retrospective | 24.8% | UK and Switzerland | Age, sex, BMI, diabetes, cirrhosis | [29] |
| PNPLA3 I148M (rs738409, C > G) | 471 with NAFLD | Cohort study, propspective | 34.4 % (F3–F4) | Italy | Age, BMI, platelet count, albumin, IFG/diabetes, fibrosis F3–F4 | [30] |
| TM6SF2 E167K (rs58542926, C >T) | 511 with liver disease (44% alcohol) 228 controls | Case-control, retrospective | 100% | Italy | NA | [36] |
| MBOAT7 (rs641738, C > T) | 132 with NAFLD 633 controls | Case-control, retrospective | 27.5% (F3–F4) | Italy | Age, sex, obesity, diabetes, fibrosis F3–F4, PNPLA3, TM6SF2 | [39] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennisi, G.; Celsa, C.; Giammanco, A.; Spatola, F.; Petta, S. The Burden of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: Screening Issue and Future Perspectives. Int. J. Mol. Sci. 2019, 20, 5613. https://doi.org/10.3390/ijms20225613
Pennisi G, Celsa C, Giammanco A, Spatola F, Petta S. The Burden of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: Screening Issue and Future Perspectives. International Journal of Molecular Sciences. 2019; 20(22):5613. https://doi.org/10.3390/ijms20225613
Chicago/Turabian StylePennisi, Grazia, Ciro Celsa, Antonina Giammanco, Federica Spatola, and Salvatore Petta. 2019. "The Burden of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: Screening Issue and Future Perspectives" International Journal of Molecular Sciences 20, no. 22: 5613. https://doi.org/10.3390/ijms20225613
APA StylePennisi, G., Celsa, C., Giammanco, A., Spatola, F., & Petta, S. (2019). The Burden of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: Screening Issue and Future Perspectives. International Journal of Molecular Sciences, 20(22), 5613. https://doi.org/10.3390/ijms20225613