Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
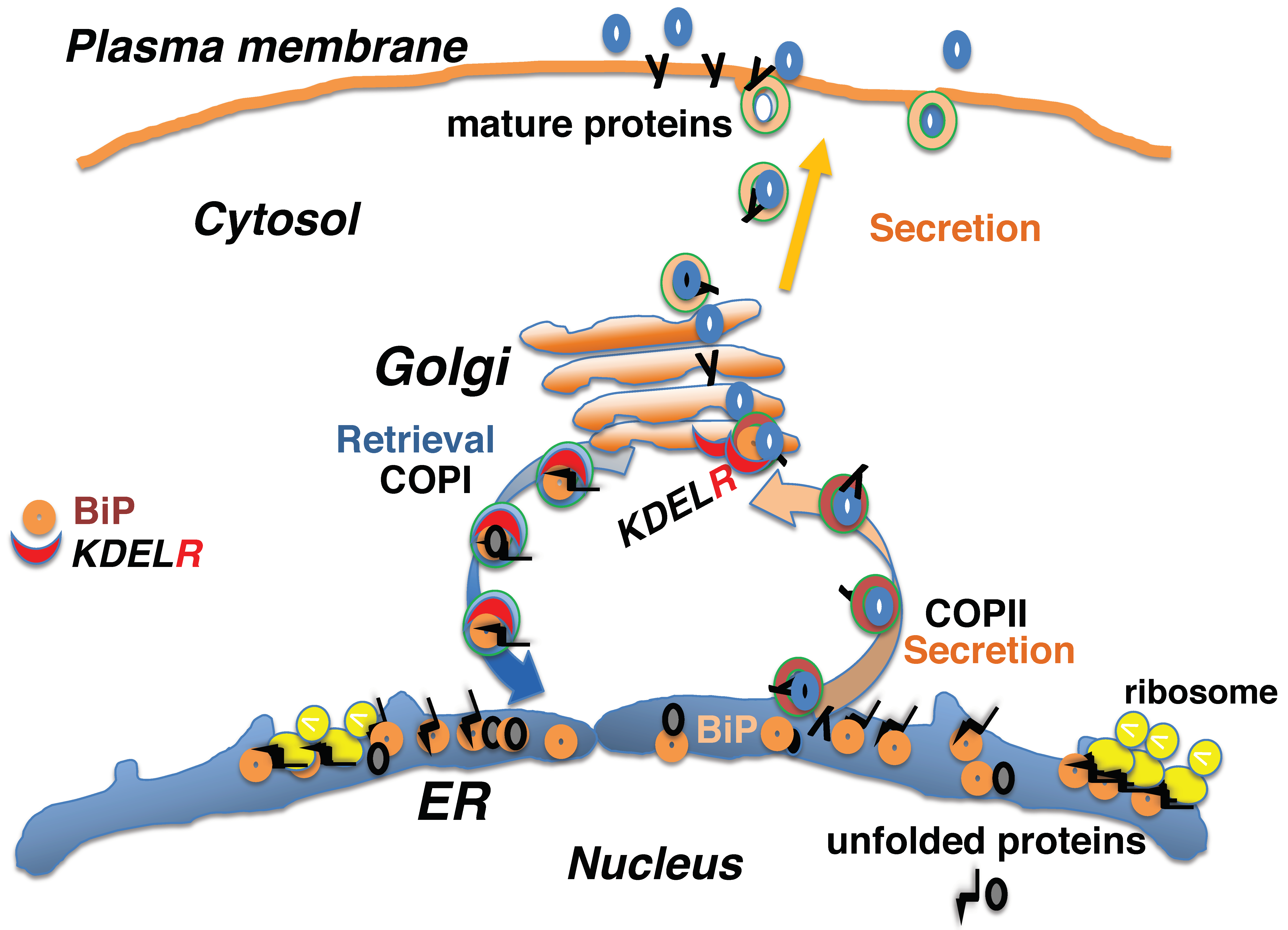
2. Protein Quality Control in the Early Secretory Pathway Is Mediated by the KDEL Retrieval System
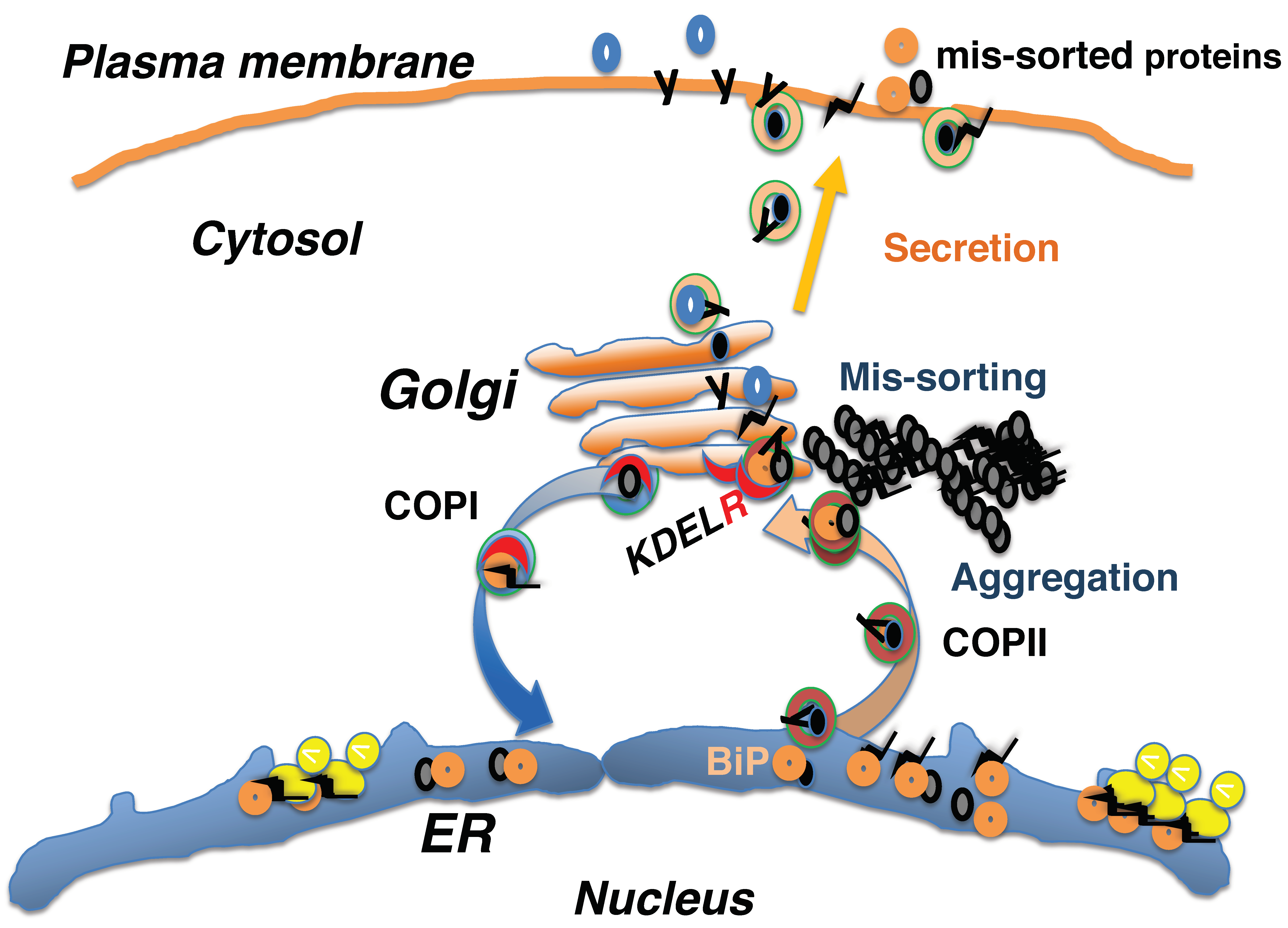
3. Mis-Sorting of KDEL Sequence-Containing Proteins in the Early Secretory System In Vivo
4. Impairment of Retrieval by the KDEL Receptor Causes Myeloproliferative Neoplasms
5. Defective Proteostasis Associated with the Retrieval Pathway Leads to Neurodegeneration
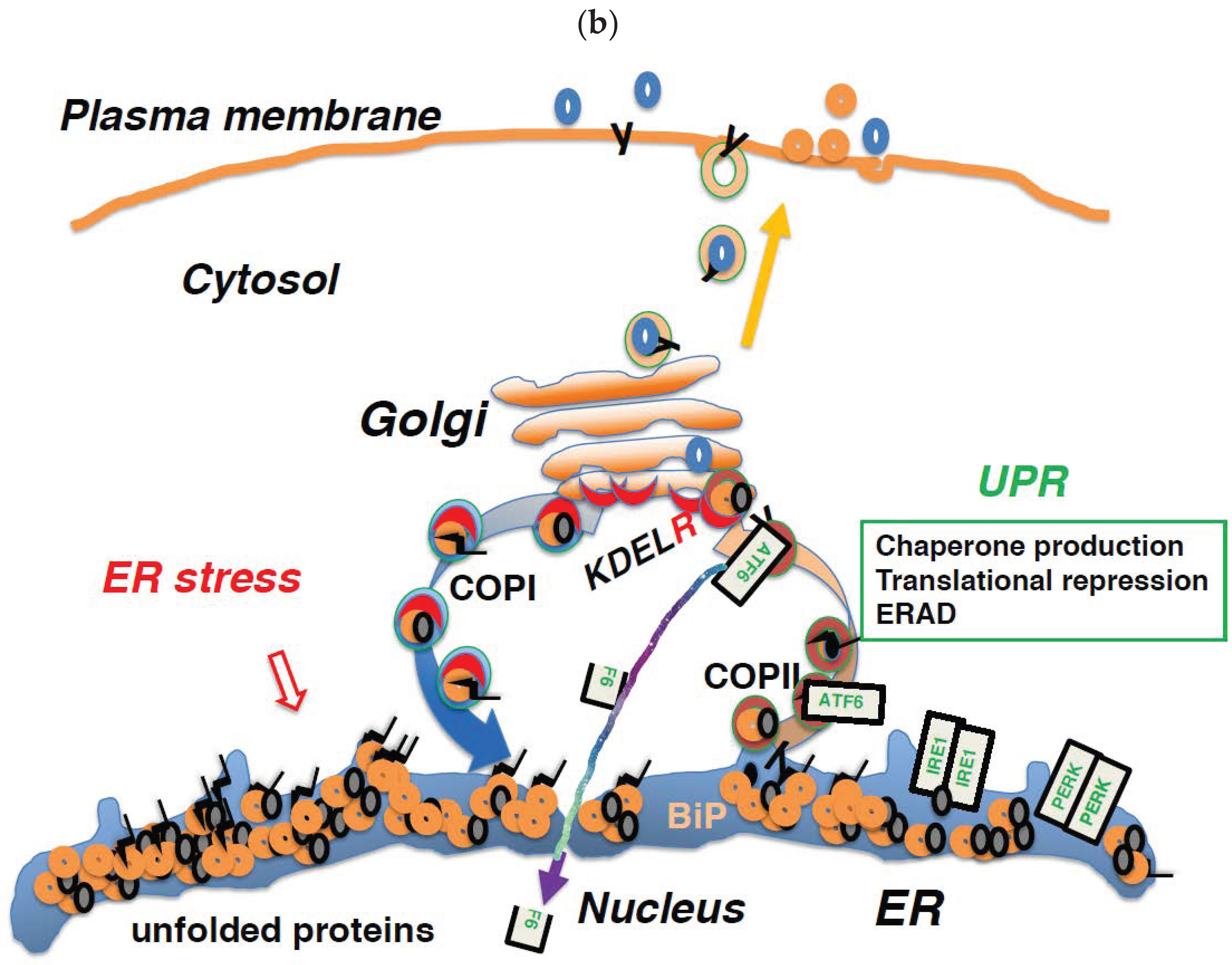
6. Regulation of Membrane Trafficking and the UPR are Linked through the KDEL Receptor
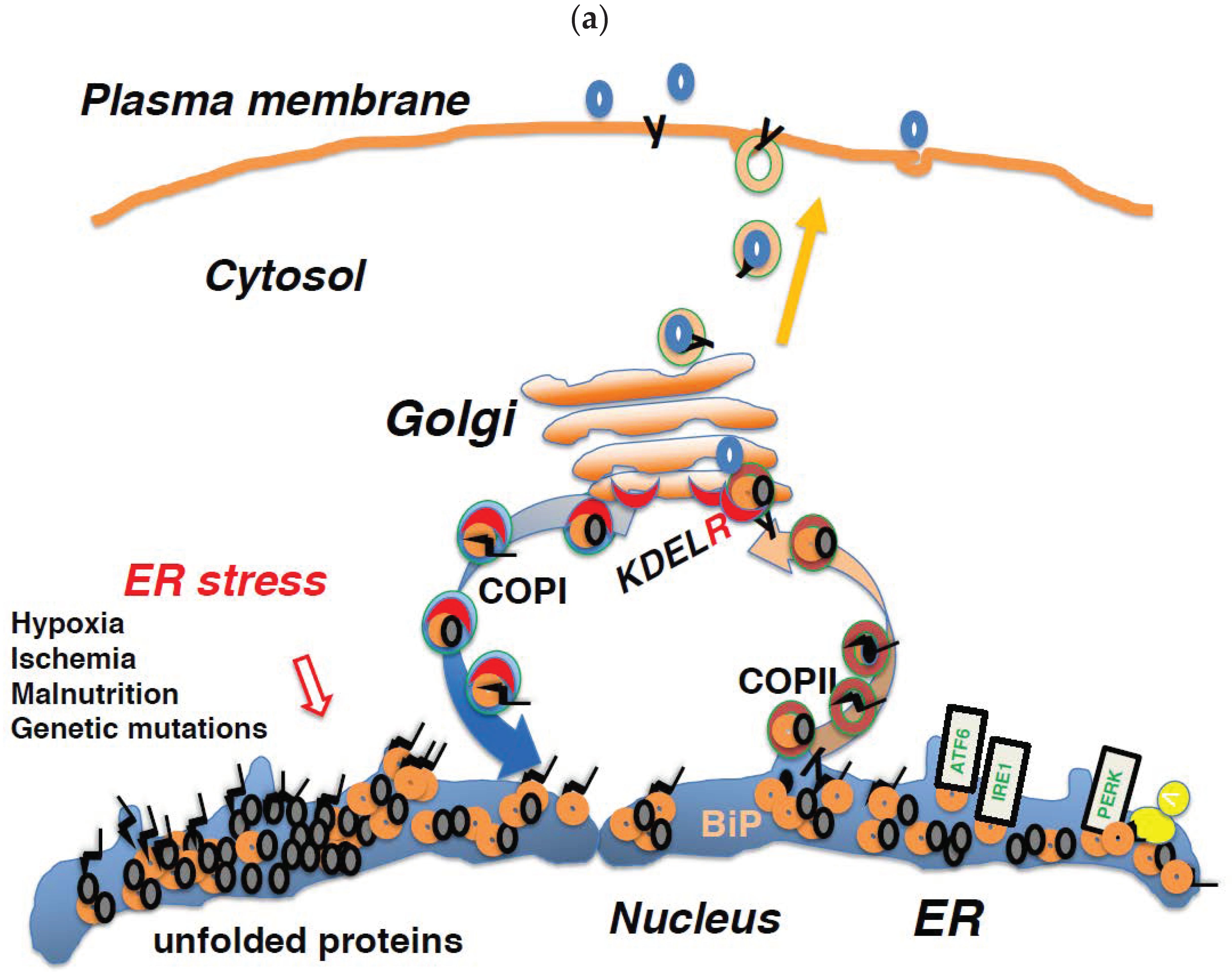
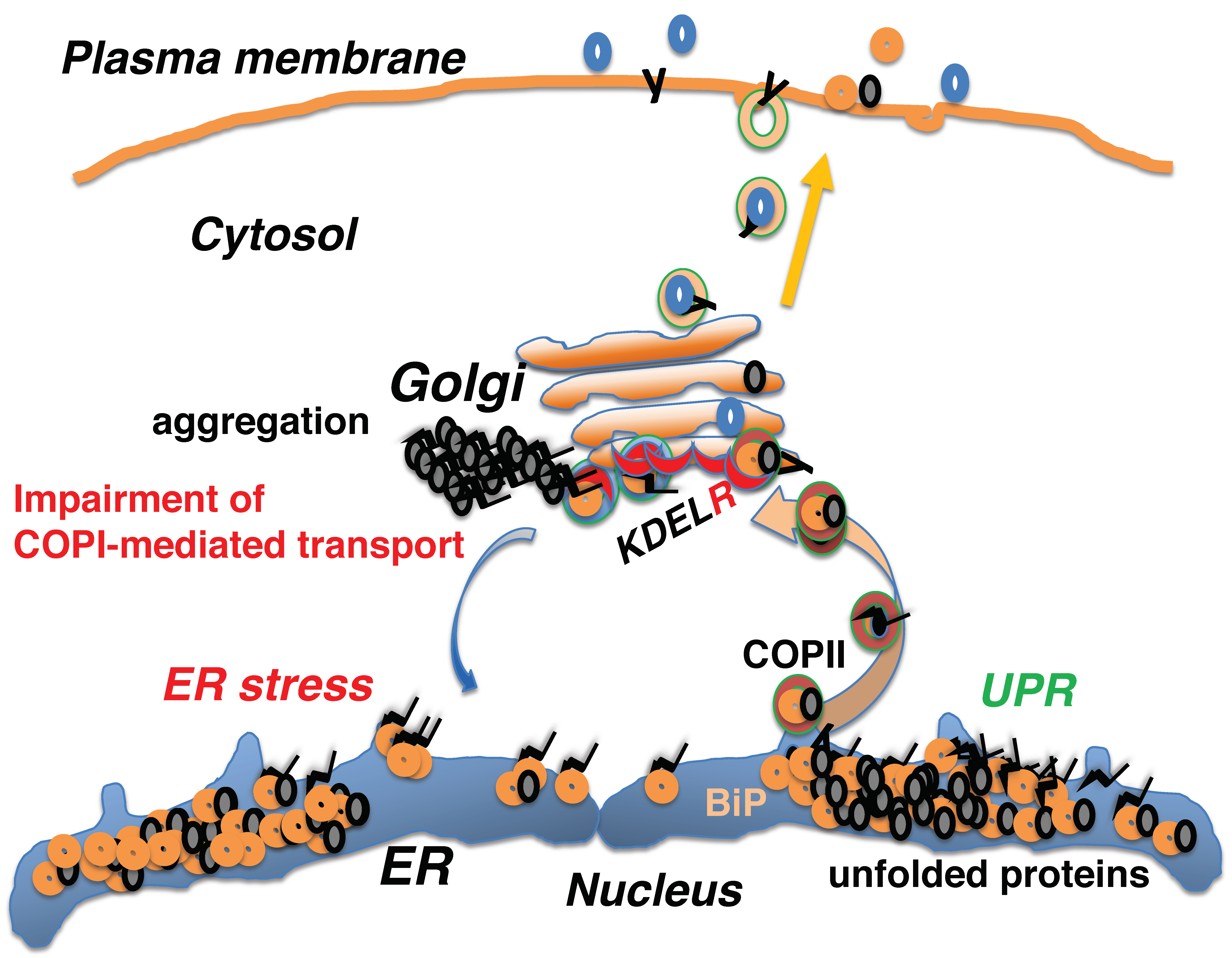
7. Impairment of COPI-Mediated Retrograde Transport Induces ER Stress
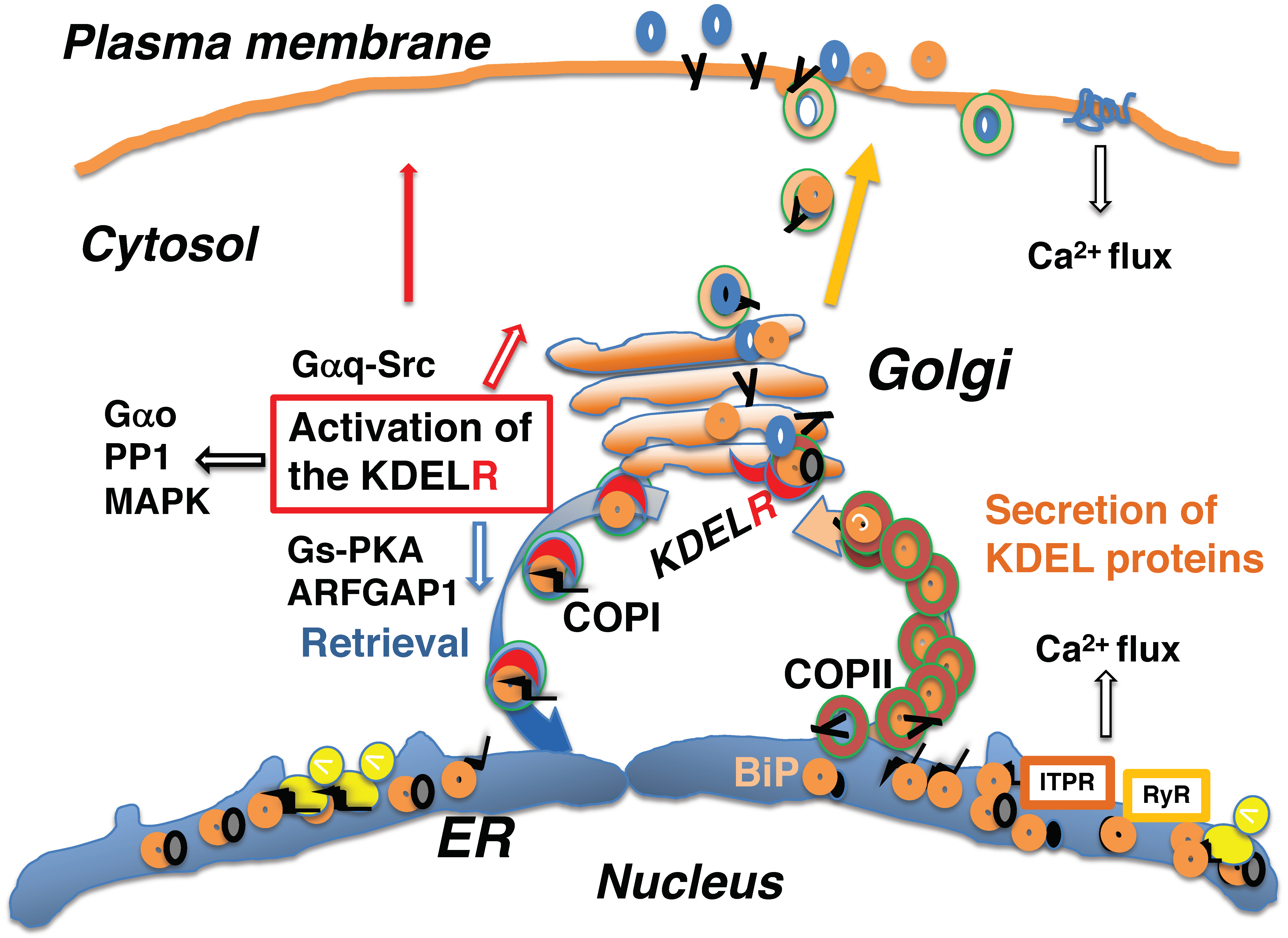
8. Activation of the KDEL Receptor May Occur Acutely or Constantly
9. The KDEL Receptor Signaling Pathway Mediates Various Functions
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| GRP94 | Glucose regulatory protein 94 |
| BiP | Binding immunoglobulin protein |
| KDEL | Lys-Asp-Glu-Leu |
| COPII | Coat protein II |
| COPI | Coat protein I |
| TCR | T cell antigen receptor |
| SP | Surfactant protein |
| CHOP | C/EBP homologous protein |
| CR | Cajal–Retzius |
| CALR | Calreticulin |
| MPNs | Myeloproliferative neoplasms |
| TpoR | Thrombopoietin receptor |
| JAK2 | Janus kinase 2 |
| UPR | Unfolded protein response |
| ERN1 | Endoplasmic reticulum to nucleus signaling 1 |
| PERK | Protein kinase RNA (PKR)-like ER kinase |
| ATF6 | Activating transcription factor 6 |
| EIF2A | Eukaryotic translation initiation factor 2A |
| XBP1 | X-box-binding protein 1 |
| PP1 | Protein phosphatase 1 |
| MAPK | Mitogen-activated protein kinase |
| JNKs | c-Jun amino-terminal kinases |
| ARFGAP1 | ADP ribosylation factor GTPase activating protein 1 |
| PKA | Protein kinase A |
| ITPR | Inositol 1,4,5-triphosphate receptor |
| ATP2A1 | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1 |
| GPCRs | G protein coupled receptors |
| ECM | Extracellular matrix |
References
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, T.A.; Li, L.; Park, E. Structural and mechanistic insights into protein translocation. Annu. Rev. Cell. Dev. Biol. 2017, 33, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Cancino, J.; Capalbo, A.; Di Campli, A.; Giannotta, M.; Rizzo, R.; Jung, J.E.; Di Martino, R.; Persico, M.; Heinklein, P.; Sallese, M.; et al. Control systems of membrane transport at the interface between the endoplasmic reticulum and the Golgi. Dev. Cell. 2014, 30, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Arakel, E.C.; Schwappach, B. Formation of COPI-coated vesicles at a glance. J. Cell. Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell. Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef]
- Wilson, D.W.; Lewis, M.J.; Pelham, H.R. pH-dependent binding of KDEL to its receptor in vitro. J. Biol. Chem. 1993, 268, 7465–7468. [Google Scholar]
- Brauer, P.; Parker, J.L.; Gerondopoulos, A.; Zimmermann, I.; Seeger, M.A.; Barr, F.A.; Newstead, S. Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 2019, 363, 1103–1107. [Google Scholar] [CrossRef]
- Yamamoto, K.; Fujii, R.; Toyofuku, Y.; Saito, T.; Koseki, H.; Hsu, V.W.; Aoe, T. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001, 20, 3082–3091. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Komita, M.; Aoe, T. The role of BiP retrieval by the KDEL receptor in the early secretory pathway and its effect on protein quality control and neurodegeneration. Front. Mol. Neurosci. 2017, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Aoe, T.; Cukierman, E.; Lee, A.; Cassel, D.; Peters, P.J.; Hsu, V.W. The KDEL receptor, ERD2, regulates intracellular traffic by recruiting a GTPase-activating protein for ARF1. EMBO J. 1997, 16, 7305–7316. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Hamada, H.; Shinkai, H.; Kohno, Y.; Koseki, H.; Aoe, T. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J. Biol. Chem. 2003, 278, 34525–34532. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Muniz, M.; Hidalgo, J.; Vega, L.; Martin, M.E.; Velasco, A. The retrieval function of the KDEL receptor requires PKA phosphorylation of its C-terminus. Mol. Biol. Cell. 2003, 14, 4114–4125. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Mazelin, L.; Pechoux-Longin, C.; Malhotra, V.; Jurdic, P. Src regulates Golgi structure and KDEL receptor-dependent retrograde transport to the endoplasmic reticulum. J. Biol. Chem. 2003, 278, 46601–46606. [Google Scholar] [CrossRef] [PubMed]
- Giannotta, M.; Ruggiero, C.; Grossi, M.; Cancino, J.; Capitani, M.; Pulvirenti, T.; Consoli, G.M.; Geraci, C.; Fanelli, F.; Luini, A.; et al. The KDEL receptor couples to Galphaq/11 to activate Src kinases and regulate transport through the Golgi. EMBO J. 2012, 31, 2869–2881. [Google Scholar] [CrossRef]
- Kamimura, D.; Katsunuma, K.; Arima, Y.; Atsumi, T.; Jiang, J.J.; Bando, H.; Meng, J.; Sabharwal, L.; Stofkova, A.; Nishikawa, N.; et al. KDEL receptor 1 regulates T-cell homeostasis via PP1 that is a key phosphatase for ISR. Nat. Commun. 2015, 6, 7474. [Google Scholar] [CrossRef]
- Solis, G.P.; Bilousov, O.; Koval, A.; Luchtenborg, A.M.; Lin, C.; Katanaev, V.L. Golgi-resident galphao promotes protrusive membrane dynamics. Cell 2017, 170, 1258. [Google Scholar] [CrossRef]
- Hamada, H.; Suzuki, M.; Yuasa, S.; Mimura, N.; Shinozuka, N.; Takada, Y.; Nishino, T.; Nakaya, H.; Koseki, H.; Aoe, T. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol. Cell. Biol. 2004, 24, 8007–8017. [Google Scholar] [CrossRef]
- Mimura, N.; Hamada, H.; Kashio, M.; Jin, H.; Toyama, Y.; Kimura, K.; Iida, M.; Goto, S.; Saisho, H.; Toshimori, K.; et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007, 14, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Yuasa, S.; Soma, M.; Jin, H.; Kimura, K.; Goto, S.; Koseki, H.; Aoe, T. Altered quality control in the endoplasmic reticulum causes cortical dysplasia in knock-in mice expressing a mutant BiP. Mol. Cell. Bio. 2008, 28, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Jin, H.; Ogawa, M.; Aoe, T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem. Biophys. Res. Commun. 2008, 366, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Mimura, N.; Kashio, M.; Koseki, H.; Aoe, T. Late-onset of spinal neurodegeneration in knock-in mice expressing a mutant BiP. PLoS ONE 2014, 9, e112837. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Komita, M.; Aoe, T. Decreased protein quality control promotes the cognitive dysfunction associated with aging and environmental insults. Front. Neurosci. 2018, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Huppa, J.B.; Ploegh, H.L. In vitro translation and assembly of a complete T cell receptor-CD3 complex. J. Exp. Med. 1997, 186, 393–403. [Google Scholar] [CrossRef]
- Hammond, C.; Braakman, I.; Helenius, A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. USA 1994, 91, 913–917. [Google Scholar] [CrossRef]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.A.; You, S.; Yoon, H.J.; Kim, D.H.; Kim, H.S.; Lee, K.; Ahn, J.H.; Hwang, D.; Lee, A.S.; Kim, K.J.; et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J. Exp. Med. 2012, 209, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Panayi, G.S.; Corrigall, V.M. Immunoglobulin heavy-chain-binding protein (BiP): A stress protein that has the potential to be a novel therapy for rheumatoid arthritis. Biochem. Soc. Trans. 2014, 42, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Dolfe, L.; Winblad, B.; Johansson, J.; Presto, J. BRICHOS binds to a designed amyloid-forming β-protein and reduces proteasomal inhibition and aggresome formation. Biochem. J. 2016, 473, 167–178. [Google Scholar] [CrossRef]
- Knight, S.D.; Presto, J.; Linse, S.; Johansson, J. The BRICHOS domain, amyloid fibril formation, and their relationship. Biochemistry 2013, 52, 7523–7531. [Google Scholar] [CrossRef]
- Beers, M.F.; Mulugeta, S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu. Rev. Physiol. 2005, 67, 663–696. [Google Scholar] [CrossRef]
- Peca, D.; Boldrini, R.; Johannson, J.; Shieh, J.T.; Citti, A.; Petrini, S.; Salerno, T.; Cazzato, S.; Testa, R.; Messina, F.; et al. Clinical and ultrastructural spectrum of diffuse lung disease associated with surfactant protein C mutations. Eur. J. Hum. Genet. 2015, 23, 1033–1041. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Lu, H.Y.; Chen, X.Q.; Tang, W.; Wang, Q.X.; Zhang, J. GRP78 silencing enhances hyperoxia-induced alveolar epithelial cell apoptosis via CHOP pathway. Mol. Med. Rep. 2017, 16, 1493–1501. [Google Scholar] [CrossRef][Green Version]
- Klymenko, O.; Huehn, M.; Wilhelm, J.; Wasnick, R.; Shalashova, I.; Ruppert, C.; Henneke, I.; Hezel, S.; Guenther, K.; Mahavadi, P.; et al. Regulation and role of the ER stress transcription factor CHOP in alveolar epithelial type-II cells. J. Mol. Med. 2019, 97, 973–990. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Kubo, K.I.; Nakajima, K. Reelin and neuropsychiatric disorders. Front. Cell Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Merlinsky, T.R.; Levine, R.L.; Pronier, E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clin. Cancer Res. 2019, 25, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, N.; Araki, M.; Yang, Y.; Hayashi, E.; Imai, M.; Edahiro, Y.; Hironaka, Y.; Mizukami, Y.; Kihara, Y.; Takei, H.; et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 2019. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019. [Google Scholar] [CrossRef]
- Brandvold, K.R.; Morimoto, R.I. The chemical biology of molecular chaperones—Implications for modulation of proteostasis. J. Mol. Biol. 2015, 427, 2931–2947. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lavail, M.M. Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 2010, 664, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.; Kramer, P.; LaMorticella, D.M.; Murphey, W.; Lovrien, E.W.; Weleber, R.G. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum. Mol. Genet. 1998, 7, 471–474. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef]
- Tang, B.S.; Zhao, G.H.; Luo, W.; Xia, K.; Cai, F.; Pan, Q.; Zhang, R.X.; Zhang, F.F.; Liu, X.M.; Chen, B.; et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 2005, 116, 222–224. [Google Scholar] [CrossRef]
- He, M.; Guo, H.; Yang, X.; Zhou, L.; Zhang, X.; Cheng, L.; Zeng, H.; Hu, F.B.; Tanguay, R.M.; Wu, T. Genetic variations in HSPA8 gene associated with coronary heart disease risk in a Chinese population. PLoS ONE 2010, 5, e9684. [Google Scholar] [CrossRef]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Higuchi, M.; Yoshiyama, Y.; Ishihara, T.; Forman, M.S.; Martinez, D.; Joyce, S.; Trojanowski, J.Q.; Lee, V.M. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J. Neurosci. 2004, 24, 4657–4667. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Komita, M.; Jin, H.; Aoe, T. The effect of endoplasmic reticulum stress on neurotoxicity caused by inhaled anesthetics. Anesth. Analg. 2013, 117, 1197–1204. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded- protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Chen, L.; Li, Q.; She, T.; Li, H.; Yue, Y.; Gao, S.; Yan, T.; Liu, S.; Ma, J.; Wang, Y. IRE1alpha-XBP1 signaling pathway, a potential therapeutic target in multiple myeloma. Leuk. Res. 2016, 49, 7–12. [Google Scholar] [CrossRef]
- Trychta, K.A.; Back, S.; Henderson, M.J.; Harvey, B.K. KDEL receptors are differentially regulated to maintain the ER proteome under calcium deficiency. Cell Rep. 2018, 25, 1829–1840. [Google Scholar] [CrossRef]
- Watkin, L.B.; Jessen, B.; Wiszniewski, W.; Vece, T.J.; Jan, M.; Sha, Y.; Thamsen, M.; Santos-Cortez, R.L.; Lee, K.; Gambin, T.; et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat. Genet 2015, 47, 654–660. [Google Scholar] [CrossRef]
- Aoe, T.; Lee, A.J.; van Donselaar, E.; Peters, P.J.; Hsu, V.W. Modulation of intracellular transport by transported proteins: Insight from regulation of COPI-mediated transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1624–1629. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Zucchi, R.; Ronca-Testoni, S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 1997, 49, 1–51. [Google Scholar] [PubMed]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium. 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Thastrup, O.; Cullen, P.J.; Drobak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Bajpayee, N.S. Molecular mechanisms of go signaling. Neurosignals 2009, 17, 23–41. [Google Scholar] [CrossRef]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How ligands Illuminate GPCR molecular pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef]
- Park, S.Y.; Yang, J.S.; Schmider, A.B.; Soberman, R.J.; Hsu, V.W. Coordinated regulation of bidirectional COPI transport at the Golgi by CDC42. Nature 2015, 521, 529–532. [Google Scholar] [CrossRef]
- Abdel Malek, M.A.; Jagannathan, S.; Malek, E.; Sayed, D.M.; Elgammal, S.A.; Abd El-Azeem, H.G.; Thabet, N.M.; Driscoll, J.J. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015, 6, 3098–3110. [Google Scholar] [CrossRef]
- Wang, P.; Li, B.; Zhou, L.; Fei, E.; Wang, G. The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins. Neuroscience 2011, 190, 43–55. [Google Scholar] [CrossRef]
- Tapia, D.; Jimenez, T.; Zamora, C.; Espinoza, J.; Rizzo, R.; Gonzalez-Cardenas, A.; Fuentes, D.; Hernandez, S.; Cavieres, V.A.; Soza, A.; et al. KDEL receptor regulates secretion by lysosome relocation- and autophagy-dependent modulation of lipid-droplet turnover. Nat. Commun. 2019, 10, 735. [Google Scholar] [CrossRef]
- Ruggiero, C.; Grossi, M.; Fragassi, G.; Di Campli, A.; Di Ilio, C.; Luini, A.; Sallese, M. The KDEL receptor signalling cascade targets focal adhesion kinase on focal adhesions and invadopodia. Oncotarget 2018, 9, 10228–10246. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.; Fragassi, G.; Grossi, M.; Picciani, B.; Di Martino, R.; Capitani, M.; Buccione, R.; Luini, A.; Sallese, M. A Golgi-based KDELR-dependent signalling pathway controls extracellular matrix degradation. Oncotarget 2015, 6, 3375–3393. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokubun, H.; Jin, H.; Aoe, T. Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex. Int. J. Mol. Sci. 2019, 20, 5614. https://doi.org/10.3390/ijms20225614
Kokubun H, Jin H, Aoe T. Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex. International Journal of Molecular Sciences. 2019; 20(22):5614. https://doi.org/10.3390/ijms20225614
Chicago/Turabian StyleKokubun, Hiroshi, Hisayo Jin, and Tomohiko Aoe. 2019. "Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex" International Journal of Molecular Sciences 20, no. 22: 5614. https://doi.org/10.3390/ijms20225614
APA StyleKokubun, H., Jin, H., & Aoe, T. (2019). Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex. International Journal of Molecular Sciences, 20(22), 5614. https://doi.org/10.3390/ijms20225614