Immunobiology of Atherosclerosis: A Complex Net of Interactions



{kind=link}
{kind=link}
Abstract
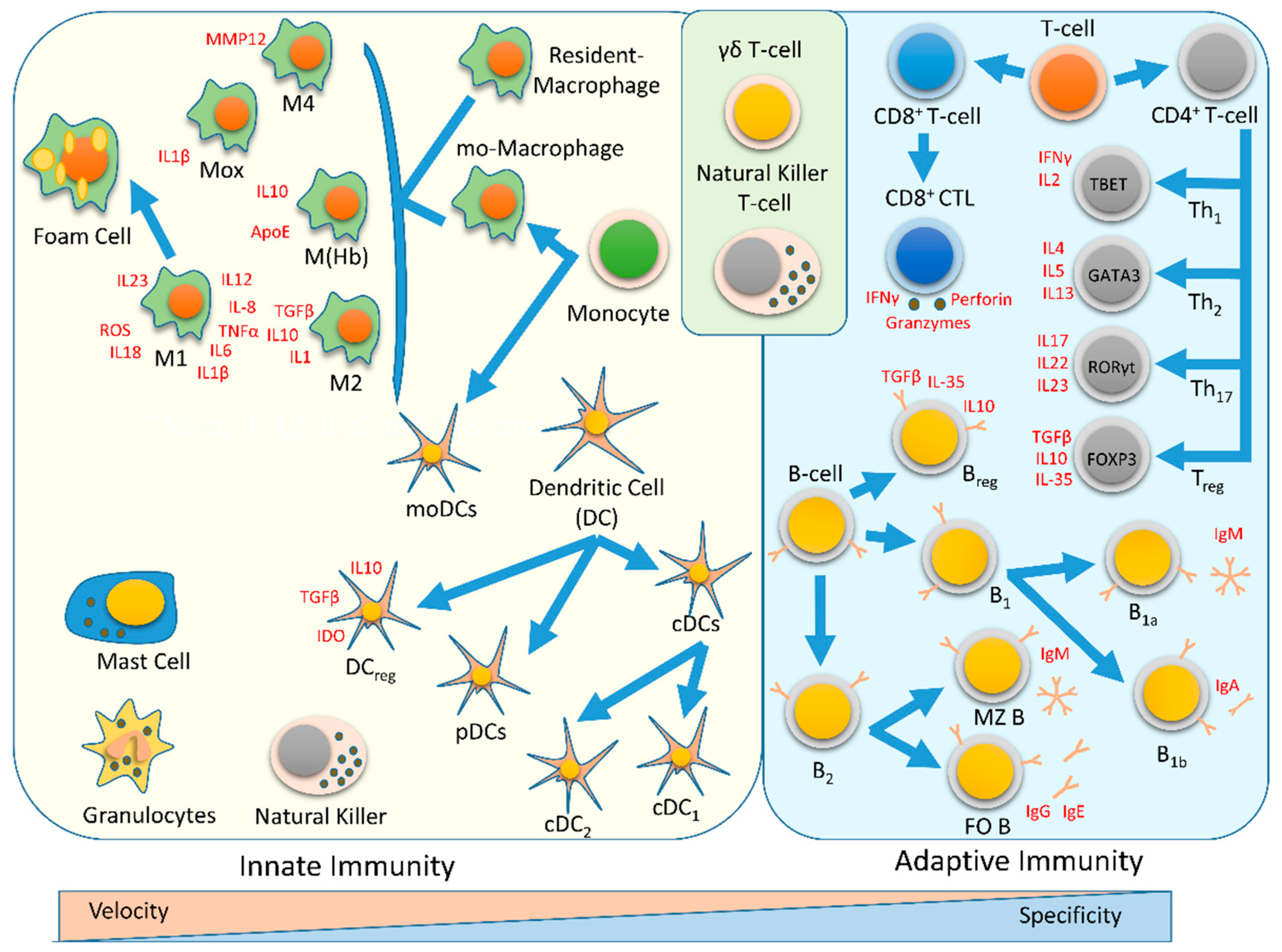
1. Immune system
2. Atherosclerosis’s Epidemiology
3. Atherosclerosis’s Pathophysiology
4. Innate Immunity
4.1. Monocytes
4.2. Macrophages
4.3. Foam Cells
4.4. Dendritic Cells
4.4.1. cDCs
4.4.2. pDCs
4.4.3. Regulatory DCs
5. Adaptive Immunity
5.1. CD4+ T Cell
5.1.1. Th1
5.1.2. Th2
5.1.3. Th17
5.1.4. Tregs
5.2. CD8 T Cells
5.3. B-Cells
6. Discussion
Funding
Acknowledgments
Conflicts of Interest
References
- Shimada, K. Immune System and Atherosclerotic Disease: Heterogeneity of Leukocyte Subsets Participating in the Pathogenesis of Atherosclerosis. Circ. J. 2009, 73, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Moroni, F.; Magnoni, M.; Camici, P.G. The Role of T and B Cells in Human Atherosclerosis and Atherothrombosis. Clin. Exp. Immunol. 2015, 179, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Trogan, E.; Ginsberg, M.; Grigaux, C.; Tian, J.; Miyata, M. Decreased Atherosclerosis in Mice Deficient in Both Macrophage Colony-Stimulating Factor (op) and Apolipoprotein E. Proc. Natl. Acad. Sci. USA 1995, 92, 8264–8268. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Leung, C.; Schindler, C. Lymphocytes are Important in Early Atherosclerosis. J. Clin. Investig. 2001, 108, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Reardon, C.A.; Blachowicz, L.; Lukens, J.; Nissenbaum, M.; Getz, G.S. Genetic Background Selectively Influences Innominate Artery Atherosclerosis: Immune System Deficiency as a Probe. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1449–1454. [Google Scholar] [CrossRef]
- Dansky, H.M.; Charlton, S.A.; Harper, M.M.; Smith, J.D. T and B Lymphocytes Play a Minor Role in Atherosclerotic Plaque Formation in the Apolipoprotein E-Deficient Mouse. Proc. Natl. Acad. Sci. USA 1997, 94, 4642–4646. [Google Scholar] [CrossRef]
- Zhou, X.; Nicoletti, A.; Elhage, R.; Hansson, G.K. Transfer of CD4(+) T Cells Aggravates Atherosclerosis in Immunodeficient Apolipoprotein E knockout Mice. Circulation 2000, 102, 2919–2922. [Google Scholar] [CrossRef]
- Reardon, C.A.; Blachowicz, L.; White, T.; Cabana, V.; Wang, Y.; Lukens, J.; Bluestone, J.; Getz, G.S. Effect of Immune Deficiency on Lipoproteins and Atherosclerosis in Male Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1011–1016. [Google Scholar] [CrossRef]
- Daugherty, A.; Pure, E.; Delfel-Butteiger, D.; Chen, S.; Leferovich, J.; Roselaar, S.E.; Rader, D.J. The Effects of Total Lymphocyte Deficiency on the Extent of Atherosclerosis in Apolipoprotein E-/-mice. J. Clin. Investig. 1997, 100, 1575–1580. [Google Scholar] [CrossRef]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune Effector Mechanisms Implicated in Atherosclerosis: From Mice to Humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, Regional and National Incidence, Prevalence, and Years Lived with Disability for 354 Diseases and Injuries for 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar]
- Berenson, G.S.; Srinivasan, S.R.; Bao, W.; Newman, W.P., 3rd; Tracy, R.E.; Wattigney, W.A. Association between Multiple Cardiovascular Risk Factors and Atherosclerosis in Children and Young Adults. The Bogalusa Heart Study. N. Engl. J. Med. 1998, 338, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and Plaque Vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-Reactive Protein: A Critical Update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.M.; Murphy, P.A.; Vella, A.T. Activated T-Effector seeds: Cultivating Atherosclerotic Plaque through Alternative Activation. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1354–H1365. [Google Scholar] [CrossRef]
- Boren, J.; Olin, K.; Lee, I.; Chait, A.; Wight, T.N.; Innerarity, T.L. Identification of the Principal Proteoglycan-Binding Site in LDL. A Single-Point Mutation in Apo-B100 Severely Affects Proteoglycan Interaction without Affecting LDL Receptor Binding. J. Clin. Investig. 1998, 101, 2658–2664. [Google Scholar] [CrossRef]
- Kwon, G.P.; Schroeder, J.L.; Amar, M.J.; Remaley, A.T.; Balaban, R.S. Contribution of Macromolecular Structure to the Retention of Low-Density Lipoprotein at Arterial Branch Points. Circulation 2008, 117, 2919–2927. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis–An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Hansson, G.K. Anti-Inflammatory Therapies for Atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis (*). Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Matsumoto, T.; Matsubara, Y.; Harada, Y.; Kyuragi, R.; Koga, J.I.; Egashira, K.; Nakashima, Y.; Yonemitsu, Y.; Maehara, Y. BubR1 Insufficiency Results in Decreased Macrophage Proliferation and Attenuated Atherogenesis in Apolipoprotein E-Deficient Mice. J. Am. Heart Assoc. 2016, 5, e004081. [Google Scholar] [CrossRef] [PubMed]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling: Molecular, Cellular and Vascular Behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Kaazempur-Mofrad, M.R.; Natarajan, S.; Zhang, Y.; Vaughn, S.; Blackman, B.R.; Kamm, R.D.; Garcia-Cardena, G.; Gimbrone, M.A., Jr. Distinct Endothelial Phenotypes Evoked by Arterial Waveforms Derived from Atherosclerosis-Susceptible and -Resistant Regions of Human Vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 14871–14876. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Lipton, B.A.; Parthasarathy, S.; Ord, V.A.; Clinton, S.K.; Libby, P.; Rosenfeld, M.E. Components of the Protein Fraction of Oxidized Low Density Lipoprotein Stimulate Interleukin-1 Alpha Production by Rabbit Arterial Macrophage-Derived foam Cells. J. Lipid. Res. 1995, 36, 2232–2242. [Google Scholar]
- Kranzhofer, R.; Schmidt, J.; Pfeiffer, C.A.; Hagl, S.; Libby, P.; Kubler, W. Angiotensin Induces Inflammatory Activation of Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1623–1629. [Google Scholar] [CrossRef]
- Libby, P.; Ordovas, J.M.; Auger, K.R.; Robbins, A.H.; Birinyi, L.K.; Dinarello, C.A. Endotoxin and Tumor Necrosis Factor Induce Interleukin-1 Gene Expression in Adult Human Vascular Endothelial Cells. Am. J. Pathol. 1986, 124, 179–185. [Google Scholar]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and Atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Mannarino, E.; Pirro, M. Endothelial Injury and Repair: A Novel Theory for Atherosclerosis. Angiology 2008, 59, 69S–72S. [Google Scholar] [CrossRef] [PubMed]
- Clinton, S.K.; Underwood, R.; Hayes, L.; Sherman, M.L.; Kufe, D.W.; Libby, P. Macrophage Colony-Stimulating Factor Gene Expression in Vascular Cells and in Experimental and Human Atherosclerosis. Am. J. Pathol. 1992, 140, 301–316. [Google Scholar] [PubMed]
- Galkina, E.; Ley, K. Vascular Adhesion Molecules in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Leducq Transatlantic Network on Atherothrombosis. Inflammation in Atherosclerosis: From Pathophysiology to Practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Pannu, P.S.; Francis, G.A. Contribution of Monocyte-Derived Macrophages and Smooth Muscle Cells to Arterial Foam Cell Formation. Cardiovasc. Res. 2012, 95, 165–172. [Google Scholar] [CrossRef]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate Sensing of Oxidation-Specific Epitopes in Health and Disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- Song, M.; Xu, S.; Zhong, A.; Zhang, J. Crosstalk between Macrophage and T Cell in Atherosclerosis: Potential Therapeutic Targets for Cardiovascular Diseases. Clin. Immunol. 2019, 202, 11–17. [Google Scholar] [CrossRef]
- Worbs, T.; Hammerschmidt, S.I.; Forster, R. Dendritic Cell Migration in Health and Disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef]
- Gil-Pulido, J.; Zernecke, A. Antigen-Presenting Dendritic Cells in Atherosclerosis. Eur. J. Pharmacol. 2017, 816, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The Role of B Cells in Atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Schonbeck, U.; Yan, Z.Q. Innate and Adaptive Immunity in the Pathogenesis of Atherosclerosis. Circ. Res. 2002, 91, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleki, F.; Gheibi Hayat, S.M.; Bianconi, V.; Johnston, T.P.; Sahebkar, A. Atherosclerosis and Immunity: A Perspective. Trends Cardiovasc. Med. 2019, 29, 363–371. [Google Scholar] [CrossRef]
- Frostegard, J.; Ulfgren, A.K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine Expression in Advanced Human Atherosclerotic Plaques: Dominance of Pro-Inflammatory (Th1) and Macrophage-Stimulating Cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional Accumulations of T Cells, Macrophages and Smooth Muscle Cells in the Human Atherosclerotic Plaque. Arteriosclerosis 1986, 6, 131–138. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Kaartinen, M.; Paavonen, T. Infiltrates of Activated Mast Cells at the Site of Coronary Atheromatous Erosion or Rupture in Myocardial Infarction. Circulation 1995, 92, 1084–1088. [Google Scholar] [CrossRef]
- Obikane, H.; Abiko, Y.; Ueno, H.; Kusumi, Y.; Esumi, M.; Mitsumata, M. Effect of Endothelial Cell Proliferation on Atherogenesis: A Role of p21(Sdi/Cip/Waf1) in Monocyte Adhesion to Endothelial Cells. Atherosclerosis 2010, 212, 116–122. [Google Scholar] [CrossRef]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in Atherosclerosis: Transition from Theory to Practice. Circ. J. 2010, 74, 213–220. [Google Scholar] [CrossRef]
- Verma, S.; Wang, C.H.; Li, S.H.; Dumont, A.S.; Fedak, P.W.; Badiwala, M.V.; Dhillon, B.; Weisel, R.D.; Li, R.K.; Mickle, D.A.; et al. A Self-Fulfilling Prophecy: C-Reactive Protein Attenuates Nitric Oxide Production and Inhibits Angiogenesis. Circulation 2002, 106, 913–919. [Google Scholar] [CrossRef]
- Quillard, T.; Tesmenitsky, Y.; Croce, K.; Travers, R.; Shvartz, E.; Koskinas, K.C.; Sukhova, G.K.; Aikawa, E.; Aikawa, M.; Libby, P. Selective Inhibition of Matrix Metalloproteinase-13 Increases Collagen Content of Established Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Sukhova, G.K.; Aikawa, M.; Canner, J.; Gerdes, N.; Tang, S.M.; Shi, G.P.; Apte, S.S.; Libby, P. Matrix-Metalloproteinase-14 Deficiency in Bone-Marrow-Derived Cells Promotes Collagen Accumulation in Mouse Atherosclerotic Plaques. Circulation 2008, 117, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Wu, Q.; Muszynski, M.; Hansson, G.K.; Libby, P. Apoptosis of Vascular Smooth Muscle Cells Induced by In Vitro Stimulation with Interferon-Gamma, Tumor Necrosis Factor-Alpha and Interleukin-1 Beta. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Houtkamp, M.A.; de Boer, O.J.; van der Loos, C.M.; van der Wal, A.C.; Becker, A.E. Adventitial Infiltrates Associated with Advanced Atherosclerotic Plaques: Structural Organization Suggests Generation of Local Humoral Immune Responses. J. Pathol. 2001, 193, 263–269. [Google Scholar] [CrossRef]
- Moos, M.P.; John, N.; Grabner, R.; Nossmann, S.; Gunther, B.; Vollandt, R.; Funk, C.D.; Kaiser, B.; Habenicht, A.J. The Lamina Adventitia is the Major Site of Immune Cell Accumulation in Standard Chow-fed Apolipoprotein E-Deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2386–2391. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Gordon, S. Monocyte Heterogeneity and Innate Immunity. Immunity 2003, 19, 2–4. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A. The Origin and Kinetics of Mononuclear Phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Camici, P.G.; Rimoldi, O.E.; Gaemperli, O.; Libby, P. Non-Invasive Anatomic and Functional Imaging of Vascular Inflammation and Unstable Plaque. Eur. Heart J. 2012, 33, 1309–1317. [Google Scholar] [CrossRef]
- Combadiere, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined Inhibition of CCL2, CX3CR1 and CCR5 Abrogates Ly6C(hi) and Ly6C(lo) Monocytosis and Almost Abolishes Atherosclerosis in Hypercholesterolemic Mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef]
- Takeya, M.; Yoshimura, T.; Leonard, E.J.; Takahashi, K. Detection of Monocyte Chemoattractant Protein-1 in Human Atherosclerotic Lesions by an Anti-Monocyte Chemoattractant Protein-1 Monoclonal Antibody. Hum. Pathol. 1993, 24, 534–539. [Google Scholar] [CrossRef]
- Yla-Herttuala, S.; Lipton, B.A.; Rosenfeld, M.E.; Sarkioja, T.; Yoshimura, T.; Leonard, E.J.; Witztum, J.L.; Steinberg, D. Expression of Monocyte Chemoattractant Protein 1 in Macrophage-Rich Areas of Human and Rabbit Atherosclerotic Lesions. Proc. Natl. Acad. Sci. USA 1991, 88, 5252–5256. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kakkar, V.; Lu, X. Impact of MCP-1 in Atherosclerosis. Curr. Pharm. Des. 2014, 20, 4580–4588. [Google Scholar] [CrossRef]
- Boisvert, W.A.; Rose, D.M.; Johnson, K.A.; Fuentes, M.E.; Lira, S.A.; Curtiss, L.K.; Terkeltaub, R.A. Up-Regulated Expression of the CXCR2 Ligand KC/GRO-Alpha in Atherosclerotic Lesions Plays a Central Role in Macrophage Accumulation and Lesion Progression. Am. J. Pathol. 2006, 168, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Liehn, E.A.; Gao, J.L.; Kuziel, W.A.; Murphy, P.M.; Weber, C. Deficiency in CCR5 but not CCR1 Protects Against Neointima Formation in Atherosclerosis-Prone Mice: Involvement of IL-10. Blood 2006, 107, 4240–4243. [Google Scholar] [CrossRef]
- Koenen, R.R.; von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting Functional Interactions between Platelet Chemokines Inhibits Atherosclerosis in Hyperlipidemic Mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef]
- Moroni, F.; Ammirati, E.; Norata, G.D.; Magnoni, M.; Camici, P.G. The Role of Monocytes and Macrophages in Human Atherosclerosis, Plaque Neoangiogenesis and Atherothrombosis. Mediat. Inflamm. 2019, 2019, 7434376. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. The CD14+ CD16+ Blood Monocytes: Their Role in Infection and Inflammation. J. Leukoc. Biol. 2007, 81, 584–592. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of Monocytes and Dendritic Cells in Blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Rogacev, K.S.; Cremers, B.; Zawada, A.M.; Seiler, S.; Binder, N.; Ege, P.; Grosse-Dunker, G.; Heisel, I.; Hornof, F.; Jeken, J.; et al. CD14++CD16+ Monocytes Independently Predict Cardiovascular Events: A Cohort Study of 951 Patients Referred for Elective Coronary Angiography. J. Am. Coll. Cardiol. 2012, 60, 1512–1520. [Google Scholar] [CrossRef]
- Sala, F.; Cutuli, L.; Grigore, L.; Pirillo, A.; Chiesa, G.; Catapano, A.L.; Norata, G.D. Prevalence of Classical CD14++/CD16- but not of Intermediate CD14++/CD16+ Monocytes in Hypoalphalipoproteinemia. Int. J. Cardiol. 2013, 168, 2886–2889. [Google Scholar] [CrossRef] [PubMed]
- Belge, K.U.; Dayyani, F.; Horelt, A.; Siedlar, M.; Frankenberger, M.; Frankenberger, B.; Espevik, T.; Ziegler-Heitbrock, L. The Proinflammatory CD14+CD16+DR++ Monocytes are a Major Source of TNF. J. Immunol. 2002, 168, 3536–3542. [Google Scholar] [CrossRef] [PubMed]
- Boltjes, A.; van Wijk, F. Human Dendritic Cell Functional Specialization in Steady-State and Inflammation. Front. Immunol. 2014, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Victora, G.D.; Schwickert, T.A.; Guermonprez, P.; Meredith, M.M.; Yao, K.; Chu, F.F.; Randolph, G.J.; Rudensky, A.Y.; Nussenzweig, M. In Vivo Analysis of Dendritic Cell Development and Homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, H.; Wong, W.C.; Sem, X.; Han, H.; Ong, S.M.; Tan, Y.C.; Yeap, W.H.; Gan, C.S.; Ng, K.Q.; et al. Identification of Novel Functional Differences in Monocyte Subsets Using Proteomic and Transcriptomic Methods. J. Proteome Res. 2009, 8, 4028–4038. [Google Scholar] [CrossRef]
- Evans, H.G.; Gullick, N.J.; Kelly, S.; Pitzalis, C.; Lord, G.M.; Kirkham, B.W.; Taams, L.S. In Vivo Activated Monocytes from the Site of Inflammation in Humans Specifically Promote Th17 Responses. Proc. Natl. Acad. Sci. USA 2009, 106, 6232–6237. [Google Scholar] [CrossRef]
- Serbina, N.V.; Jia, T.; Hohl, T.M.; Pamer, E.G. Monocyte-Mediated Defense against Microbial Pathogens. Annu. Rev. Immunol. 2008, 26, 421–452. [Google Scholar] [CrossRef]
- Avraham-Davidi, I.; Yona, S.; Grunewald, M.; Landsman, L.; Cochain, C.; Silvestre, J.S.; Mizrahi, H.; Faroja, M.; Strauss-Ayali, D.; Mack, M.; et al. On-Site Education of VEGF-Recruited Monocytes Improves Their Performance as Angiogenic and Arteriogenic Accessory Cells. J. Exp. Med. 2013, 210, 2611–2625. [Google Scholar] [CrossRef]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi Monocytes Dominate Hypercholesterolemia-Associated Monocytosis and Give Rise to Macrophages in Atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef]
- Justo-Junior, A.S.; Villarejos, L.M.; Lima, X.T.V.; Nadruz, W., Jr.; Sposito, A.C.; Mamoni, R.L.; Abdalla, R.; Fernandes, J.L.; Oliveira, R.T.D.; Blotta, M. Monocytes of Patients with Unstable Angina Express High Levels of Chemokine and Pattern-Recognition Receptors. Cytokine 2019, 113, 61–67. [Google Scholar] [CrossRef]
- Ozaki, Y.; Imanishi, T.; Hosokawa, S.; Nishiguchi, T.; Taruya, A.; Tanimoto, T.; Kuroi, A.; Yamano, T.; Matsuo, Y.; Ino, Y.; et al. Association of Toll-Like Receptor 4 on Human Monocyte Subsets and Vulnerability Characteristics of Coronary Plaque as Assessed by 64-Slice Multidetector Computed Tomography. Circ. J. 2017, 81, 837–845. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A Major Role for VCAM-1, but not ICAM-1, in Early Atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef]
- Sundell, C.L.; Somers, P.K.; Meng, C.Q.; Hoong, L.K.; Suen, K.L.; Hill, R.R.; Landers, L.K.; Chapman, A.; Butteiger, D.; Jones, M.; et al. AGI-1067: A Multifunctional Phenolic Antioxidant, Lipid Modulator, Anti-Inflammatory and Antiatherosclerotic Agent. J. Pharmacol. Exp. Ther. 2003, 305, 1116–1123. [Google Scholar] [CrossRef]
- Qiao, J.H.; Tripathi, J.; Mishra, N.K.; Cai, Y.; Tripathi, S.; Wang, X.P.; Imes, S.; Fishbein, M.C.; Clinton, S.K.; Libby, P.; et al. Role of Macrophage Colony-Stimulating Factor in Atherosclerosis: Studies of Osteopetrotic Mice. Am. J. Pathol. 1997, 150, 1687–1699. [Google Scholar]
- Nicola, N.A.; Metcalf, D. Specificity of Action of Colony-Stimulating Factors in the Differentiation of Granulocytes and Macrophages. Ciba Found. Symp. 1986, 118, 7–28. [Google Scholar]
- Woollard, K.J.; Geissmann, F. Monocytes in Atherosclerosis: Subsets and Functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef]
- Zhou, X.; Hansson, G.K. Detection of B Cells and Proinflammatory Cytokines in Atherosclerotic Plaques of Hypercholesterolaemic Apolipoprotein E Knockout Mice. Scand. J. Immunol. 1999, 50, 25–30. [Google Scholar] [CrossRef]
- Randolph, G.J. Mechanisms that Regulate Macrophage Burden in Atherosclerosis. Circ. Res. 2014, 114, 1757–1771. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte Subsets Differentially Employ CCR2, CCR5, and CX3CR1 to Accumulate within Atherosclerotic Plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local Proliferation Dominates Lesional Macrophage Accumulation in Atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Li, Q.; Park, K.; Xia, Y.; Matsumoto, M.; Qi, W.; Fu, J.; Yokomizo, H.; Khamaisi, M.; Wang, X.; Rask-Madsen, C.; et al. Regulation of Macrophage Apoptosis and Atherosclerosis by Lipid-Induced PKCdelta Isoform Activation. Circ. Res. 2017, 121, 1153–1167. [Google Scholar] [CrossRef]
- Shashkin, P.; Dragulev, B.; Ley, K. Macrophage Differentiation to Foam Cells. Curr. Pharm. Des. 2005, 11, 3061–3072. [Google Scholar] [CrossRef]
- Seimon, T.; Tabas, I. Mechanisms and Consequences of Macrophage Apoptosis in Atherosclerosis. J. Lipid. Res. 2009, 50 (Suppl.), S382–S387. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in Atherosclerotic Lesions: Malfunctioning Regulatory Pathways and Control Mechanisms. Pharmacol. Ther. 2018, 188, 12–25. [Google Scholar] [CrossRef]
- Thorp, E.; Subramanian, M.; Tabas, I. The Role of Macrophages and Dendritic Cells in the Clearance of Apoptotic Cells in Advanced Atherosclerosis. Eur. J. Immunol. 2011, 41, 2515–2518. [Google Scholar] [CrossRef]
- Thorp, E.; Tabas, I. Mechanisms and Consequences of Efferocytosis in Advanced Atherosclerosis. J. Leukoc. Biol. 2009, 86, 1089–1095. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage Activation and Polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Porcheray, F.; Viaud, S.; Rimaniol, A.C.; Leone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage Activation Switching: An Asset for the Resolution of Inflammation. Clin. Exp. Immunol. 2005, 142, 481–489. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef]
- Feig, J.E.; Rong, J.X.; Shamir, R.; Sanson, M.; Vengrenyuk, Y.; Liu, J.; Rayner, K.; Moore, K.; Garabedian, M.; Fisher, E.A. HDL Promotes Rapid Atherosclerosis Regression in Mice and Alters Inflammatory Properties of Plaque Monocyte-Derived Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7166–7171. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Joseph, P.; Tawakol, A. Imaging Atherosclerosis with Positron Emission Tomography. Eur. Heart J. 2016, 37, 2974–2980. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Phan, A.T.; Goldrath, A.W.; Glass, C.K. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity 2017, 46, 714–729. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and Human Dendritic Cell Subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef]
- Waldo, S.W.; Li, Y.; Buono, C.; Zhao, B.; Billings, E.M.; Chang, J.; Kruth, H.S. Heterogeneity of Human Macrophages in Culture and in Atherosclerotic Plaques. Am. J. Pathol. 2008, 172, 1112–1126. [Google Scholar] [CrossRef]
- Hoeve, M.A.; Savage, N.D.; de Boer, T.; Langenberg, D.M.; de Waal Malefyt, R.; Ottenhoff, T.H.; Verreck, F.A. Divergent Effects of IL-12 and IL-23 on the Production of IL-17 by Human T Cells. Eur. J. Immunol. 2006, 36, 661–670. [Google Scholar] [CrossRef]
- Brocheriou, I.; Maouche, S.; Durand, H.; Braunersreuther, V.; Le Naour, G.; Gratchev, A.; Koskas, F.; Mach, F.; Kzhyshkowska, J.; Ninio, E. Antagonistic Regulation of Macrophage Phenotype by M-CSF and GM-CSF: Implication in Atherosclerosis. Atherosclerosis 2011, 214, 316–324. [Google Scholar] [CrossRef]
- Burgess, A.W.; Metcalf, D. The Nature and Action of Granulocyte-Macrophage Colony Stimulating Factors. Blood 1980, 56, 947–958. [Google Scholar] [CrossRef]
- Gasson, J.C. Molecular Physiology of Granulocyte-Macrophage Colony-Stimulating Factor. Blood 1991, 77, 1131–1145. [Google Scholar] [CrossRef]
- Plenz, G.; Koenig, C.; Severs, N.J.; Robenek, H. Smooth Muscle Cells Express Granulocyte-Macrophage Colony-Stimulating Factor in the Undiseased and Atherosclerotic Human Coronary Artery. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2489–2499. [Google Scholar] [CrossRef]
- Lacave-Lapalun, J.V.; Benderitter, M.; Linard, C. Flagellin or Lipopolysaccharide Treatment Modified Macrophage Populations after Colorectal Radiation of Rats. J. Pharmacol. Exp. Ther. 2013, 346, 75–85. [Google Scholar] [CrossRef]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-Producing Type 1 Macrophages Promote but IL-10-Producing Type 2 Macrophages Subvert Immunity to (myco)Bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef]
- Mosser, D.M. The Many Faces of Macrophage Activation. J. Leukoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Sobenin, I.A.; Orekhov, A.N. Macrophage Phenotypic Plasticity in Atherosclerosis: The Associated Features and the Peculiarities of the Expression of Inflammatory Genes. Int. J. Cardiol. 2015, 184, 436–445. [Google Scholar] [CrossRef]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Changes in Transcriptome of Macrophages in Atherosclerosis. J. Cell. Mol. Med. 2015, 19, 1163–1173. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-like Phenotype is Independent of Interleukin-4 Receptor Alpha (IL-4Ralpha) Signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Anderson, C.F.; Gerber, J.S.; Mosser, D.M. Modulating Macrophage Function with IgG Immune Complexes. J. Endotoxin Res. 2002, 8, 477–481. [Google Scholar] [CrossRef]
- Kadl, A.; Sharma, P.R.; Chen, W.; Agrawal, R.; Meher, A.K.; Rudraiah, S.; Grubbs, N.; Sharma, R.; Leitinger, N. Oxidized Phospholipid-Induced Inflammation is Mediated by Toll-Like Receptor 2. Free Radic. Biol. Med. 2011, 51, 1903–1909. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a Novel Macrophage Phenotype that Develops in Response to Atherogenic Phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Marques, L.; Negre-Salvayre, A.; Costa, L.; Canonne-Hergaux, F. Iron Gene Expression Profile in Atherogenic Mox Macrophages. Biochim. Biophys. Acta 2016, 1862, 1137–1146. [Google Scholar] [CrossRef]
- Liberale, L.; Dallegri, F.; Montecucco, F.; Carbone, F. Pathophysiological Relevance of Macrophage Subsets in Atherogenesis. Thromb. Haemost. 2017, 117, 7–18. [Google Scholar] [CrossRef]
- Gill, N.; Leng, Y.; Romero, R.; Xu, Y.; Panaitescu, B.; Miller, D.; Arif, A.; Mumuni, S.; Qureshi, F.; Hsu, C.D.; et al. The Immunophenotype of Decidual Macrophages in Acute Atherosis. Am. J. Reprod. Immunol. 2019, 81, e13098. [Google Scholar] [CrossRef]
- Kockx, M.M.; Cromheeke, K.M.; Knaapen, M.W.; Bosmans, J.M.; De Meyer, G.R.; Herman, A.G.; Bult, H. Phagocytosis and Macrophage Activation Associated with Hemorrhagic Microvessels in Human Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Macrophages and Systemic Iron Homeostasis. J. Innate Immun. 2012, 4, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J.; Johns, M.; Kampfer, T.; Nguyen, A.T.; Game, L.; Schaer, D.J.; Mason, J.C.; Haskard, D.O. Activating Transcription Factor 1 Directs Mhem Atheroprotective Macrophages through Coordinated Iron Handling and Foam Cell Protection. Circ. Res. 2012, 110, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Finn, A.V. The Role of Iron Metabolism as a Mediator of Macrophage Inflammation and Lipid Handling in Atherosclerosis. Front. Pharmacol. 2014, 5, 195. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Moller, H.J.; Moestrup, S.K. Hemoglobin and Heme Scavenger Receptors. Antioxid. Redox Signal. 2010, 12, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Huo, Y.; Johns, M.; Piper, E.; Mason, J.C.; Carling, D.; Haskard, D.O.; Boyle, J.J. 5’-AMP-Activated Protein Kinase-Activating Transcription Factor 1 Cascade Modulates Human Monocyte-Derived Macrophages to Atheroprotective Functions in Response to Heme or Metformin. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2470–2480. [Google Scholar] [CrossRef]
- Boyle, J.J. Heme and Haemoglobin Direct Macrophage Mhem Phenotype and Counter Foam Cell Formation in Areas of Intraplaque Haemorrhage. Curr. Opin. Lipidol. 2012, 23, 453–461. [Google Scholar] [CrossRef]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary Intraplaque Hemorrhage Evokes a Novel Atheroprotective Macrophage Phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef]
- Pitsilos, S.; Hunt, J.; Mohler, E.R.; Prabhakar, A.M.; Poncz, M.; Dawicki, J.; Khalapyan, T.Z.; Wolfe, M.L.; Fairman, R.; Mitchell, M.; et al. Platelet Factor 4 Localization in Carotid Atherosclerotic Plaques: Correlation with Clinical Parameters. Thromb. Haemost. 2003, 90, 1112–1120. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Ley, K. CXCL4 in Atherosclerosis: Possible Roles in Monocyte Arrest and Macrophage Foam Cell Formation. Thromb. Haemost. 2007, 98, 917–918. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage Subsets in Atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC Chemokine Ligand 4 Induces a Unique Transcriptome in Monocyte-Derived Macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Okuyucu, D.; Akhavanpoor, M.; Zhao, L.; Wangler, S.; Hakimi, M.; Doesch, A.; Dengler, T.J.; Katus, H.A.; Gleissner, C.A. A Human Ex Vivo Atherosclerotic Plaque Model to Study Lesion Biology. J. Vis. Exp. 2014, 87. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, F.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage Phenotypes and Their Modulation in Atherosclerosis. Circ. J. 2014, 78, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages and Immune Cells in Atherosclerosis: Recent Advances and Novel Concepts. Basic Res. Cardiol. 2015, 110, 34. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-Inflammatory Properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Erbel, C.; Bockler, D.; Katus, H.A.; Ley, K. CXCL4 Downregulates the Atheroprotective Hemoglobin Receptor CD163 in Human Macrophages. Circ. Res. 2010, 106, 203–211. [Google Scholar] [CrossRef]
- Stoger, J.L.; Gijbels, M.J.; van der Velden, S.; Manca, M.; van der Loos, C.M.; Biessen, E.A.; Daemen, M.J.; Lutgens, E.; de Winther, M.P. Distribution of Macrophage Polarization Markers in Human Atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human Atherosclerotic Plaque Alternative Macrophages Display Low Cholesterol Handling but High Phagocytosis Because of Distinct Activities of the PPARgamma and LXRalpha Pathways. Circ. Res. 2011, 108, 985–995. [Google Scholar] [CrossRef]
- Pirro, M.; Schillaci, G.; Savarese, G.; Gemelli, F.; Mannarino, M.R.; Siepi, D.; Bagaglia, F.; Mannarino, E. Attenuation of inflammation with short-term dietary intervention is associated with a reduction of arterial stiffness in subjects with hypercholesterolaemia. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Singer, K.; Lumeng, C.N. Adipose tissue macrophages: Phenotypic plasticity and diversity in lean and obese states. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Llodra, J.; Angeli, V.; Liu, J.; Trogan, E.; Fisher, E.A.; Randolph, G.J. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. USA 2004, 101, 11779–11784. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharea, A.; Lee, M.K.; Moore, X.L.; Fang, L.; Sviridov, D.; Chin-Dusting, J.; Andrews, K.L.; Murphy, A.J. Native LDL promotes differentiation of human monocytes to macrophages with an inflammatory phenotype. Thromb Haemost 2016, 115, 762–772. [Google Scholar]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1beta by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem Biophys Res Commun 2012, 425, 121–126. [Google Scholar] [CrossRef]
- van Tits, L.J.; Stienstra, R.; van Lent, P.L.; Netea, M.G.; Joosten, L.A.; Stalenhoef, A.F. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: A crucial role for Kruppel-like factor 2. Atherosclerosis 2011, 214, 345–349. [Google Scholar] [CrossRef]
- Hirata, Y.; Tabata, M.; Kurobe, H.; Motoki, T.; Akaike, M.; Nishio, C.; Higashida, M.; Mikasa, H.; Nakaya, Y.; Takanashi, S.; et al. Coronary atherosclerosis is associated with macrophage polarization in epicardial adipose tissue. J. Am. Coll. Cardiol. 2011, 58, 248–255. [Google Scholar] [CrossRef]
- Hirata, Y.; Kurobe, H.; Akaike, M.; Chikugo, F.; Hori, T.; Bando, Y.; Nishio, C.; Higashida, M.; Nakaya, Y.; Kitagawa, T.; et al. Enhanced inflammation in epicardial fat in patients with coronary artery disease. Int. Heart J. 2011, 52, 139–142. [Google Scholar] [CrossRef]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Wootton, R.; Lewis, B. Selective retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo. Molecular size as a determinant of fractional loss from the intima-inner media. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.; Wootton, R.; Nordestgaard, B.G.; Baskerville, P.; Lumley, J.S.; La Ville, A.E.; Quiney, J.; Lewis, B. Quantitative studies of transfer in vivo of low density, Sf 12-60, and Sf 60-400 lipoproteins between plasma and arterial intima in humans. Arterioscler. Thromb. 1991, 11, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Houde, M.; Van Eck, M. Escaping the atherogenic trap: Preventing LDL fusion and binding in the intima. Atherosclerosis 2018, 275, 376–378. [Google Scholar] [CrossRef]
- Oorni, K.; Pentikainen, M.O.; Ala-Korpela, M.; Kovanen, P.T. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: Molecular mechanisms and effects on matrix interactions. J. Lipid Res. 2000, 41, 1703–1714. [Google Scholar]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial Barrier and Its Abnormalities in Cardiovascular Disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef]
- Hutchins, P.M.; Heinecke, J.W. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr. Opin. Lipidol. 2015, 26, 388–393. [Google Scholar] [CrossRef]
- Lao, K.H.; Zeng, L.; Xu, Q. Endothelial and smooth muscle cell transformation in atherosclerosis. Curr. Opin. Lipidol. 2015, 26, 449–456. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V. Monocyte recruitment and foam cell formation in atherosclerosis. Micron 2006, 37, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.L.; Scherer, P.E.; Lodish, H.F.; Krieger, M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J. Biol. Chem. 1994, 269, 21003–21009. [Google Scholar]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar]
- Kodama, T.; Freeman, M.; Rohrer, L.; Zabrecky, J.; Matsudaira, P.; Krieger, M. Type I macrophage scavenger receptor contains alpha-helical and collagen-like coiled coils. Nature 1990, 343, 531–535. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef]
- Coller, S.P.; Paulnock, D.M. Signaling pathways initiated in macrophages after engagement of type A scavenger receptors. J. Leukoc. Biol. 2001, 70, 142–148. [Google Scholar]
- Agrawal, S.; Febbraio, M.; Podrez, E.; Cathcart, M.K.; Stark, G.R.; Chisolm, G.M. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation 2007, 115, 2939–2947. [Google Scholar] [CrossRef]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef]
- Kume, N.; Moriwaki, H.; Kataoka, H.; Minami, M.; Murase, T.; Sawamura, T.; Masaki, T.; Kita, T. Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Ann. N. Y. Acad. Sci. 2000, 902, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Early steps in reverse cholesterol transport: Cholesteryl ester hydrolase and other hydrolases. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Chroni, A.; Tietge, U.J.; Zanotti, I.; Escola-Gil, J.C.; Bernini, F. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 2015, 224, 181–206. [Google Scholar] [PubMed]
- da Silva, R.F.; Lappalainen, J.; Lee-Rueckert, M.; Kovanen, P.T. Conversion of human M-CSF macrophages into foam cells reduces their proinflammatory responses to classical M1-polarizing activation. Atherosclerosis 2016, 248, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Zhang, Q.H.; Chen, Z.; He, Z.J.; Yi, G.H. Oxidized low-density lipoprotein attenuated desmoglein 1 and desmocollin 2 expression via LOX-1/Ca(2+)/PKC-beta signal in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2015, 468, 380–386. [Google Scholar] [CrossRef] [PubMed]
- van Nieuw Amerongen, G.P.; Vermeer, M.A.; Negre-Aminou, P.; Lankelma, J.; Emeis, J.J.; van Hinsbergh, V.W. Simvastatin improves disturbed endothelial barrier function. Circulation 2000, 102, 2803–2809. [Google Scholar] [CrossRef]
- Kasa, A.; Csortos, C.; Verin, A.D. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015, 3, e974448. [Google Scholar] [CrossRef]
- Syed, S.E.; Trinnaman, B.; Martin, S.; Major, S.; Hutchinson, J.; Magee, A.I. Molecular interactions between desmosomal cadherins. Biochem. J. 2002, 362, 317–327. [Google Scholar] [CrossRef]
- Ben, J.; Zhu, X.; Zhang, H.; Chen, Q. Class A1 scavenger receptors in cardiovascular diseases. Br. J. Pharmacol. 2015, 172, 5523–5530. [Google Scholar] [CrossRef]
- Murphy, J.E.; Tedbury, P.R.; Homer-Vanniasinkam, S.; Walker, J.H.; Ponnambalam, S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis 2005, 182, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Freeman, M.W. Scavenger receptors in atherosclerosis: Beyond lipid uptake. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Makinen, P.I.; Lappalainen, J.P.; Heinonen, S.E.; Leppanen, P.; Lahteenvuo, M.T.; Aarnio, J.V.; Heikkila, J.; Turunen, M.P.; Yla-Herttuala, S. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc. Res. 2010, 88, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Y.; Cai, Y.; Mao, D.D.; Qi, Y.F.; Tang, C.; Xu, Q.; Zhu, Y.; Xu, M.J.; Wang, X. Increased stability of phosphatase and tensin homolog by intermedin leading to scavenger receptor A inhibition of macrophages reduces atherosclerosis in apolipoprotein E-deficient mice. J. Mol. Cell. Cardiol. 2012, 53, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kurihara, Y.; Takeya, M.; Kamada, N.; Kataoka, M.; Jishage, K.; Ueda, O.; Sakaguchi, H.; Higashi, T.; Suzuki, T.; et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 1997, 386, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Takeya, M.; Suzuki, H.; Hakamata, H.; Kodama, T.; Horiuchi, S.; Gordon, S.; van der Laan, L.J.; Kraal, G.; Ishibashi, S.; et al. Role of macrophage scavenger receptors in diet-induced atherosclerosis in mice. Lab. Investig. 1998, 78, 423–434. [Google Scholar] [PubMed]
- Babaev, V.R.; Gleaves, L.A.; Carter, K.J.; Suzuki, H.; Kodama, T.; Fazio, S.; Linton, M.F. Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2593–2599. [Google Scholar] [CrossRef]
- Hashizume, M.; Mihara, M. Blockade of IL-6 and TNF-alpha inhibited oxLDL-induced production of MCP-1 via scavenger receptor induction. Eur. J. Pharmacol. 2012, 689, 249–254. [Google Scholar] [CrossRef]
- Zhao, J.F.; Ching, L.C.; Huang, Y.C.; Chen, C.Y.; Chiang, A.N.; Kou, Y.R.; Shyue, S.K.; Lee, T.S. Molecular mechanism of curcumin on the suppression of cholesterol accumulation in macrophage foam cells and atherosclerosis. Mol. Nutr. Food Res. 2012, 56, 691–701. [Google Scholar] [CrossRef]
- Van Berkel, T.J.; Van Eck, M.; Herijgers, N.; Fluiter, K.; Nion, S. Scavenger receptor classes A and B. Their roles in atherogenesis and the metabolism of modified LDL and HDL. Ann. N. Y. Acad. Sci. 2000, 902, 113–126. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.S.; Barrett, L.; Little, C.; Grant, M.D. Harnessing CD36 to rein in inflammation. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Hrboticky, N.; Draude, G.; Hapfelmeier, G.; Lorenz, R.; Weber, P.C. Lovastatin decreases the receptor-mediated degradation of acetylated and oxidized LDLs in human blood monocytes during the early stage of differentiation into macrophages. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1267–1275. [Google Scholar] [CrossRef]
- Fuhrman, B.; Koren, L.; Volkova, N.; Keidar, S.; Hayek, T.; Aviram, M. Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis 2002, 164, 179–185. [Google Scholar] [CrossRef]
- Geloen, A.; Helin, L.; Geeraert, B.; Malaud, E.; Holvoet, P.; Marguerie, G. CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis. PLoS ONE 2012, 7, e37633. [Google Scholar] [CrossRef] [PubMed]
- Mansor, L.S.; Sousa Fialho, M.D.L.; Yea, G.; Coumans, W.A.; West, J.A.; Kerr, M.; Carr, C.A.; Luiken, J.; Glatz, J.F.C.; Evans, R.D.; et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc. Res. 2017, 113, 737–748. [Google Scholar] [CrossRef]
- Mimche, P.N.; Brady, L.M.; Keeton, S.; Fenne, D.S.; King, T.P.; Quicke, K.M.; Hudson, L.E.; Lamb, T.J. Expression of the Receptor Tyrosine Kinase EphB2 on Dendritic Cells Is Modulated by Toll-Like Receptor Ligation but Is Not Required for T Cell Activation. PLoS ONE 2015, 10, e0138835. [Google Scholar] [CrossRef]
- Li, L.; Sawamura, T.; Renier, G. Glucose enhances human macrophage LOX-1 expression: Role for LOX-1 in glucose-induced macrophage foam cell formation. Circ. Res. 2004, 94, 892–901. [Google Scholar] [CrossRef]
- Gao, D.; Pararasa, C.; Dunston, C.R.; Bailey, C.J.; Griffiths, H.R. Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic. Biol. Med. 2012, 53, 796–806. [Google Scholar] [CrossRef]
- Li, X.Y.; Wang, C.; Xiang, X.R.; Chen, F.C.; Yang, C.M.; Wu, J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol. Rep. 2013, 30, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Bae, J.Y.; Kim, D.S.; Li, J.; Kim, J.L.; Lee, Y.J.; Kang, Y.H. Dietary compound quercitrin dampens VEGF induction and PPARgamma activation in oxidized LDL-exposed murine macrophages: Association with scavenger receptor CD36. J. Agric. Food Chem. 2010, 58, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.T.; Cao, Y.; Wang, T.Q.; Wang, L.J.; Guo, J.; Zhou, X.S.; Xu, S.W.; Liu, W.H.; Liu, P.Q.; Huang, H.Q. Tanshinone IIA attenuates atherosclerosis in ApoE(-/-) mice through down-regulation of scavenger receptor expression. Eur. J. Pharmacol. 2011, 650, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; Quiles, J.L.; Ramirez-Tortosa, C.L.; Ochoa-Herrera, J.; Perez-Lopez, P.; Pulido-Moran, M.; Ramirez-Tortosa, M.C. Squalene ameliorates atherosclerotic lesions through the reduction of CD36 scavenger receptor expression in macrophages. Mol. Nutr. Food Res. 2012, 56, 733–740. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Moore, K.J.; Kunjathoor, V.V.; Koehn, S.L.; Manning, J.J.; Tseng, A.A.; Silver, J.M.; McKee, M.; Freeman, M.W. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J. Clin. Investig. 2005, 115, 2192–2201. [Google Scholar] [CrossRef]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Moriwaki, H.; Murase, T.; Sawamura, T.; Masaki, T.; Hashimoto, N.; Kita, T. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999, 99, 3110–3117. [Google Scholar] [CrossRef]
- Schaeffer, D.F.; Riazy, M.; Parhar, K.S.; Chen, J.H.; Duronio, V.; Sawamura, T.; Steinbrecher, U.P. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J. Lipid Res. 2009, 50, 1676–1684. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef]
- Inoue, K.; Arai, Y.; Kurihara, H.; Kita, T.; Sawamura, T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ. Res. 2005, 97, 176–184. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Shmookler Reis, R.J.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef]
- Ishino, S.; Mukai, T.; Kume, N.; Asano, D.; Ogawa, M.; Kuge, Y.; Minami, M.; Kita, T.; Shiomi, M.; Saji, H. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability--analysis in hypercholesterolemic rabbits. Atherosclerosis 2007, 195, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Kuge, Y.; Kume, N.; Ishino, S.; Takai, N.; Ogawa, Y.; Mukai, T.; Minami, M.; Shiomi, M.; Saji, H. Prominent lectin-like oxidized low density lipoprotein (LDL) receptor-1 (LOX-1) expression in atherosclerotic lesions is associated with tissue factor expression and apoptosis in hypercholesterolemic rabbits. Biol. Pharm. Bull. 2008, 31, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Fazio, S.; Major, A.S.; Swift, L.L.; Gleaves, L.A.; Accad, M.; Linton, M.F.; Farese, R.V., Jr. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J. Clin. Investig. 2001, 107, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Accad, M.; Smith, S.J.; Newland, D.L.; Sanan, D.A.; King, L.E., Jr.; Linton, M.F.; Fazio, S.; Farese, R.V., Jr. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J. Clin. Investig. 2000, 105, 711–719. [Google Scholar] [CrossRef][Green Version]
- Perrey, S.; Legendre, C.; Matsuura, A.; Guffroy, C.; Binet, J.; Ohbayashi, S.; Tanaka, T.; Ortuno, J.C.; Matsukura, T.; Laugel, T.; et al. Preferential pharmacological inhibition of macrophage ACAT increases plaque formation in mouse and rabbit models of atherogenesis. Atherosclerosis 2001, 155, 359–370. [Google Scholar] [CrossRef]
- Chang, C.C.; Sakashita, N.; Ornvold, K.; Lee, O.; Chang, E.T.; Dong, R.; Lin, S.; Lee, C.Y.; Strom, S.C.; Kashyap, R.; et al. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J. Biol. Chem. 2000, 275, 28083–28092. [Google Scholar]
- Cheng, B.; Wan, J.; Wang, Y.; Mei, C.; Liu, W.; Ke, L.; He, P. Ghrelin inhibits foam cell formation via simultaneously down-regulating the expression of acyl-coenzyme A:cholesterol acyltransferase 1 and up-regulating adenosine triphosphate-binding cassette transporter A1. Cardiovasc. Pathol. 2010, 19, e159–e166. [Google Scholar] [CrossRef]
- Nagashima, M.; Watanabe, T.; Terasaki, M.; Tomoyasu, M.; Nohtomi, K.; Kim-Kaneyama, J.; Miyazaki, A.; Hirano, T. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia 2011, 54, 2649–2659. [Google Scholar] [CrossRef]
- Darsalia, V.; Larsson, M.; Klein, T.; Patrone, C. The high need for trials assessing functional outcome after stroke rather than stroke prevention with GLP-1 agonists and DPP-4 inhibitors. Cardiovasc. Diabetol. 2018, 17, 32. [Google Scholar] [CrossRef]
- Ge, J.; Zhai, W.; Cheng, B.; He, P.; Qi, B.; Lu, H.; Zeng, Y.; Chen, X. Insulin induces human acyl-coenzyme A: Cholesterol acyltransferase1 gene expression via MAP kinases and CCAAT/enhancer-binding protein alpha. J. Cell. Biochem. 2013, 114, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Hongo, S.; Watanabe, T.; Arita, S.; Kanome, T.; Kageyama, H.; Shioda, S.; Miyazaki, A. Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E474–E482. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Osuga, J.; Isshiki, M.; Sekiya, M.; Okazaki, H.; Takase, S.; Takanashi, M.; Ohta, K.; Kumagai, M.; Nishi, M.; et al. Targeting of neutral cholesterol ester hydrolase to the endoplasmic reticulum via its N-terminal sequence. J. Lipid Res. 2010, 51, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Song, J.; Chow, W.N.; St Clair, R.W.; Rudel, L.L.; Ghosh, S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J. Clin. Investig. 2007, 117, 2983–2992. [Google Scholar] [CrossRef]
- Igarashi, M.; Osuga, J.; Uozaki, H.; Sekiya, M.; Nagashima, S.; Takahashi, M.; Takase, S.; Takanashi, M.; Li, Y.; Ohta, K.; et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ. Res. 2010, 107, 1387–1395. [Google Scholar] [CrossRef]
- Sekiya, M.; Osuga, J.; Nagashima, S.; Ohshiro, T.; Igarashi, M.; Okazaki, H.; Takahashi, M.; Tazoe, F.; Wada, T.; Ohta, K.; et al. Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab. 2009, 10, 219–228. [Google Scholar] [CrossRef]
- Sakai, K.; Igarashi, M.; Yamamuro, D.; Ohshiro, T.; Nagashima, S.; Takahashi, M.; Enkhtuvshin, B.; Sekiya, M.; Okazaki, H.; Osuga, J.; et al. Critical role of neutral cholesteryl ester hydrolase 1 in cholesteryl ester hydrolysis in murine macrophages. J. Lipid Res. 2014, 55, 2033–2040. [Google Scholar] [CrossRef]
- Sekiya, M.; Osuga, J.; Igarashi, M.; Okazaki, H.; Ishibashi, S. The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J. Atheroscler Thromb 2011, 18, 359–364. [Google Scholar] [CrossRef]
- Zhao, Y.; Pennings, M.; Vrins, C.L.; Calpe-Berdiel, L.; Hoekstra, M.; Kruijt, J.K.; Ottenhoff, R.; Hildebrand, R.B.; van der Sluis, R.; Jessup, W.; et al. Hypocholesterolemia, foam cell accumulation, but no atherosclerosis in mice lacking ABC-transporter A1 and scavenger receptor BI. Atherosclerosis 2011, 218, 314–322. [Google Scholar] [CrossRef]
- Joyce, C.W.; Wagner, E.M.; Basso, F.; Amar, M.J.; Freeman, L.A.; Shamburek, R.D.; Knapper, C.L.; Syed, J.; Wu, J.; Vaisman, B.L.; et al. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J. Biol. Chem. 2006, 281, 33053–33065. [Google Scholar] [CrossRef]
- Baldan, A.; Pei, L.; Lee, R.; Tarr, P.; Tangirala, R.K.; Weinstein, M.M.; Frank, J.; Li, A.C.; Tontonoz, P.; Edwards, P.A. Impaired development of atherosclerosis in hyperlipidemic Ldlr-/- and ApoE-/- mice transplanted with Abcg1-/- bone marrow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Meurs, I.; Lammers, B.; Zhao, Y.; Out, R.; Hildebrand, R.B.; Hoekstra, M.; Van Berkel, T.J.; Van Eck, M. The effect of ABCG1 deficiency on atherosclerotic lesion development in LDL receptor knockout mice depends on the stage of atherogenesis. Atherosclerosis 2012, 221, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yancey, P.G.; Su, Y.R.; Babaev, V.R.; Zhang, Y.; Fazio, S.; Linton, M.F. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2003, 108, 2258–2263. [Google Scholar] [CrossRef]
- Bennett, D.J.; Cooke, A.J.; Edwards, A.S. Non-steroidal LXR agonists; an emerging therapeutic strategy for the treatment of atherosclerosis. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 21–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Moon, J.; Cho, Y.; Chung, J.H.; Shin, M.J. Quercetin up-regulates expressions of peroxisome proliferator-activated receptor gamma, liver X receptor alpha, and ATP binding cassette transporter A1 genes and increases cholesterol efflux in human macrophage cell line. Nutr. Res. 2013, 33, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Ayaori, M.; Terao, Y.; Hisada, T.; Iizuka, M.; Takiguchi, S.; Uto-Kondo, H.; Yakushiji, E.; Nakaya, K.; Sasaki, M.; et al. Proteasomal inhibition promotes ATP-binding cassette transporter A1 (ABCA1) and ABCG1 expression and cholesterol efflux from macrophages in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1980–1987. [Google Scholar] [CrossRef]
- Tang, C.K.; Tang, G.H.; Yi, G.H.; Wang, Z.; Liu, L.S.; Wan, S.; Yuan, Z.H.; He, X.S.; Yang, J.H.; Ruan, C.G.; et al. Effect of apolipoprotein A-I on ATP binding cassette transporter A1 degradation and cholesterol efflux in THP-1 macrophage-derived foam cells. Acta Biochim. Biophys. Sin. (Shanghai) 2004, 36, 218–226. [Google Scholar] [CrossRef]
- Rousselle, A.; Qadri, F.; Leukel, L.; Yilmaz, R.; Fontaine, J.F.; Sihn, G.; Bader, M.; Ahluwalia, A.; Duchene, J. CXCL5 limits macrophage foam cell formation in atherosclerosis. J. Clin. Investig. 2013, 123, 1343–1347. [Google Scholar] [CrossRef]
- Santamarina-Fojo, S.; Remaley, A.T.; Neufeld, E.B.; Brewer, H.B., Jr. Regulation and intracellular trafficking of the ABCA1 transporter. J. Lipid Res. 2001, 42, 1339–1345. [Google Scholar]
- Wang, Y.; Oram, J.F. Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a protein kinase C delta pathway. J. Lipid Res. 2007, 48, 1062–1068. [Google Scholar] [CrossRef]
- Ku, C.S.; Park, Y.; Coleman, S.L.; Lee, J. Unsaturated fatty acids repress expression of ATP binding cassette transporter A1 and G1 in RAW 264.7 macrophages. J. Nutr. Biochem. 2012, 23, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Jiang, H.L.; Chen, W.J.; Yin, K.; Zhao, G.J.; Mo, Z.C.; Ouyang, X.P.; Lv, Y.C.; Jiang, Z.S.; Zhang, D.W.; et al. Interleukin-18 and interleukin-12 together downregulate ATP-binding cassette transporter A1 expression through the interleukin-18R/nuclear factor-kappaB signaling pathway in THP-1 macrophage-derived foam cells. Circ. J. 2012, 76, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; Hoang, M.H.; Yeo, S.K.; Jia, Y.; Lee, S.J. Induction of ABCA1 and ABCG1 expression by the liver X receptor modulator cineole in macrophages. Bioorg Med. Chem. Lett. 2013, 23, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Helal, O.; Berrougui, H.; Loued, S.; Khalil, A. Extra-virgin olive oil consumption improves the capacity of HDL to mediate cholesterol efflux and increases ABCA1 and ABCG1 expression in human macrophages. Br. J. Nutr. 2013, 109, 1844–1855. [Google Scholar] [CrossRef]
- Wang, D.; Xia, M.; Yan, X.; Li, D.; Wang, L.; Xu, Y.; Jin, T.; Ling, W. Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA-10b. Circ. Res. 2012, 111, 967–981. [Google Scholar] [CrossRef]
- Uto-Kondo, H.; Ayaori, M.; Ogura, M.; Nakaya, K.; Ito, M.; Suzuki, A.; Takiguchi, S.; Yakushiji, E.; Terao, Y.; Ozasa, H.; et al. Coffee consumption enhances high-density lipoprotein-mediated cholesterol efflux in macrophages. Circ. Res. 2010, 106, 779–787. [Google Scholar] [CrossRef]
- Kammerer, I.; Ringseis, R.; Biemann, R.; Wen, G.; Eder, K. 13-hydroxy linoleic acid increases expression of the cholesterol transporters ABCA1, ABCG1 and SR-BI and stimulates apoA-I-dependent cholesterol efflux in RAW264.7 macrophages. Lipids Health Dis. 2011, 10, 222. [Google Scholar] [CrossRef]
- Voloshyna, I.; Hai, O.; Littlefield, M.J.; Carsons, S.; Reiss, A.B. Resveratrol mediates anti-atherogenic effects on cholesterol flux in human macrophages and endothelium via PPARgamma and adenosine. Eur. J. Pharmacol. 2013, 698, 299–309. [Google Scholar] [CrossRef]
- Tang, S.L.; Chen, W.J.; Yin, K.; Zhao, G.J.; Mo, Z.C.; Lv, Y.C.; Ouyang, X.P.; Yu, X.H.; Kuang, H.J.; Jiang, Z.S.; et al. PAPP-A negatively regulates ABCA1, ABCG1 and SR-B1 expression by inhibiting LXRalpha through the IGF-I-mediated signaling pathway. Atherosclerosis 2012, 222, 344–354. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Libby, P.; Aikawa, M.; Schonbeck, U. Cholesterol and atherosclerosis. Biochim. Biophys. Acta 2000, 1529, 299–309. [Google Scholar] [CrossRef]
- Sottero, B.; Gamba, P.; Longhi, M.; Robbesyn, F.; Abuja, P.M.; Schaur, R.J.; Poli, G.; Leonarduzzi, G. Expression and synthesis of TGFbeta1 is induced in macrophages by 9-oxononanoyl cholesterol, a major cholesteryl ester oxidation product. Biofactors 2005, 24, 209–216. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.; Duffy, M.M.; Mooney, D.; James, W.G.; Griffin, M.D.; Fitzgerald, D.J.; Belton, O. IL-10 mediates the immunoregulatory response in conjugated linoleic acid-induced regression of atherosclerosis. FASEB J. 2013, 27, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Zernecke, A. Dendritic cells in atherosclerosis: Evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef]
- Dutertre, C.A.; Wang, L.F.; Ginhoux, F. Aligning bona fide dendritic cell populations across species. Cell. Immunol. 2014, 291, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Helft, J.; Chow, A.; Hashimoto, D.; Mortha, A.; Agudo-Cantero, J.; Bogunovic, M.; Gautier, E.L.; Miller, J.; Leboeuf, M.; et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 2012, 36, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. Macrophages as APC and the dendritic cell myth. J. Immunol. 2008, 181, 5829–5835. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Haidari, M.; Zhu, S.N.; Chen, M.; Guha, D.; Cybulsky, M.I. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 2006, 203, 2073–2083. [Google Scholar] [CrossRef]
- Paulson, K.E.; Zhu, S.N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 2010, 106, 383–390. [Google Scholar] [CrossRef]
- Choi, J.H.; Cheong, C.; Dandamudi, D.B.; Park, C.G.; Rodriguez, A.; Mehandru, S.; Velinzon, K.; Jung, I.H.; Yoo, J.Y.; Oh, G.T.; et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 2011, 35, 819–831. [Google Scholar] [CrossRef]
- Choi, J.H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 2009, 206, 497–505. [Google Scholar] [CrossRef]
- Roufaiel, M.; Gracey, E.; Siu, A.; Zhu, S.N.; Lau, A.; Ibrahim, H.; Althagafi, M.; Tai, K.; Hyduk, S.J.; Cybulsky, K.O.; et al. CCL19-CCR7-dependent reverse transendothelial migration of myeloid cells clears Chlamydia muridarum from the arterial intima. Nat. Immunol. 2016, 17, 1263–1272. [Google Scholar] [CrossRef]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Martino, M.; Sutherland, C.L.; Gold, M.R.; Ricciardi-Castagnoli, P. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 1998, 188, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Pflicke, H.; Sixt, M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 2009, 206, 2925–2935. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.F.; Lukacs-Kornek, V.; Kuligowski, M.P.; Pitcher, L.A.; Degn, S.E.; Kim, Y.A.; Cloninger, M.J.; Martinez-Pomares, L.; Gordon, S.; Turley, S.J.; et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 2010, 11, 427–434. [Google Scholar] [CrossRef] [PubMed]
- MacRitchie, N.; Grassia, G.; Noonan, J.; Cole, J.E.; Hughes, C.E.; Schroeder, J.; Benson, R.A.; Cochain, C.; Zernecke, A.; Guzik, T.J.; et al. The aorta can act as a site of naive CD4+ T cell priming. Cardiovasc Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Meiler, S.; Doring, Y.; Koch, M.; Drechsler, M.; Megens, R.T.; Rowinska, Z.; Bidzhekov, K.; Fecher, C.; Ribechini, E.; et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Investig. 2011, 121, 2898–2910. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Garcia, Z.; Chodaczek, G.; Landau, M.; McArdle, S.; Scott, S.R.; von Vietinghoff, S.; Galkina, E.; Miller, Y.I.; Acton, S.T.; et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J. Clin. Investig. 2012, 122, 3114–3126. [Google Scholar] [CrossRef]
- Sun, J.; Hartvigsen, K.; Chou, M.Y.; Zhang, Y.; Sukhova, G.K.; Zhang, J.; Lopez-Ilasaca, M.; Diehl, C.J.; Yakov, N.; Harats, D.; et al. Deficiency of antigen-presenting cell invariant chain reduces atherosclerosis in mice. Circulation 2010, 122, 808–820. [Google Scholar] [CrossRef]
- Wigren, M.; Rattik, S.; Yao Mattisson, I.; Tomas, L.; Gronberg, C.; Soderberg, I.; Alm, R.; Sundius, L.; Ljungcrantz, I.; Bjorkbacka, H.; et al. Lack of Ability to Present Antigens on Major Histocompatibility Complex Class II Molecules Aggravates Atherosclerosis in ApoE(-/-) Mice. Circulation 2019, 139, 2554–2566. [Google Scholar] [CrossRef]
- Subramanian, M.; Thorp, E.; Hansson, G.K.; Tabas, I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Investig. 2013, 123, 179–188. [Google Scholar] [CrossRef]
- Loschko, J.; Schreiber, H.A.; Rieke, G.J.; Esterhazy, D.; Meredith, M.M.; Pedicord, V.A.; Yao, K.H.; Caballero, S.; Pamer, E.G.; Mucida, D.; et al. Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J. Exp. Med. 2016, 213, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Agouridis, A.P.; Elisaf, M.; Milionis, H.J. An overview of lipid abnormalities in patients with inflammatory bowel disease. Ann. Gastroenterol. 2011, 24, 181–187. [Google Scholar] [PubMed]
- Lievens, D.; Habets, K.L.; Robertson, A.K.; Laouar, Y.; Winkels, H.; Rademakers, T.; Beckers, L.; Wijnands, E.; Boon, L.; Mosaheb, M.; et al. Abrogated transforming growth factor beta receptor II (TGFbetaRII) signalling in dendritic cells promotes immune reactivity of T cells resulting in enhanced atherosclerosis. Eur. Heart J. 2013, 34, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.M.; Sluimer, J.C.; Koch, M.; Theelen, T.L.; Manthey, H.D.; Busch, M.; Caballero-Franco, C.; Vogel, F.; Cochain, C.; Pelisek, J.; et al. Deficiency of HIF1alpha in Antigen-Presenting Cells Aggravates Atherosclerosis and Type 1 T-Helper Cell Responses in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Alberts-Grill, N.; Engelbertsen, D.; Bu, D.; Foks, A.; Grabie, N.; Herter, J.M.; Kuperwaser, F.; Chen, T.; Destefano, G.; Jarolim, P.; et al. Dendritic Cell KLF2 Expression Regulates T Cell Activation and Proatherogenic Immune Responses. J. Immunol. 2016, 197, 4651–4662. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Robbins, P.D. Tolerogenic dendritic cells for autoimmune disease and transplantation. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), iii90–iii96. [Google Scholar] [CrossRef]
- Mok, M.Y. Tolerogenic dendritic cells: Role and therapeutic implications in systemic lupus erythematosus. Int. J. Rheum. Dis 2015, 18, 250–259. [Google Scholar] [CrossRef]
- Habets, K.L.; van Puijvelde, G.H.; van Duivenvoorde, L.M.; van Wanrooij, E.J.; de Vos, P.; Tervaert, J.W.; van Berkel, T.J.; Toes, R.E.; Kuiper, J. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice. Cardiovasc. Res. 2010, 85, 622–630. [Google Scholar] [CrossRef]
- Hermansson, A.; Johansson, D.K.; Ketelhuth, D.F.; Andersson, J.; Zhou, X.; Hansson, G.K. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation 2011, 123, 1083–1091. [Google Scholar] [CrossRef]
- Rombouts, M.; Cools, N.; Grootaert, M.O.; de Bakker, F.; Van Brussel, I.; Wouters, A.; De Meyer, G.R.; De Winter, B.Y.; Schrijvers, D.M. Long-Term Depletion of Conventional Dendritic Cells Cannot Be Maintained in an Atherosclerotic Zbtb46-DTR Mouse Model. PLoS ONE 2017, 12, e0169608. [Google Scholar] [CrossRef]
- McKenna, H.J.; Stocking, K.L.; Miller, R.E.; Brasel, K.; De Smedt, T.; Maraskovsky, E.; Maliszewski, C.R.; Lynch, D.H.; Smith, J.; Pulendran, B.; et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Legein, B.; Janssen, E.M.; Theelen, T.L.; Gijbels, M.J.; Walraven, J.; Klarquist, J.S.; Hennies, C.M.; Wouters, K.; Seijkens, T.T.; Wijnands, E.; et al. Ablation of CD8alpha(+) dendritic cell mediated cross-presentation does not impact atherosclerosis in hyperlipidemic mice. Sci Rep. 2015, 5, 15414. [Google Scholar] [CrossRef] [PubMed]
- Gil-Pulido, J.; Cochain, C.; Lippert, M.A.; Schneider, N.; Butt, E.; Amezaga, N.; Zernecke, A. Deletion of Batf3-dependent antigen-presenting cells does not affect atherosclerotic lesion formation in mice. PLoS ONE 2017, 12, e0181947. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, X.; Duan, W.; Tian, H.; Zhu, G.; He, H.; Yao, S.; Yi, S.; Song, W.; Tang, H. Batf3-dependent CD8alpha(+) Dendritic Cells Aggravates Atherosclerosis via Th1 Cell Induction and Enhanced CCL5 Expression in Plaque Macrophages. EBioMedicine 2017, 18, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Haddad, Y.; Lahoute, C.; Clement, M.; Laurans, L.; Metghalchi, S.; Zeboudj, L.; Giraud, A.; Loyer, X.; Vandestienne, M.; Wain-Hobson, J.; et al. The Dendritic Cell Receptor DNGR-1 Promotes the Development of Atherosclerosis in Mice. Circ. Res. 2017, 121, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Daissormont, I.T.; Christ, A.; Temmerman, L.; Sampedro Millares, S.; Seijkens, T.; Manca, M.; Rousch, M.; Poggi, M.; Boon, L.; van der Loos, C.; et al. Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-cell proliferation and activity. Circ. Res. 2011, 109, 1387–1395. [Google Scholar] [CrossRef]
- Doring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef]
- Macritchie, N.; Grassia, G.; Sabir, S.R.; Maddaluno, M.; Welsh, P.; Sattar, N.; Ialenti, A.; Kurowska-Stolarska, M.; McInnes, I.B.; Brewer, J.M.; et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2569–2579. [Google Scholar] [CrossRef]
- Yun, T.J.; Lee, J.S.; Machmach, K.; Shim, D.; Choi, J.; Wi, Y.J.; Jang, H.S.; Jung, I.H.; Kim, K.; Yoon, W.K.; et al. Indoleamine 2,3-Dioxygenase-Expressing Aortic Plasmacytoid Dendritic Cells Protect against Atherosclerosis by Induction of Regulatory T Cells. Cell Metab. 2016, 23, 852–866. [Google Scholar] [CrossRef]
- Mandl, M.; Drechsler, M.; Jansen, Y.; Neideck, C.; Noels, H.; Faussner, A.; Soehnlein, O.; Weber, C.; Doring, Y. Evaluation of the BDCA2-DTR Transgenic Mouse Model in Chronic and Acute Inflammation. PLoS ONE 2015, 10, e0134176. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Murphy, D.; Maffia, P.; Masters, L.M.; Sabir, S.R.; Baker, L.L.; Cambrook, H.; Finigan, A.J.; Ait-Oufella, H.; Grassia, G.; et al. MHC Class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation 2014, 130, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, D.; Rohm, I.; Schaller, S.; Betge, S.; Pistulli, R.; Atiskova, Y.; Figulla, H.R.; Yilmaz, A. Decrease in circulating dendritic cell precursors in patients with peripheral artery disease. Mediat. Inflamm. 2015, 2015, 450957. [Google Scholar] [CrossRef] [PubMed]
- Van Vre, E.A.; Van Brussel, I.; Bosmans, J.M.; Vrints, C.J.; Bult, H. Dendritic cells in human atherosclerosis: From circulation to atherosclerotic plaques. Mediat. Inflamm. 2011, 2011, 941396. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Ming, J.Y.; Fiskesund, R.; Ninio, E.; Karabina, S.A.; Bergmark, C.; Frostegard, A.G.; Frostegard, J. Induction of dendritic cell-mediated T-cell activation by modified but not native low-density lipoprotein in humans and inhibition by annexin a5: Involvement of heat shock proteins. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 197–205. [Google Scholar] [CrossRef]
- Fehervari, Z.; Sakaguchi, S. Control of Foxp3+ CD25+CD4+ regulatory cell activation and function by dendritic cells. Int. Immunol. 2004, 16, 1769–1780. [Google Scholar] [CrossRef]
- Liu, J.; Cao, X. Regulatory dendritic cells in autoimmunity: A comprehensive review. J. Autoimmun. 2015, 63, 1–12. [Google Scholar] [CrossRef]
- Morelli, A.E.; Thomson, A.W. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 2007, 7, 610–621. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Y.H.; Wang, Y.; Huang, L.; Sandoval, H.; Liu, Y.J.; Wang, J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science 2006, 311, 1160–1164. [Google Scholar] [CrossRef]
- Selenko-Gebauer, N.; Majdic, O.; Szekeres, A.; Hofler, G.; Guthann, E.; Korthauer, U.; Zlabinger, G.; Steinberger, P.; Pickl, W.F.; Stockinger, H.; et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J. Immunol. 2003, 170, 3637–3644. [Google Scholar] [CrossRef]
- Darrasse-Jeze, G.; Deroubaix, S.; Mouquet, H.; Victora, G.D.; Eisenreich, T.; Yao, K.H.; Masilamani, R.F.; Dustin, M.L.; Rudensky, A.; Liu, K.; et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J. Exp. Med. 2009, 206, 1853–1862. [Google Scholar] [CrossRef]
- Ye, Z.S.; Huang, R.C. Selectins modify dendritic cells during atherosclerosis. Chronic Dis. Transl. Med. 2018, 4, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Torres-Aguilar, H.; Sanchez-Torres, C.; Jara, L.J.; Blank, M.; Shoenfeld, Y. IL-10/TGF-beta-treated dendritic cells, pulsed with insulin, specifically reduce the response to insulin of CD4+ effector/memory T cells from type 1 diabetic individuals. J. Clin. Immunol. 2010, 30, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, C.; Johansson, D.; Hermansson, A.; Hansson, G.K.; Zhou, X. Dendritic cells pulsed with malondialdehyde modified low density lipoprotein aggravate atherosclerosis in Apoe(-/-) mice. Atherosclerosis 2010, 209, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Frodermann, V.; van Puijvelde, G.H.; Wierts, L.; Lagraauw, H.M.; Foks, A.C.; van Santbrink, P.J.; Bot, I.; Kuiper, J.; de Jager, S.C. Oxidized low-density lipoprotein-induced apoptotic dendritic cells as a novel therapy for atherosclerosis. J. Immunol. 2015, 194, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.; Hansson, G.K. Adaptive Response of T and B Cells in Atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Bustos-Moran, E.; Blas-Rus, N.; Martin-Cofreces, N.B.; Sanchez-Madrid, F. Orchestrating Lymphocyte Polarity in Cognate Immune Cell-Cell Interactions. Int. Rev. Cell Mol. Biol. 2016, 327, 195–261. [Google Scholar] [PubMed]
- Gronberg, C.; Nilsson, J.; Wigren, M. Recent advances on CD4(+) T cells in atherosclerosis and its implications for therapy. Eur. J. Pharmacol. 2017, 816, 58–66. [Google Scholar] [CrossRef]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef]
- Rocha-Perugini, V.; Gonzalez-Granado, J.M. Nuclear envelope lamin-A as a coordinator of T cell activation. Nucleus 2014, 5, 396–401. [Google Scholar] [CrossRef]
- van Panhuys, N.; Klauschen, F.; Germain, R.N. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity 2014, 41, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Tse, K.; Tse, H.; Sidney, J.; Sette, A.; Ley, K. T cells in atherosclerosis. Int. Immunol. 2013, 25, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef]
- Gounopoulos, P.; Merki, E.; Hansen, L.F.; Choi, S.H.; Tsimikas, S. Antibodies to oxidized low density lipoprotein: Epidemiological studies and potential clinical applications in cardiovascular disease. Minerva Cardioangiol. 2007, 55, 821–837. [Google Scholar]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Lahoute, C.; Herbin, O.; Mallat, Z.; Tedgui, A. Adaptive immunity in atherosclerosis: Mechanisms and future therapeutic targets. Nat. Rev. Cardiol. 2011, 8, 348–358. [Google Scholar] [CrossRef]
- Iborra, S.; Gonzalez-Granado, J.M. In Vitro Differentiation of Naive CD4(+) T Cells: A Tool for Understanding the Development of Atherosclerosis. Methods Mol. Biol. 2015, 1339, 177–189. [Google Scholar]
- Aubry, M.C.; Riehle, D.L.; Edwards, W.D.; Maradit-Kremers, H.; Roger, V.L.; Sebo, T.J.; Gabriel, S.E. B-Lymphocytes in plaque and adventitia of coronary arteries in two patients with rheumatoid arthritis and coronary atherosclerosis: Preliminary observations. Cardiovasc. Pathol. 2004, 13, 233–236. [Google Scholar] [CrossRef]
- Ketelhuth, D.F.; Hansson, G.K. Cellular immunity, low-density lipoprotein and atherosclerosis: Break of tolerance in the artery wall. Thromb. Haemost. 2011, 106, 779–786. [Google Scholar] [CrossRef]
- Mallat, Z.; Taleb, S.; Ait-Oufella, H.; Tedgui, A. The role of adaptive T cell immunity in atherosclerosis. J. Lipid Res. 2009, 50 (Suppl.), S364–S369. [Google Scholar] [CrossRef] [PubMed]
- Laurat, E.; Poirier, B.; Tupin, E.; Caligiuri, G.; Hansson, G.K.; Bariety, J.; Nicoletti, A. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 2001, 104, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McArdle, S.; Gholami, A.; Kimura, T.; Wolf, D.; Gerhardt, T.; Miller, J.; Weber, C.; Ley, K. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ. Res. 2016, 118, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Gourdy, P.; Brouchet, L.; Jawien, J.; Fouque, M.J.; Fievet, C.; Huc, X.; Barreira, Y.; Couloumiers, J.C.; Arnal, J.F.; et al. Deleting TCR alpha beta+ or CD4+ T lymphocytes leads to opposite effects on site-specific atherosclerosis in female apolipoprotein E-deficient mice. Am. J. Pathol. 2004, 165, 2013–2018. [Google Scholar] [CrossRef]
- Zhou, X.; Robertson, A.K.; Rudling, M.; Parini, P.; Hansson, G.K. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circ. Res. 2005, 96, 427–434. [Google Scholar] [CrossRef]
- Buono, C.; Binder, C.J.; Stavrakis, G.; Witztum, J.L.; Glimcher, L.H.; Lichtman, A.H. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 2005, 102, 1596–1601. [Google Scholar] [CrossRef]
- Buono, C.; Come, C.E.; Stavrakis, G.; Maguire, G.F.; Connelly, P.W.; Lichtman, A.H. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 454–460. [Google Scholar] [CrossRef]
- Jones, K.L.; Maguire, J.J.; Davenport, A.P. Chemokine receptor CCR5: From AIDS to atherosclerosis. Br. J. Pharmacol. 2011, 162, 1453–1469. [Google Scholar] [CrossRef]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Davenport, P.; Tipping, P.G. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 2003, 163, 1117–1125. [Google Scholar] [CrossRef]
- Hauer, A.D.; Uyttenhove, C.; de Vos, P.; Stroobant, V.; Renauld, J.C.; van Berkel, T.J.; van Snick, J.; Kuiper, J. Blockade of interleukin-12 function by protein vaccination attenuates atherosclerosis. Circulation 2005, 112, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Graber, P.; Alouani, S.; Esposito, B.; Humbert, Y.; Chvatchko, Y.; Tedgui, A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 2001, 89, E41–E45. [Google Scholar] [CrossRef] [PubMed]
- Haybar, H.; Rezaeeyan, H.; Shahjahani, M.; Shirzad, R.; Saki, N. T-bet transcription factor in cardiovascular disease: Attenuation or inflammation factor? J. Cell. Physiol. 2019, 234, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Hwang, E.S. The role of protein modifications of T-bet in cytokine production and differentiation of T helper cells. J. Immunol. Res. 2014, 2014, 589672. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Sharma, S.; Edwards, J.; Feigenbaum, L.; Zhu, J. Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat. Immunol. 2015, 16, 197–206. [Google Scholar] [CrossRef]
- Whitman, S.C.; Ravisankar, P.; Elam, H.; Daugherty, A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am. J. Pathol. 2000, 157, 1819–1824. [Google Scholar] [CrossRef]
- Gupta, S.; Pablo, A.M.; Jiang, X.; Wang, N.; Tall, A.R.; Schindler, C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Investig. 1997, 99, 2752–2761. [Google Scholar] [CrossRef]
- Moss, J.W.; Ramji, D.P. Interferon-gamma: Promising therapeutic target in atherosclerosis. World J. Exp. Med. 2015, 5, 154–159. [Google Scholar] [CrossRef]
- Lim, W.S.; Timmins, J.M.; Seimon, T.A.; Sadler, A.; Kolodgie, F.D.; Virmani, R.; Tabas, I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation 2008, 117, 940–951. [Google Scholar] [CrossRef]
- Blankenberg, S.; Barbaux, S.; Tiret, L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003, 170, 191–203. [Google Scholar] [CrossRef]
- Chung, H.K.; Lee, I.K.; Kang, H.; Suh, J.M.; Kim, H.; Park, K.C.; Kim, D.W.; Kim, Y.K.; Ro, H.K.; Shong, M. Statin inhibits interferon-gamma-induced expression of intercellular adhesion molecule-1 (ICAM-1) in vascular endothelial and smooth muscle cells. Exp. Mol. Med. 2002, 34, 451–461. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harvey, E.J.; Ramji, D.P. Interferon-gamma and atherosclerosis: Pro- or anti-atherogenic? Cardiovasc. Res. 2005, 67, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Wuttge, D.M.; Zhou, X.; Sheikine, Y.; Wagsater, D.; Stemme, V.; Hedin, U.; Stemme, S.; Hansson, G.K.; Sirsjo, A. CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Zernecke, A.; Arnaud, C.; Liehn, E.A.; Steffens, S.; Shagdarsuren, E.; Bidzhekov, K.; Burger, F.; Pelli, G.; Luckow, B.; et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 373–379. [Google Scholar] [CrossRef]
- Afzal, A.R.; Kiechl, S.; Daryani, Y.P.; Weerasinghe, A.; Zhang, Y.; Reindl, M.; Mayr, A.; Weger, S.; Xu, Q.; Willeit, J. Common CCR5-del32 frameshift mutation associated with serum levels of inflammatory markers and cardiovascular disease risk in the Bruneck population. Stroke 2008, 39, 1972–1978. [Google Scholar] [CrossRef]
- Szalai, C.; Duba, J.; Prohaszka, Z.; Kalina, A.; Szabo, T.; Nagy, B.; Horvath, L.; Csaszar, A. Involvement of polymorphisms in the chemokine system in the susceptibility for coronary artery disease (CAD). Coincidence of elevated Lp(a) and MCP-1 -2518 G/G genotype in CAD patients. Atherosclerosis 2001, 158, 233–239. [Google Scholar] [CrossRef]
- Pai, J.K.; Kraft, P.; Cannuscio, C.C.; Manson, J.E.; Rexrode, K.M.; Albert, C.M.; Hunter, D.; Rimm, E.B. Polymorphisms in the CC-chemokine receptor-2 (CCR2) and -5 (CCR5) genes and risk of coronary heart disease among US women. Atherosclerosis 2006, 186, 132–139. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Candore, G.; Caruso, M.; Incalcaterra, E.; Franceschi, C.; Caruso, C. Role of polymorphisms of CC-chemokine receptor-5 gene in acute myocardial infarction and biological implications for longevity. Haematologica 2008, 93, 637–638. [Google Scholar] [CrossRef]
- Gonzalez, P.; Alvarez, R.; Batalla, A.; Reguero, J.R.; Alvarez, V.; Astudillo, A.; Cubero, G.I.; Cortina, A.; Coto, E. Genetic variation at the chemokine receptors CCR5/CCR2 in myocardial infarction. Genes Immun 2001, 2, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Sharda, S.; Gilmour, A.; Harris, V.; Singh, V.P.; Sinha, N.; Tewari, S.; Ramesh, V.; Agrawal, S.; Mastana, S. Chemokine receptor 5 (CCR5) deletion polymorphism in North Indian patients with coronary artery disease. Int. J. Cardiol. 2008, 124, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, G.; Biondi, M.L.; Turri, O.; Pateri, F.; d’Eril, G.M.; Scorza, R. Genetic control of chemokines in severe human internal carotid artery stenosis. Cytokine 2008, 41, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Apostolakis, S.; Baritaki, S.; Kochiadakis, G.E.; Igoumenidis, N.E.; Panutsopulos, D.; Spandidos, D.A. Effects of polymorphisms in chemokine ligands and receptors on susceptibility to coronary artery disease. Thromb Res. 2007, 119, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Petrkova, J.; Cermakova, Z.; Lukl, J.; Petrek, M. CC chemokine receptor 5 (CCR5) deletion polymorphism does not protect Czech males against early myocardial infarction. J. Intern. Med. 2005, 257, 564–566. [Google Scholar] [CrossRef]
- Simeoni, E.; Winkelmann, B.R.; Hoffmann, M.M.; Fleury, S.; Ruiz, J.; Kappenberger, L.; Marz, W.; Vassalli, G. Association of RANTES G-403A gene polymorphism with increased risk of coronary arteriosclerosis. Eur. Heart J. 2004, 25, 1438–1446. [Google Scholar] [CrossRef]
- van Wanrooij, E.J.; Happe, H.; Hauer, A.D.; de Vos, P.; Imanishi, T.; Fujiwara, H.; van Berkel, T.J.; Kuiper, J. HIV entry inhibitor TAK-779 attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2642–2647. [Google Scholar] [CrossRef]
- Veillard, N.R.; Kwak, B.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.; Mach, F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 2004, 94, 253–261. [Google Scholar] [CrossRef]
- Schober, A.; Manka, D.; von Hundelshausen, P.; Huo, Y.; Hanrath, P.; Sarembock, I.J.; Ley, K.; Weber, C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 2002, 106, 1523–1529. [Google Scholar] [CrossRef]
- Quinones, M.P.; Martinez, H.G.; Jimenez, F.; Estrada, C.A.; Dudley, M.; Willmon, O.; Kulkarni, H.; Reddick, R.L.; Fernandes, G.; Kuziel, W.A.; et al. CC chemokine receptor 5 influences late-stage atherosclerosis. Atherosclerosis 2007, 195, e92–e103. [Google Scholar] [CrossRef]
- Potteaux, S.; Combadiere, C.; Esposito, B.; Lecureuil, C.; Ait-Oufella, H.; Merval, R.; Ardouin, P.; Tedgui, A.; Mallat, Z. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Luckow, B.; Joergensen, J.; Chilla, S.; Li, J.P.; Henger, A.; Kiss, E.; Wieczorek, G.; Roth, L.; Hartmann, N.; Hoffmann, R.; et al. Reduced intragraft mRNA expression of matrix metalloproteinases Mmp3, Mmp12, Mmp13 and Adam8, and diminished transplant arteriosclerosis in Ccr5-deficient mice. Eur. J. Immunol. 2004, 34, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Kuziel, W.A.; Dawson, T.C.; Quinones, M.; Garavito, E.; Chenaux, G.; Ahuja, S.S.; Reddick, R.L.; Maeda, N. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis 2003, 167, 25–32. [Google Scholar] [CrossRef]
- Piconi, S.; Pocaterra, D.; Rainone, V.; Cossu, M.; Masetti, M.; Rizzardini, G.; Clerici, M.; Trabattoni, D. Maraviroc Reduces Arterial Stiffness in PI-Treated HIV-infected Patients. Sci. Rep. 2016, 6, 28853. [Google Scholar] [CrossRef]
- Cipriani, S.; Francisci, D.; Mencarelli, A.; Renga, B.; Schiaroli, E.; D’Amore, C.; Baldelli, F.; Fiorucci, S. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 2013, 127, 2114–2124. [Google Scholar] [CrossRef]
- Krohn, R.; Raffetseder, U.; Bot, I.; Zernecke, A.; Shagdarsuren, E.; Liehn, E.A.; van Santbrink, P.J.; Nelson, P.J.; Biessen, E.A.; Mertens, P.R.; et al. Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation 2007, 116, 1812–1820. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Steffens, S.; Arnaud, C.; Pelli, G.; Burger, F.; Proudfoot, A.; Mach, F. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1090–1096. [Google Scholar] [CrossRef]
- Robertson, A.K.; Rudling, M.; Zhou, X.; Gorelik, L.; Flavell, R.A.; Hansson, G.K. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J. Clin. Investig. 2003, 112, 1342–1350. [Google Scholar] [CrossRef]
- Ho, I.C.; Tai, T.S.; Pai, S.Y. GATA3 and the T-cell lineage: Essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 2009, 9, 125–135. [Google Scholar] [CrossRef]
- Engelbertsen, D.; Andersson, L.; Ljungcrantz, I.; Wigren, M.; Hedblad, B.; Nilsson, J.; Bjorkbacka, H. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 637–644. [Google Scholar] [CrossRef]
- Mantani, P.T.; Duner, P.; Bengtsson, E.; Alm, R.; Ljungcrantz, I.; Soderberg, I.; Sundius, L.; To, F.; Nilsson, J.; Bjorkbacka, H.; et al. IL-25 inhibits atherosclerosis development in apolipoprotein E deficient mice. PLoS ONE 2015, 10, e0117255. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.; McLeod, O.; Strawbridge, R.J.; Gertow, K.; Sennblad, B.; Baldassarre, D.; Veglia, F.; Deleskog, A.; Persson, J.; Leander, K.; et al. Plasma IL-5 concentration and subclinical carotid atherosclerosis. Atherosclerosis 2015, 239, 125–130. [Google Scholar] [CrossRef]
- Binder, C.J.; Hartvigsen, K.; Chang, M.K.; Miller, M.; Broide, D.; Palinski, W.; Curtiss, L.K.; Corr, M.; Witztum, J.L. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J. Clin. Investig. 2004, 114, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Engelbertsen, D.; Rattik, S.; Knutsson, A.; Bjorkbacka, H.; Bengtsson, E.; Nilsson, J. Induction of T helper 2 responses against human apolipoprotein B100 does not affect atherosclerosis in ApoE-/- mice. Cardiovasc. Res. 2014, 103, 304–312. [Google Scholar] [CrossRef]
- King, V.L.; Szilvassy, S.J.; Daugherty, A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor-/- mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 456–461. [Google Scholar] [CrossRef]
- King, V.L.; Cassis, L.A.; Daugherty, A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am. J. Pathol. 2007, 171, 2040–2047. [Google Scholar] [CrossRef]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef]
- Cornicelli, J.A.; Butteiger, D.; Rateri, D.L.; Welch, K.; Daugherty, A. Interleukin-4 augments acetylated LDL-induced cholesterol esterification in macrophages. J. Lipid Res. 2000, 41, 376–383. [Google Scholar]
- Barks, J.L.; McQuillan, J.J.; Iademarco, M.F. TNF-alpha and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J. Immunol. 1997, 159, 4532–4538. [Google Scholar]
- Lee, Y.W.; Kuhn, H.; Hennig, B.; Neish, A.S.; Toborek, M. IL-4-induced oxidative stress upregulates VCAM-1 gene expression in human endothelial cells. J. Mol. Cell. Cardiol. 2001, 33, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, T.; Arima, N.; Tanimoto, A.; Shimajiri, S.; Hamada, T.; Sasaguri, Y. A role for interleukin 4 in production of matrix metalloproteinase 1 by human aortic smooth muscle cells. Atherosclerosis 1998, 138, 247–253. [Google Scholar] [CrossRef]
- Walch, L.; Massade, L.; Dufilho, M.; Brunet, A.; Rendu, F. Pro-atherogenic effect of interleukin-4 in endothelial cells: Modulation of oxidative stress, nitric oxide and monocyte chemoattractant protein-1 expression. Atherosclerosis 2006, 187, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Vila-Caballer, M.; Gonzalez-Granado, J.M.; Zorita, V.; Abu Nabah, Y.N.; Silvestre-Roig, C.; Del Monte-Monge, A.; Molina-Sanchez, P.; Ait-Oufella, H.; Andres-Manzano, M.J.; Sanz, M.J.; et al. Disruption of the CCL1-CCR8 axis inhibits vascular Treg recruitment and function and promotes atherosclerosis in mice. J. Mol. Cell. Cardiol. 2019, 132, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hu, H.; Liu, L.; Ye, J.; Wang, Z.; Que, B.; Liu, W.; Shi, Y.; Zeng, T.; Shi, L.; et al. Interleukin-12p35 Deficiency Reverses the Th1/Th2 Imbalance, Aggravates the Th17/Treg Imbalance, and Ameliorates Atherosclerosis in ApoE-/- Mice. Mediat. Inflamm. 2019, 2019, 3152040. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhang, W.; Zhao, Q.; Zhou, J.; Wu, Y.; Liu, Y.; Yuan, Z.; Wang, L. Curcumin ameliorates atherosclerosis in apolipoprotein E deficient asthmatic mice by regulating the balance of Th2/Treg cells. Phytomedicine 2019, 52, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef]
- Selmi, C. Autoimmunity in 2018. Clin. Rev. Allergy Immunol. 2019, 56, 375–384. [Google Scholar] [CrossRef]
- Akhavanpoor, M.; Akhavanpoor, H.; Gleissner, C.A.; Wangler, S.; Doesch, A.O.; Katus, H.A.; Erbel, C. The Two Faces of Interleukin-17A in Atherosclerosis. Curr. Drug Targets 2017, 18, 863–873. [Google Scholar] [CrossRef]
- Allam, G.; Abdel-Moneim, A.; Gaber, A.M. The pleiotropic role of interleukin-17 in atherosclerosis. Biomed. Pharmacother. 2018, 106, 1412–1418. [Google Scholar] [CrossRef]
- Lu, X. The Impact of IL-17 in Atherosclerosis. Curr. Med. Chem. 2017, 24, 2345–2358. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, T. Interleukin-22, a potent target for treatment of non-autoimmune diseases. Hum. Vaccin. Immunother. 2018, 14, 2811–2819. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Tedgui, A. IL-17 in atherosclerosis: The good and the bad. Cardiovasc. Res. 2018, 114, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Prasad, K.M.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 2010, 121, 1746–1755. [Google Scholar] [CrossRef]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef]
- Weaver, C.T.; Hatton, R.D. Interplay between the TH17 and TReg cell lineages: A (co-)evolutionary perspective. Nat. Rev. Immunol. 2009, 9, 883–889. [Google Scholar] [CrossRef]
- Zuniga, L.A.; Jain, R.; Haines, C.; Cua, D.J. Th17 cell development: From the cradle to the grave. Immunol. Rev. 2013, 252, 78–88. [Google Scholar] [CrossRef]
- Krausgruber, T.; Schiering, C.; Adelmann, K.; Harrison, O.J.; Chomka, A.; Pearson, C.; Ahern, P.P.; Shale, M.; Oukka, M.; Powrie, F. T-bet is a key modulator of IL-23-driven pathogenic CD4(+) T cell responses in the intestine. Nat. Commun. 2016, 7, 11627. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.; Bakery, H.H.; Allam, G. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus. Biomed. Pharmacother. 2018, 101, 287–292. [Google Scholar] [CrossRef]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C.; Tolaini, M.; Menzel, U.; et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu Rev. Immunol 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Liuzzo, G.; Trotta, F.; Pedicino, D. Interleukin-17 in atherosclerosis and cardiovascular disease: The good, the bad, and the unknown. Eur. Heart J. 2013, 34, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Butcher, M.; Galkina, E. Current views on the functions of interleukin-17A-producing cells in atherosclerosis. Thromb. Haemost. 2011, 106, 787–795. [Google Scholar] [CrossRef]
- Erbel, C.; Chen, L.; Bea, F.; Wangler, S.; Celik, S.; Lasitschka, F.; Wang, Y.; Bockler, D.; Katus, H.A.; Dengler, T.J. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J. Immunol. 2009, 183, 8167–8175. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar]
- Madhur, M.S.; Funt, S.A.; Li, L.; Vinh, A.; Chen, W.; Lob, H.E.; Iwakura, Y.; Blinder, Y.; Rahman, A.; Quyyumi, A.A.; et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Beringer, A.; Noack, M.; Miossec, P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol. Med. 2016, 22, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Smith, E.; Stark, M.A. IL-17A-producing neutrophil-regulatory Tn lymphocytes. Immunol. Res. 2006, 34, 229–242. [Google Scholar] [CrossRef]
- Taleb, S.; Romain, M.; Ramkhelawon, B.; Uyttenhove, C.; Pasterkamp, G.; Herbin, O.; Esposito, B.; Perez, N.; Yasukawa, H.; Van Snick, J.; et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 2009, 206, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Gistera, A.; Robertson, A.K.; Andersson, J.; Ketelhuth, D.F.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci. Transl. Med. 2013, 5, 196ra100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, Q.; Guo, C.; Wang, X.; Cao, X.; Shi, Y.; Gao, F.; Ma, C.; Zhang, L. IL-17 induces apoptosis of vascular endothelial cells: A potential mechanism for human acute coronary syndrome. Clin. Immunol. 2011, 141, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Madhur, M.S.; Lob, H.E.; McCann, L.A.; Iwakura, Y.; Blinder, Y.; Guzik, T.J.; Harrison, D.G. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010, 55, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Danzaki, K.; Matsui, Y.; Ikesue, M.; Ohta, D.; Ito, K.; Kanayama, M.; Kurotaki, D.; Morimoto, J.; Iwakura, Y.; Yagita, H.; et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 273–280. [Google Scholar] [CrossRef]
- van Es, T.; van Puijvelde, G.H.; Ramos, O.H.; Segers, F.M.; Joosten, L.A.; van den Berg, W.B.; Michon, I.M.; de Vos, P.; van Berkel, T.J.; Kuiper, J. Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem. Biophys. Res. Commun. 2009, 388, 261–265. [Google Scholar] [CrossRef]
- Butcher, M.J.; Gjurich, B.N.; Phillips, T.; Galkina, E.V. The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ. Res. 2012, 110, 675–687. [Google Scholar] [CrossRef]
- Gao, Q.; Jiang, Y.; Ma, T.; Zhu, F.; Gao, F.; Zhang, P.; Guo, C.; Wang, Q.; Wang, X.; Ma, C.; et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J. Immunol. 2010, 185, 5820–5827. [Google Scholar] [CrossRef] [PubMed]
- Nordlohne, J.; von Vietinghoff, S. Interleukin 17A in atherosclerosis - Regulation and pathophysiologic effector function. Cytokine 2017, 122, 154089. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Waseem, T.C.; Galkina, E.V. Smooth Muscle Cell-Derived Interleukin-17C Plays an Atherogenic Role via the Recruitment of Proinflammatory Interleukin-17A+ T Cells to the Aorta. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Sedda, S.; Marafini, I.; Figliuzzi, M.M.; Pallone, F.; Monteleone, G. An overview of the role of innate lymphoid cells in gut infections and inflammation. Mediat. Inflamm. 2014, 2014, 235460. [Google Scholar] [CrossRef]
- Kumar, P.; Thakar, M.S.; Ouyang, W.; Malarkannan, S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013, 6, 69–82. [Google Scholar] [CrossRef]
- Brockmann, L.; Giannou, A.D.; Gagliani, N.; Huber, S. Regulation of TH17 Cells and Associated Cytokines in Wound Healing, Tissue Regeneration, and Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1033. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Karow, M.; Flavell, R.A. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007, 27, 647–659. [Google Scholar] [CrossRef]
- Gong, F.; Wu, J.; Zhou, P.; Zhang, M.; Liu, J.; Liu, Y.; Lu, X.; Liu, Z. Interleukin-22 Might Act as a Double-Edged Sword in Type 2 Diabetes and Coronary Artery Disease. Mediat. Inflamm 2016, 2016, 8254797. [Google Scholar] [CrossRef]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef]
- Xia, Q.; Xiang, X.; Patel, S.; Puranik, R.; Xie, Q.; Bao, S. Characterisation of IL-22 and interferon-gamma-inducible chemokines in human carotid plaque. Int. J. Cardiol. 2012, 154, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Chellan, B.; Yan, L.; Sontag, T.J.; Reardon, C.A.; Hofmann Bowman, M.A. IL-22 is induced by S100/calgranulin and impairs cholesterol efflux in macrophages by downregulating ABCG1. J. Lipid Res. 2014, 55, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Rattik, S.; Hultman, K.; Rauch, U.; Soderberg, I.; Sundius, L.; Ljungcrantz, I.; Hultgardh-Nilsson, A.; Wigren, M.; Bjorkbacka, H.; Fredrikson, G.N.; et al. IL-22 affects smooth muscle cell phenotype and plaque formation in apolipoprotein E knockout mice. Atherosclerosis 2015, 242, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Fatkhullina, A.R.; Peshkova, I.O.; Dzutsev, A.; Aghayev, T.; McCulloch, J.A.; Thovarai, V.; Badger, J.H.; Vats, R.; Sundd, P.; Tang, H.Y.; et al. An Interleukin-23-Interleukin-22 Axis Regulates Intestinal Microbial Homeostasis to Protect from Diet-Induced Atherosclerosis. Immunity 2018, 49, 943–957. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Apostolou, I.; von Boehmer, H. In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. 2004, 199, 1401–1408. [Google Scholar] [CrossRef]
- Ray, A.; Khare, A.; Krishnamoorthy, N.; Qi, Z.; Ray, P. Regulatory T cells in many flavors control asthma. Mucosal Immunol. 2010, 3, 216–229. [Google Scholar] [CrossRef]
- Roncarolo, M.G.; Bacchetta, R.; Bordignon, C.; Narula, S.; Levings, M.K. Type 1 T regulatory cells. Immunol. Rev. 2001, 182, 68–79. [Google Scholar] [CrossRef]
- Bocian, K.; Kiernozek, E.; Domagala-Kulawik, J.; Korczak-Kowalska, G.; Stelmaszczyk-Emmel, A.; Drela, N. Expanding Diversity and Common Goal of Regulatory T and B Cells. I: Origin, Phenotype, Mechanisms. Arch. Immunol. Ther. Exp. (Warsz) 2017, 65, 501–520. [Google Scholar] [CrossRef]
- Weiner, H.L. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol. Rev. 2001, 182, 207–214. [Google Scholar] [CrossRef]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.M.; Jack, R.S.; Wunderlich, F.T.; Bruning, J.C.; Muller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.Z.; Sasaki, N.; Hirata, K.I. Regulatory T Cell Immunity in Atherosclerosis. Acta Med. Indones 2017, 49, 63–68. [Google Scholar] [PubMed]
- Arce-Sillas, A.; Alvarez-Luquin, D.D.; Tamaya-Dominguez, B.; Gomez-Fuentes, S.; Trejo-Garcia, A.; Melo-Salas, M.; Cardenas, G.; Rodriguez-Ramirez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1720827. [Google Scholar] [CrossRef] [PubMed]
- Tselios, K.; Sarantopoulos, A.; Gkougkourelas, I.; Boura, P. T regulatory cells: A promising new target in atherosclerosis. Crit. Rev. Immunol. 2014, 34, 389–397. [Google Scholar] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Gondek, D.C.; Lu, L.F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef]
- Garin, M.I.; Chu, C.C.; Golshayan, D.; Cernuda-Morollon, E.; Wait, R.; Lechler, R.I. Galectin-1: A key effector of regulation mediated by CD4+CD25+ T cells. Blood 2007, 109, 2058–2065. [Google Scholar] [CrossRef]
- Ren, X.; Ye, F.; Jiang, Z.; Chu, Y.; Xiong, S.; Wang, Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ. 2007, 14, 2076–2084. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef]
- Mallat, Z.; Besnard, S.; Duriez, M.; Deleuze, V.; Emmanuel, F.; Bureau, M.F.; Soubrier, F.; Esposito, B.; Duez, H.; Fievet, C.; et al. Protective role of interleukin-10 in atherosclerosis. Circ. Res. 1999, 85, e17–e24. [Google Scholar] [CrossRef] [PubMed]
- Potteaux, S.; Esposito, B.; van Oostrom, O.; Brun, V.; Ardouin, P.; Groux, H.; Tedgui, A.; Mallat, Z. Leukocyte-derived interleukin 10 is required for protection against atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Gojova, A.; Brun, V.; Esposito, B.; Fournier, N.; Cottrez, F.; Tedgui, A.; Groux, H. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation 2003, 108, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Hamilton, K.; Vignali, D.A. Interleukin-35: Expanding Its Job Profile. J. Interferon Cytokine Res. 2015, 35, 499–512. [Google Scholar] [CrossRef]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef]
- Sha, X.; Meng, S.; Li, X.; Xi, H.; Maddaloni, M.; Pascual, D.W.; Shan, H.; Jiang, X.; Wang, H.; Yang, X.F. Interleukin-35 Inhibits Endothelial Cell Activation by Suppressing MAPK-AP-1 Pathway. J. Biol. Chem. 2015, 290, 19307–19318. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Yang, W.Y.; Wang, H.; Yang, X. IL-35, as a newly proposed homeostasis-associated molecular pattern, plays three major functions including anti-inflammatory initiator, effector, and blocker in cardiovascular diseases. Cytokine 2017, 122, 154076. [Google Scholar] [CrossRef]
- Vignali, D.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef]
- Collison, L.W.; Vignali, D.A. Interleukin-35: Odd one out or part of the family? Immunol. Rev. 2008, 226, 248–262. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Olson, B.M.; Sullivan, J.A.; Burlingham, W.J. Interleukin 35: A key mediator of suppression and the propagation of infectious tolerance. Front. Immunol. 2013, 4, 315. [Google Scholar] [CrossRef] [PubMed]
- Kempe, S.; Heinz, P.; Kokai, E.; Devergne, O.; Marx, N.; Wirth, T. Epstein-barr virus-induced gene-3 is expressed in human atheroma plaques. Am. J. Pathol. 2009, 175, 440–447. [Google Scholar] [CrossRef]
- Lin, Y.; Huang, Y.; Lu, Z.; Luo, C.; Shi, Y.; Zeng, Q.; Cao, Y.; Liu, L.; Wang, X.; Ji, Q. Decreased plasma IL-35 levels are related to the left ventricular ejection fraction in coronary artery diseases. PLoS ONE 2012, 7, e52490. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhu, J.; Chen, Y.; Wang, Q.; Pan, Y.; Yu, Q.; Zhou, B.; Zhu, H. IL-35 improves Treg-mediated immune suppression in atherosclerotic mice. Exp. Ther. Med. 2016, 12, 2469–2476. [Google Scholar] [CrossRef]
- Hirase, T.; Hara, H.; Miyazaki, Y.; Ide, N.; Nishimoto-Hazuku, A.; Fujimoto, H.; Saris, C.J.; Yoshida, H.; Node, K. Interleukin 27 inhibits atherosclerosis via immunoregulation of macrophages in mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H420–H429. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, E.K.; Kim, G.; Lloyd, K.M.; Saris, C.J.; von Vietinghoff, S.; Kronenberg, M.; Ley, K. Interleukin-27 receptor limits atherosclerosis in Ldlr-/- mice. Circ. Res. 2012, 111, 1274–1285. [Google Scholar] [CrossRef]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef]
- Kobie, J.J.; Shah, P.R.; Yang, L.; Rebhahn, J.A.; Fowell, D.J.; Mosmann, T.R. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J. Immunol. 2006, 177, 6780–6786. [Google Scholar] [CrossRef]
- Borsellino, G.; Kleinewietfeld, M.; Di Mitri, D.; Sternjak, A.; Diamantini, A.; Giometto, R.; Hopner, S.; Centonze, D.; Bernardi, G.; Dell’Acqua, M.L.; et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood 2007, 110, 1225–1232. [Google Scholar] [CrossRef]
- Zarek, P.E.; Huang, C.T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 2008, 111, 251–259. [Google Scholar] [CrossRef] [PubMed]
- de Boer, O.J.; van der Meer, J.J.; Teeling, P.; van der Loos, C.M.; van der Wal, A.C. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS ONE 2007, 2, e779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mao, S.; Zhan, Z.; Yu, K.; He, C.; Wang, C. Effect of hyperlipidemia on Foxp3 expression in apolipoprotein E-knockout mice. J. Cardiovasc. Med. (Hagerstown) 2014, 15, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.X.; Guo, B.B.; Liu, Q.; Li, Y.K.; Yang, Z.; Feng, W.J.; Mo, Z.C. Regulatory T cells as a new therapeutic target for atherosclerosis. Acta Pharmacol. Sin. 2018, 39, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Yamashita, T.; Kasahara, K.; Takeda, M.; Hirata, K. Regulatory T cells and tolerogenic dendritic cells as critical immune modulators in atherogenesis. Curr. Pharm Des. 2015, 21, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Lahl, K.; Loddenkemper, C.; Drouin, C.; Freyer, J.; Arnason, J.; Eberl, G.; Hamann, A.; Wagner, H.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007, 204, 57–63. [Google Scholar] [CrossRef]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gistera, A.; Strodthoff, D.; Ketelhuth, D.F.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Investig. 2013, 123, 1323–1334. [Google Scholar] [CrossRef]
- Steffens, S.; Burger, F.; Pelli, G.; Dean, Y.; Elson, G.; Kosco-Vilbois, M.; Chatenoud, L.; Mach, F. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation 2006, 114, 1977–1984. [Google Scholar] [CrossRef]
- Sasaki, N.; Yamashita, T.; Takeda, M.; Shinohara, M.; Nakajima, K.; Tawa, H.; Usui, T.; Hirata, K. Oral anti-CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation 2009, 120, 1996–2005. [Google Scholar] [CrossRef]
- Dinh, T.N.; Kyaw, T.S.; Kanellakis, P.; To, K.; Tipping, P.; Toh, B.H.; Bobik, A.; Agrotis, A. Cytokine therapy with interleukin-2/anti-interleukin-2 monoclonal antibody complexes expands CD4+CD25+Foxp3+ regulatory T cells and attenuates development and progression of atherosclerosis. Circulation 2012, 126, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Sasaki, N.; Yamashita, T.; Kita, T.; Yodoi, K.; Sasaki, Y.; Takeda, M.; Hirata, K. CD3 antibody and IL-2 complex combination therapy inhibits atherosclerosis by augmenting a regulatory immune response. J. Am. Heart Assoc. 2014, 3, e000719. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Yamashita, T.; Sasaki, N.; Kasahara, K.; Sasaki, Y.; Yodoi, K.; Takeda, M.; Nakajima, K.; Hirata, K. Regression of atherosclerosis with anti-CD3 antibody via augmenting a regulatory T-cell response in mice. Cardiovasc. Res. 2014, 102, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Yamashita, T.; Kasahara, K.; Fukunaga, A.; Yamaguchi, T.; Emoto, T.; Yodoi, K.; Matsumoto, T.; Nakajima, K.; Kita, T.; et al. UVB Exposure Prevents Atherosclerosis by Regulating Immunoinflammatory Responses. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 66–74. [Google Scholar] [CrossRef]
- Zhu, Z.F.; Meng, K.; Zhong, Y.C.; Qi, L.; Mao, X.B.; Yu, K.W.; Zhang, W.; Zhu, P.F.; Ren, Z.P.; Wu, B.W.; et al. Impaired circulating CD4+ LAP+ regulatory T cells in patients with acute coronary syndrome and its mechanistic study. PLoS ONE 2014, 9, e88775. [Google Scholar] [CrossRef]
- Zhang, W.C.; Wang, J.; Shu, Y.W.; Tang, T.T.; Zhu, Z.F.; Xia, N.; Nie, S.F.; Liu, J.; Zhou, S.F.; Li, J.J.; et al. Impaired thymic export and increased apoptosis account for regulatory T cell defects in patients with non-ST segment elevation acute coronary syndrome. J. Biol. Chem. 2012, 287, 34157–34166. [Google Scholar] [CrossRef]
- Wigren, M.; Bjorkbacka, H.; Andersson, L.; Ljungcrantz, I.; Fredrikson, G.N.; Persson, M.; Bryngelsson, C.; Hedblad, B.; Nilsson, J. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2000–2004. [Google Scholar] [CrossRef]
- Ammirati, E.; Cianflone, D.; Banfi, M.; Vecchio, V.; Palini, A.; De Metrio, M.; Marenzi, G.; Panciroli, C.; Tumminello, G.; Anzuini, A.; et al. Circulating CD4+CD25hiCD127lo regulatory T-Cell levels do not reflect the extent or severity of carotid and coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1832–1841. [Google Scholar] [CrossRef]
- Mor, A.; Luboshits, G.; Planer, D.; Keren, G.; George, J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur Heart J. 2006, 27, 2530–2537. [Google Scholar] [CrossRef]
- Emoto, T.; Sasaki, N.; Yamashita, T.; Kasahara, K.; Yodoi, K.; Sasaki, Y.; Matsumoto, T.; Mizoguchi, T.; Hirata, K. Regulatory/effector T-cell ratio is reduced in coronary artery disease. Circ. J. 2014, 78, 2935–2941. [Google Scholar] [CrossRef]
- Kalia, V.; Sarkar, S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2-A Balancing Act. Front. Immunol. 2018, 9, 2987. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Invest. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed]
- van Duijn, J.; Kuiper, J.; Slutter, B. The many faces of CD8+ T cells in atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Kolbus, D.; Ljungcrantz, I.; Soderberg, I.; Alm, R.; Bjorkbacka, H.; Nilsson, J.; Fredrikson, G.N. TAP1-deficiency does not alter atherosclerosis development in Apoe-/- mice. PLoS ONE 2012, 7, e33932. [Google Scholar] [CrossRef]
- Fyfe, A.I.; Qiao, J.H.; Lusis, A.J. Immune-deficient mice develop typical atherosclerotic fatty streaks when fed an atherogenic diet. J. Clin. Investig. 1994, 94, 2516–2520. [Google Scholar] [CrossRef]
- Chyu, K.Y.; Zhao, X.; Dimayuga, P.C.; Zhou, J.; Li, X.; Yano, J.; Lio, W.M.; Chan, L.F.; Kirzner, J.; Trinidad, P.; et al. CD8+ T cells mediate the athero-protective effect of immunization with an ApoB-100 peptide. PLoS ONE 2012, 7, e30780. [Google Scholar] [CrossRef]
- Gewaltig, J.; Kummer, M.; Koella, C.; Cathomas, G.; Biedermann, B.C. Requirements for CD8 T-cell migration into the human arterial wall. Hum. Pathol 2008, 39, 1756–1762. [Google Scholar] [CrossRef]
- van Duijn, J.; van Elsas, M.; Benne, N.; Depuydt, M.; Wezel, A.; Smeets, H.; Bot, I.; Jiskoot, W.; Kuiper, J.; Slutter, B. CD39 identifies a microenvironment-specific anti-inflammatory CD8(+) T-cell population in atherosclerotic lesions. Atherosclerosis 2019, 285, 71–78. [Google Scholar] [CrossRef]
- Barquera, S.; Pedroza-Tobias, A.; Medina, C.; Hernandez-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global Overview of the Epidemiology of Atherosclerotic Cardiovascular Disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef]
- Kolbus, D.; Ramos, O.H.; Berg, K.E.; Persson, J.; Wigren, M.; Bjorkbacka, H.; Fredrikson, G.N.; Nilsson, J. CD8+ T cell activation predominate early immune responses to hypercholesterolemia in Apoe(-)(/)(-) mice. BMC Immunol. 2010, 11, 58. [Google Scholar] [CrossRef]
- Olofsson, P.S.; Soderstrom, L.A.; Wagsater, D.; Sheikine, Y.; Ocaya, P.; Lang, F.; Rabu, C.; Chen, L.; Rudling, M.; Aukrust, P.; et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation 2008, 117, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Winship, A.; Tay, C.; Kanellakis, P.; Hosseini, H.; Cao, A.; Li, P.; Tipping, P.; Bobik, A.; Toh, B.H. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation 2013, 127, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.G.; Ji, Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R.; Mandal, M.; Ochiai, K.; Singh, H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 2014, 14, 69–80. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 2005, 310, 1510–1512. [Google Scholar] [CrossRef]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol 2009, 9, 767–777. [Google Scholar] [CrossRef]
- Hardy, R.R.; Kincade, P.W.; Dorshkind, K. The protean nature of cells in the B lymphocyte lineage. Immunity 2007, 26, 703–714. [Google Scholar] [CrossRef]
- Hardy, R.R. B-1 B cells: Development, selection, natural autoantibody and leukemia. Curr. Opin. Immunol. 2006, 18, 547–555. [Google Scholar] [CrossRef]
- Baumgarth, N. B-1 Cell Heterogeneity and the Regulation of Natural and Antigen-Induced IgM Production. Front. Immunol. 2016, 7, 324. [Google Scholar] [CrossRef]
- Choi, Y.S.; Dieter, J.A.; Rothaeusler, K.; Luo, Z.; Baumgarth, N. B-1 cells in the bone marrow are a significant source of natural IgM. Eur. J. Immunol. 2012, 42, 120–129. [Google Scholar] [CrossRef]
- Berland, R.; Wortis, H.H. Origins and functions of B-1 cells with notes on the role of CD5. Annu. Rev. Immunol. 2002, 20, 253–300. [Google Scholar] [CrossRef] [PubMed]
- Srikakulapu, P.; McNamara, C.A. B cells and atherosclerosis. Am. J. Physiol Heart Circ. Physiol. 2017, 312, H1060–H1067. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Shukla, S.; Lyszkiewicz, M.; Krey, M.; Viegas, N.; Duber, S.; Weiss, S. Somatic hypermutation in peritoneal B1b cells. Mol. Immunol. 2009, 46, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Brennecke, A.M.; Agarwal, S.; Krey, M.; Duber, S.; Weiss, S. An intrinsic propensity of murine peritoneal B1b cells to switch to IgA in presence of TGF-beta and retinoic acid. PLoS ONE 2013, 8, e82121. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.T.; Roghanian, A.; Cragg, M.S. B cells--masters of the immunoverse. Int. J. Biochem. Cell Biol. 2011, 43, 280–285. [Google Scholar] [CrossRef]
- Hamel, K.M.; Liarski, V.M.; Clark, M.R. Germinal center B-cells. Autoimmunity 2012, 45, 333–347. [Google Scholar] [CrossRef]
- Shen, P.; Fillatreau, S. Antibody-independent functions of B cells: A focus on cytokines. Nat. Rev. Immunol. 2015, 15, 441–451. [Google Scholar] [CrossRef]
- Lund, F.E. Cytokine-producing B lymphocytes-key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef]
- Lund, F.E.; Garvy, B.A.; Randall, T.D.; Harris, D.P. Regulatory roles for cytokine-producing B cells in infection and autoimmune disease. Curr. Dir. Autoimmun. 2005, 8, 25–54. [Google Scholar]
- Parekh, V.V.; Prasad, D.V.; Banerjee, P.P.; Joshi, B.N.; Kumar, A.; Mishra, G.C. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8+ T cells: Role of TGF-beta 1. J. Immunol. 2003, 170, 5897–5911. [Google Scholar] [CrossRef]
- Tian, J.; Zekzer, D.; Hanssen, L.; Lu, Y.; Olcott, A.; Kaufman, D.L. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J. Immunol. 2001, 167, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Bouaziz, J.D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008, 28, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, G.; Nicoletti, A.; Poirier, B.; Hansson, G.K. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J. Clin. Investig. 2002, 109, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Major, A.S.; Fazio, S.; Linton, M.F. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1892–1898. [Google Scholar] [CrossRef]
- Doran, A.C.; Lipinski, M.J.; Oldham, S.N.; Garmey, J.C.; Campbell, K.A.; Skaflen, M.D.; Cutchins, A.; Lee, D.J.; Glover, D.K.; Kelly, K.A.; et al. B-cell aortic homing and atheroprotection depend on Id3. Circ. Res. 2012, 110, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W.; Miller, E.; Witztum, J.L. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 821–825. [Google Scholar] [CrossRef]
- Ameli, S.; Hultgardh-Nilsson, A.; Regnstrom, J.; Calara, F.; Yano, J.; Cercek, B.; Shah, P.K.; Nilsson, J. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1074–1079. [Google Scholar] [CrossRef]
- Sage, A.P.; Tsiantoulas, D.; Baker, L.; Harrison, J.; Masters, L.; Murphy, D.; Loinard, C.; Binder, C.J.; Mallat, Z. BAFF receptor deficiency reduces the development of atherosclerosis in mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1573–1576. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Khan, A.; Dumouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Hosseini, H.; Kanellakis, P.; Gadowski, T.; MacKay, F.; Tipping, P.; Bobik, A.; Toh, B.H. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS ONE 2012, 7, e29371. [Google Scholar] [CrossRef]
- Kyaw, T.; Cui, P.; Tay, C.; Kanellakis, P.; Hosseini, H.; Liu, E.; Rolink, A.G.; Tipping, P.; Bobik, A.; Toh, B.H. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(-/-) mice. PLoS ONE 2013, 8, e60430. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Herbin, O.; Bouaziz, J.D.; Binder, C.J.; Uyttenhove, C.; Laurans, L.; Taleb, S.; Van Vre, E.; Esposito, B.; Vilar, J.; et al. B cell depletion reduces the development of atherosclerosis in mice. J. Exp. Med. 2010, 207, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Sage, A.P.; Goderle, L.; Ozsvar-Kozma, M.; Murphy, D.; Porsch, F.; Pasterkamp, G.; Menche, J.; Schneider, P.; Mallat, Z.; et al. B Cell-Activating Factor Neutralization Aggravates Atherosclerosis. Circulation 2018, 138, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Horkko, S.; Bird, D.A.; Miller, E.; Itabe, H.; Leitinger, N.; Subbanagounder, G.; Berliner, J.A.; Friedman, P.; Dennis, E.A.; Curtiss, L.K.; et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J. Clin. Investig. 1999, 103, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Malik, T.H.; Ehrenstein, M.R.; Boyle, J.J.; Botto, M.; Haskard, D.O. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2009, 120, 417–426. [Google Scholar] [CrossRef]
- Hosseini, H.; Li, Y.; Kanellakis, P.; Tay, C.; Cao, A.; Liu, E.; Peter, K.; Tipping, P.; Toh, B.H.; Bobik, A.; et al. Toll-Like Receptor (TLR)4 and MyD88 are Essential for Atheroprotection by Peritoneal B1a B Cells. J. Am. Heart Assoc. 2016, 5, e002947. [Google Scholar] [CrossRef]
- Rosenfeld, S.M.; Perry, H.M.; Gonen, A.; Prohaska, T.A.; Srikakulapu, P.; Grewal, S.; Das, D.; McSkimming, C.; Taylor, A.M.; Tsimikas, S.; et al. B-1b Cells Secrete Atheroprotective IgM and Attenuate Atherosclerosis. Circ. Res. 2015, 117, e28–e39. [Google Scholar] [CrossRef]
- Bagchi-Chakraborty, J.; Francis, A.; Bray, T.; Masters, L.; Tsiantoulas, D.; Nus, M.; Harrison, J.; Broekhuizen, M.; Leggat, J.; Clatworthy, M.R.; et al. B Cell Fcgamma Receptor IIb Modulates Atherosclerosis in Male and Female Mice by Controlling Adaptive Germinal Center and Innate B-1-Cell Responses. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1379–1389. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Mauri, C.; Gray, D.; Mushtaq, N.; Londei, M. Prevention of arthritis by interleukin 10-producing B cells. J. Exp. Med. 2003, 197, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Rattik, S.; Mantani, P.T.; Yao Mattisson, I.; Ljungcrantz, I.; Sundius, L.; Bjorkbacka, H.; Terrinoni, M.; Lebens, M.; Holmgren, J.; Nilsson, J.; et al. B cells treated with CTB-p210 acquire a regulatory phenotype in vitro and reduce atherosclerosis in apolipoprotein E deficient mice. Vascul. Pharmacol. 2018, 111, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.C.; Cross, A.J.; Cole, J.E.; Blair, P.A.; Leib, C.; Goddard, M.E.; Rosser, E.C.; Park, I.; Hultgardh Nilsson, A.; Nilsson, J.; et al. B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb. Haemost. 2015, 114, 835–847. [Google Scholar] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Investigators, C. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Everett, B.M.; Pradhan, A.D.; Solomon, D.H.; Paynter, N.; Macfadyen, J.; Zaharris, E.; Gupta, M.; Clearfield, M.; Libby, P.; Hasan, A.A.; et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: A test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 2013, 166, 199–207. [Google Scholar] [CrossRef]
- Palmer, R.D.; Vaccarezza, M. New Promises and Challenges on Inflammation and Atherosclerosis: Insights From CANTOS and CIRT Trials. Front. Cardiovasc. Med. 2019, 6, 90. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. https://doi.org/10.3390/ijms20215293
Herrero-Fernandez B, Gomez-Bris R, Somovilla-Crespo B, Gonzalez-Granado JM. Immunobiology of Atherosclerosis: A Complex Net of Interactions. International Journal of Molecular Sciences. 2019; 20(21):5293. https://doi.org/10.3390/ijms20215293
Chicago/Turabian StyleHerrero-Fernandez, Beatriz, Raquel Gomez-Bris, Beatriz Somovilla-Crespo, and Jose Maria Gonzalez-Granado. 2019. "Immunobiology of Atherosclerosis: A Complex Net of Interactions" International Journal of Molecular Sciences 20, no. 21: 5293. https://doi.org/10.3390/ijms20215293
APA StyleHerrero-Fernandez, B., Gomez-Bris, R., Somovilla-Crespo, B., & Gonzalez-Granado, J. M. (2019). Immunobiology of Atherosclerosis: A Complex Net of Interactions. International Journal of Molecular Sciences, 20(21), 5293. https://doi.org/10.3390/ijms20215293