Production of IFNβ by Conventional Dendritic Cells after Stimulation with Viral Compounds and IFNβ-Independent IFNAR1-Signaling Pathways are Associated with Aggravation of Polymicrobial Sepsis


Abstract
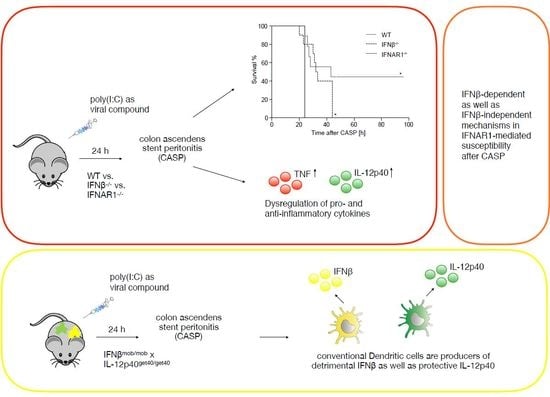
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
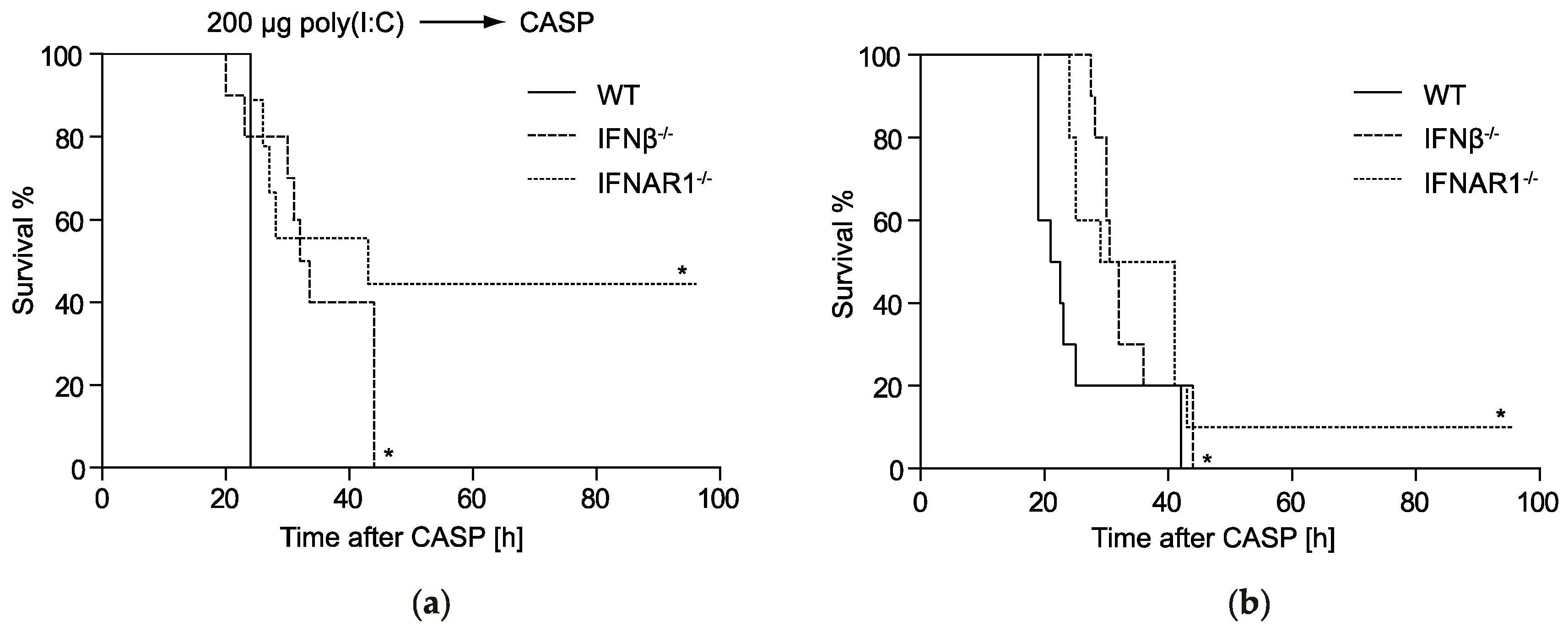
2.1. IFNβ Deficiency Increases Early Survival in Polymicrobial Peritonitis
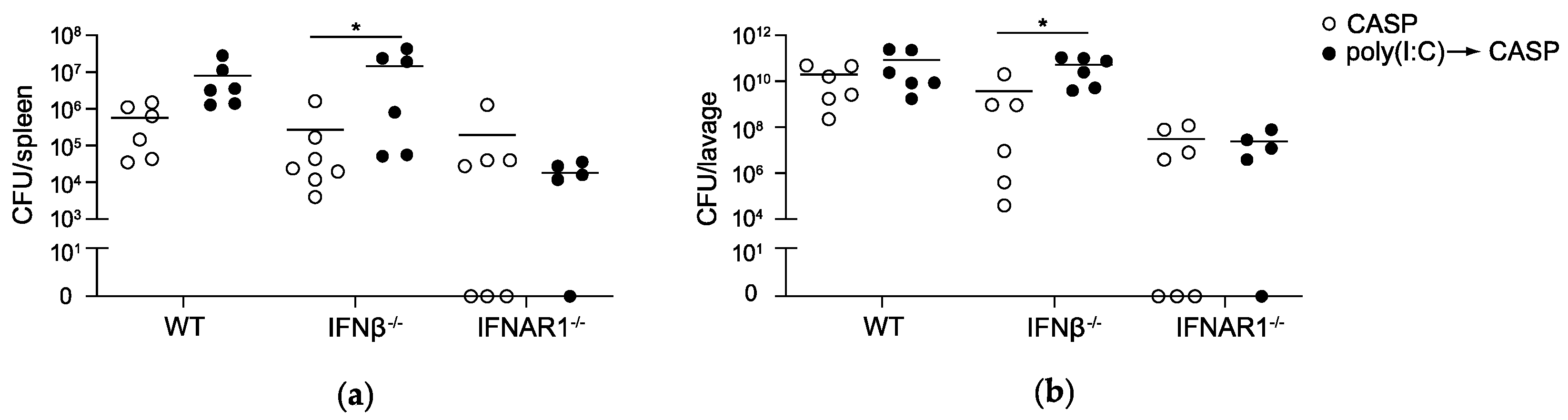
2.2. IFNAR1 Deficiency, but Not IFNβ Deficiency Prevents an Increase in Bacterial Counts Early after Poly(I:C) Sensitization
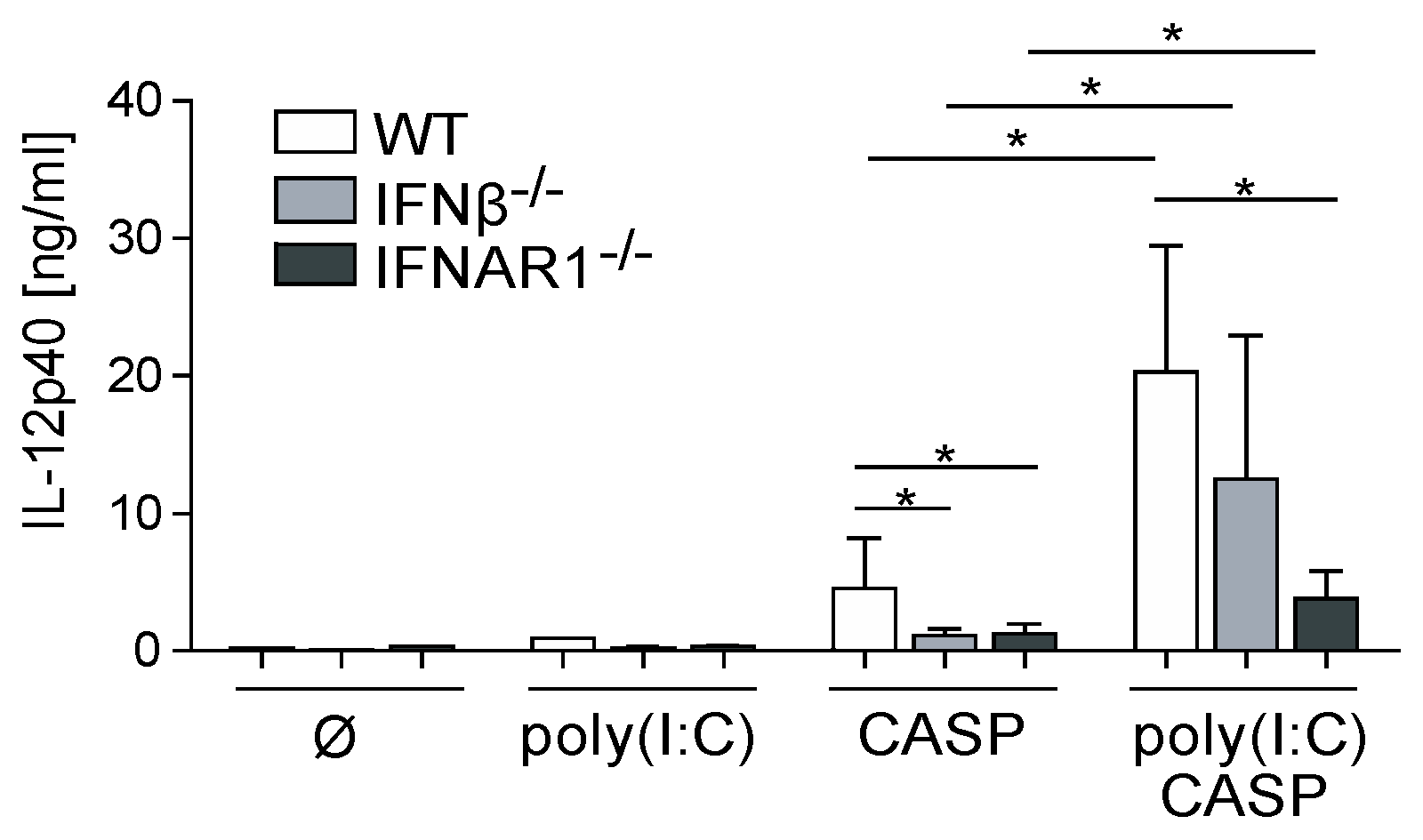
2.3. Dysregulated IL-12p40 Production during Septic Peritonitis Following Poly(I:C) Treatment
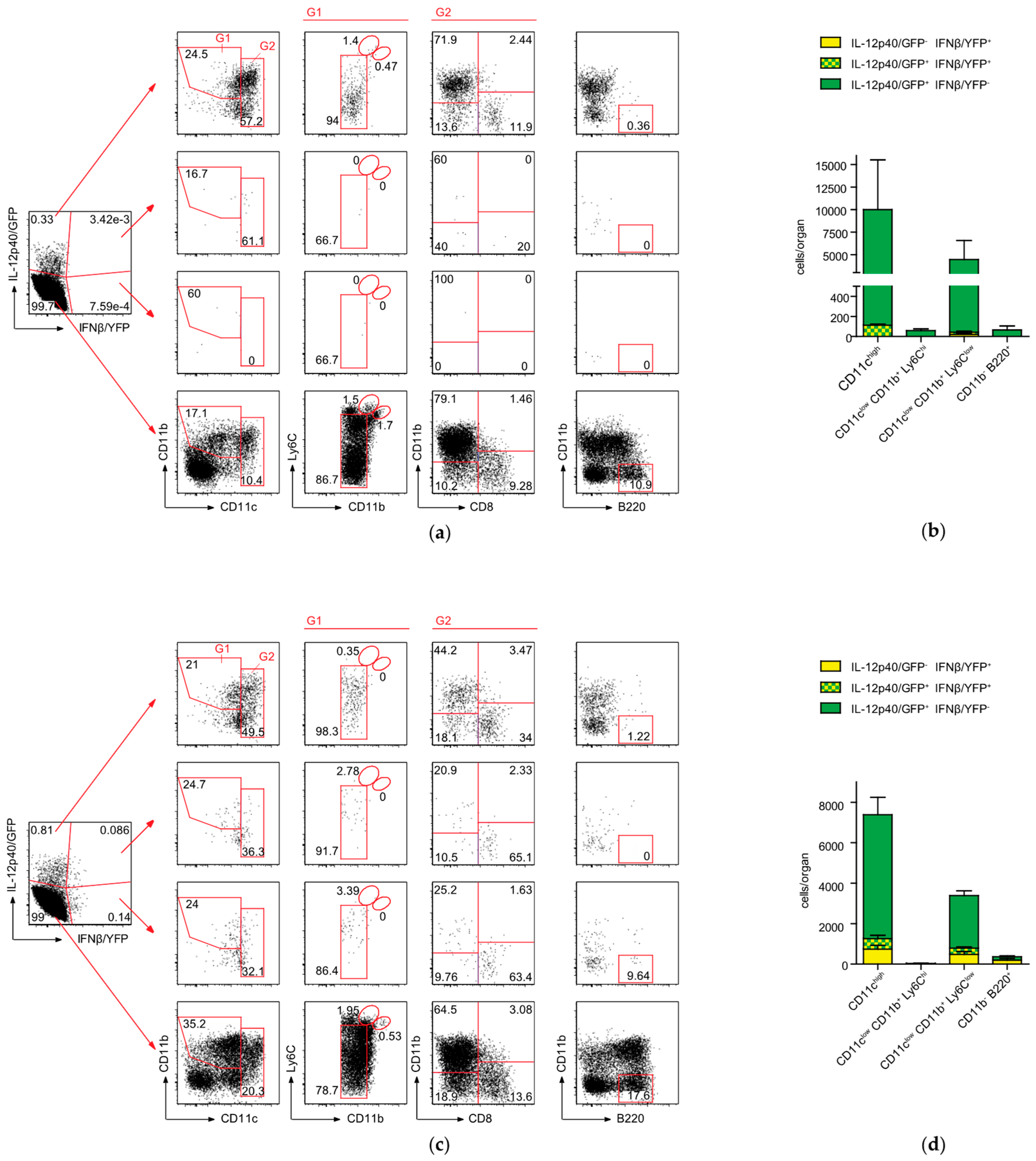
2.4. Conventional DCs Represent the Primary Source of IFNβ and IL-12p40 After CASP
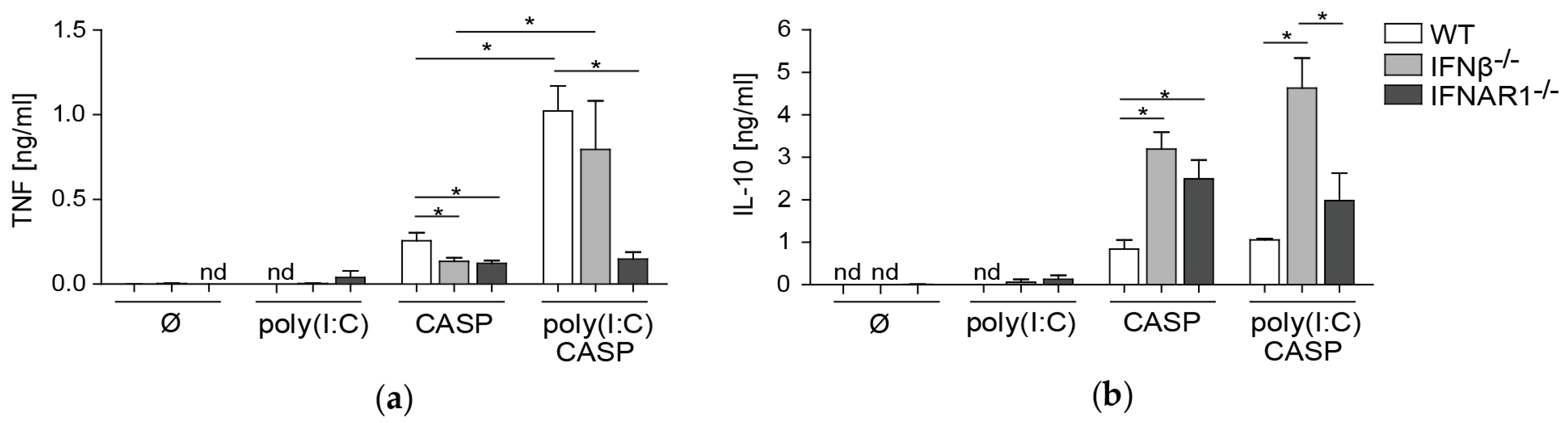
2.5. TNF and IL-10 Levels in the Peritoneal Lavage during Septic Peritonitis Are Differentially Modulated Following Poly(I:C) Challenge
3. Discussion
4. Materials and Methods
4.1. Mice, Colon Ascendens Stent Peritonitis (CASP), Poly(I:C) Stimulation, and Listeria Monocytogenes Infection
4.2. Bacterial Counts
4.3. Cytokine Production
4.4. Antibodies
4.5. FACS Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CASP | Colon Ascendens Stent Peritonitis |
| cDC | conventional Dendritic Cell |
| CFU | Colony Forming Unit |
| DC | Dendritic Cell |
| ELISA | Enzyme Linked Immunosorbent Assay |
| GFP | Green Fluorescent Protein |
| IFN | Interferon |
| IFNAR | Type I IFN Receptor |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| pDC | plasmacytoid Dendritic Cell |
| poly(I:C) | polyinosinic polycytidylic acid |
| SD | Standard Deviation |
| SEM | Standard Error of Mean |
| TNF | Tumor Necrosis Factor |
| WT | Wildtype |
| YFP | Yellow Fluorescent Protein |
References
- Dellinger, R.P.; Levy, M.M.; Carlet, J.M.; Bion, J.; Parker, M.M.; Jaeschke, R.; Reinhart, K.; Angus, D.C.; Brun-Buisson, C.; Beale, R.; et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008, 34, 17–60. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Liu, V.X.; Iwashyna, T.J.; Brunkhorst, F.M.; Rea, T.D.; Scherag, A.; Rubenfeld, G.; Kahn, J.M.; Shankar-Hari, M.; Singer, M.; et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Shankar-Hari, M.; Phillips, G.S.; Levy, M.L.; Seymour, C.W.; Liu, V.X.; Deutschman, C.S.; Angus, D.C.; Rubenfeld, G.D.; Singer, M.; Sepsis Definitions Task, F. Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 775–787. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef]
- van der Poll, T.; Opal, S.M. Host-pathogen interactions in sepsis. Lancet Infect. Dis. 2008, 8, 32–43. [Google Scholar] [CrossRef]
- Rackov, G.; Shokri, R.; De Mon, M.A.; Martinez, A.C.; Balomenos, D. The Role of IFN-beta during the Course of Sepsis Progression and Its Therapeutic Potential. Front. Immunol. 2017, 8, 493. [Google Scholar] [CrossRef]
- Zantl, N.; Uebe, A.; Neumann, B.; Wagner, H.; Siewert, J.R.; Holzmann, B.; Heidecke, C.D.; Pfeffer, K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect. Immun. 1998, 66, 2300–2309. [Google Scholar]
- Entleutner, M.; Traeger, T.; Westerholt, A.; Holzmann, B.; Stier, A.; Pfeffer, K.; Maier, S.; Heidecke, C.D. Impact of interleukin-12, oxidative burst, and iNOS on the survival of murine fecal peritonitis. Int J. Colorectal Dis. 2006, 21, 64–70. [Google Scholar] [CrossRef]
- Beadling, C.; Slifka, M.K. How do viral infections predispose patients to bacterial infections? Curr. Opin. Infect. Dis. 2004, 17, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Huys, L.; Van Hauwermeiren, F.; Dejager, L.; Dejonckheere, E.; Lienenklaus, S.; Weiss, S.; Leclercq, G.; Libert, C. Type I interferon drives tumor necrosis factor-induced lethal shock. J. Exp. Med. 2009, 206, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Weighardt, H.; Kaiser-Moore, S.; Schlautkotter, S.; Rossmann-Bloeck, T.; Schleicher, U.; Bogdan, C.; Holzmann, B. Type I IFN modulates host defense and late hyperinflammation in septic peritonitis. J. Immunol. 2006, 177, 5623–5630. [Google Scholar] [CrossRef] [PubMed]
- Navarini, A.A.; Recher, M.; Lang, K.S.; Georgiev, P.; Meury, S.; Bergthaler, A.; Flatz, L.; Bille, J.; Landmann, R.; Odermatt, B.; et al. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc. Natl. Acad. Sci. USA 2006, 103, 15535–15539. [Google Scholar] [CrossRef] [PubMed]
- Doughty, L.A.; Carlton, S.; Galen, B.; Cooma-Ramberan, I.; Chung, C.S.; Ayala, A. Activation of common antiviral pathways can potentiate inflammatory responses to septic shock. Shock 2006, 26, 187–194. [Google Scholar] [CrossRef]
- Hensler, T.; Heidecke, C.D.; Hecker, H.; Heeg, K.; Bartels, H.; Zantl, N.; Wagner, H.; Siewert, J.R.; Holzmann, B. Increased susceptibility to postoperative sepsis in patients with impaired monocyte IL-12 production. J. Immunol. 1998, 161, 2655–2659. [Google Scholar] [PubMed]
- Scheu, S.; Dresing, P.; Locksley, R.M. Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo. Proc. Natl. Acad. Sci. USA. 2008, 105, 20416–20421. [Google Scholar] [CrossRef]
- Reinhardt, R.L.; Hong, S.; Kang, S.J.; Wang, Z.E.; Locksley, R.M. Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J. Immunol. 2006, 177, 1618–1627. [Google Scholar] [CrossRef]
- Bauer, J.; Dress, R.J.; Schulze, A.; Dresing, P.; Ali, S.; Deenen, R.; Alferink, J.; Scheu, S. Cutting Edge: IFN-beta Expression in the Spleen Is Restricted to a Subpopulation of Plasmacytoid Dendritic Cells Exhibiting a Specific Immune Modulatory Transcriptome Signature. J. Immunol. 2016, 196, 4447–4451. [Google Scholar] [CrossRef]
- Dresing, P.; Borkens, S.; Kocur, M.; Kropp, S.; Scheu, S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon beta in murine listeriosis. PLoS ONE 2010, 5, e15567. [Google Scholar] [CrossRef]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Leiner, I.; Dorothee, G.; Brandl, K.; Pamer, E.G. MyD88 and Type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J. Immunol. 2009, 183, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Coordinating innate immune cells to optimize microbial killing. Immunity 2008, 29, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Riche, F.C.; Cholley, B.P.; Panis, Y.H.; Laisne, M.J.; Briard, C.G.; Graulet, A.M.; Gueris, J.L.; Valleur, P.D. Inflammatory cytokine response in patients with septic shock secondary to generalized peritonitis. Crit. Care Med. 2000, 28, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Stamme, C.; Bundschuh, D.S.; Hartung, T.; Gebert, U.; Wollin, L.; Nusing, R.; Wendel, A.; Uhlig, S. Temporal sequence of pulmonary and systemic inflammatory responses to graded polymicrobial peritonitis in mice. Infect. Immun. 1999, 67, 5642–5650. [Google Scholar] [PubMed]
- Sethi, S. Bacterial pneumonia. Managing a deadly complication of influenza in older adults with comorbid disease. Geriatrics 2002, 57, 56–61. [Google Scholar]
- Jarstrand, C.; Tunevall, G. The influence of bacterial superinfection on the clinical course of influenza. Studies from the influenza epidemics in Stockholm during the winters 1969-70 and 1971-72. Scand. J. Infect. Dis. 1975, 7, 243–247. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Dejager, L.; Vandevyver, S.; Ballegeer, M.; Van Wonterghem, E.; An, L.L.; Riggs, J.; Kolbeck, R.; Libert, C. Pharmacological inhibition of type I interferon signaling protects mice against lethal sepsis. J. Infect. Dis. 2014, 209, 960–970. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Vivian, J.P.; Nguyen, T.K.; Mangan, N.E.; Gould, J.A.; Braniff, S.J.; Zaker-Tabrizi, L.; Fung, K.Y.; Forster, S.C.; Beddoe, T.; et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- McCartney, S.; Vermi, W.; Gilfillan, S.; Cella, M.; Murphy, T.L.; Schreiber, R.D.; Murphy, K.M.; Colonna, M. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J. Exp. Med. 2009, 206, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Emmanuilidis, K.; Weighardt, H.; Maier, S.; Gerauer, K.; Fleischmann, T.; Zheng, X.X.; Hancock, W.W.; Holzmann, B.; Heidecke, C.D. Critical role of Kupffer cell-derived IL-10 for host defense in septic peritonitis. J. Immunol. 2001, 167, 3919–3927. [Google Scholar] [CrossRef] [PubMed]
- Song, G.Y.; Chung, C.S.; Chaudry, I.H.; Ayala, A. What is the role of interleukin 10 in polymicrobial sepsis: Anti-inflammatory agent or immunosuppressant? Surgery 1999, 126, 378–383. [Google Scholar] [CrossRef]
- Solodova, E.; Jablonska, J.; Weiss, S.; Lienenklaus, S. Production of IFN-beta during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS ONE 2011, 6, e18543. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson, L.; Blumenthal, R.; Eloranta, M.L.; Engel, H.; Alm, G.; Weiss, S.; Leanderson, T. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr. Biol. 1998, 8, 223–226. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Hertzog, P.J.; Holland, K.A.; Sumarsono, S.H.; Tymms, M.J.; Hamilton, J.A.; Whitty, G.; Bertoncello, I.; Kola, I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc. Natl. Acad. Sci. USA 1995, 92, 11284–11288. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howe, M.; Bauer, J.; Schulze, A.; Kropp, S.; Locksley, R.M.; Alferink, J.; Weighardt, H.; Scheu, S. Production of IFNβ by Conventional Dendritic Cells after Stimulation with Viral Compounds and IFNβ-Independent IFNAR1-Signaling Pathways are Associated with Aggravation of Polymicrobial Sepsis. Int. J. Mol. Sci. 2019, 20, 4410. https://doi.org/10.3390/ijms20184410
Howe M, Bauer J, Schulze A, Kropp S, Locksley RM, Alferink J, Weighardt H, Scheu S. Production of IFNβ by Conventional Dendritic Cells after Stimulation with Viral Compounds and IFNβ-Independent IFNAR1-Signaling Pathways are Associated with Aggravation of Polymicrobial Sepsis. International Journal of Molecular Sciences. 2019; 20(18):4410. https://doi.org/10.3390/ijms20184410
Chicago/Turabian StyleHowe, Magdalena, Jens Bauer, Anja Schulze, Sonja Kropp, Richard M. Locksley, Judith Alferink, Heike Weighardt, and Stefanie Scheu. 2019. "Production of IFNβ by Conventional Dendritic Cells after Stimulation with Viral Compounds and IFNβ-Independent IFNAR1-Signaling Pathways are Associated with Aggravation of Polymicrobial Sepsis" International Journal of Molecular Sciences 20, no. 18: 4410. https://doi.org/10.3390/ijms20184410
APA StyleHowe, M., Bauer, J., Schulze, A., Kropp, S., Locksley, R. M., Alferink, J., Weighardt, H., & Scheu, S. (2019). Production of IFNβ by Conventional Dendritic Cells after Stimulation with Viral Compounds and IFNβ-Independent IFNAR1-Signaling Pathways are Associated with Aggravation of Polymicrobial Sepsis. International Journal of Molecular Sciences, 20(18), 4410. https://doi.org/10.3390/ijms20184410