The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma


Abstract
1. Introduction
2. Results
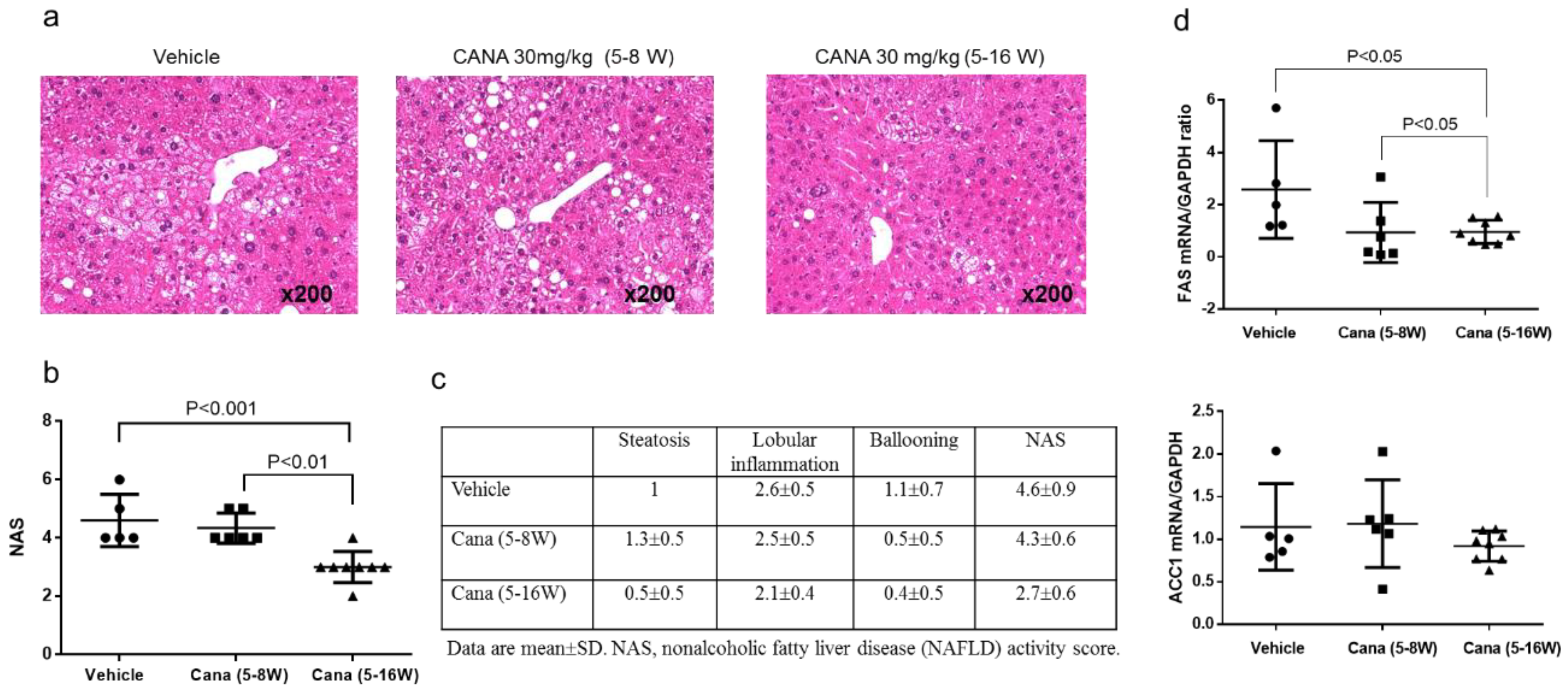
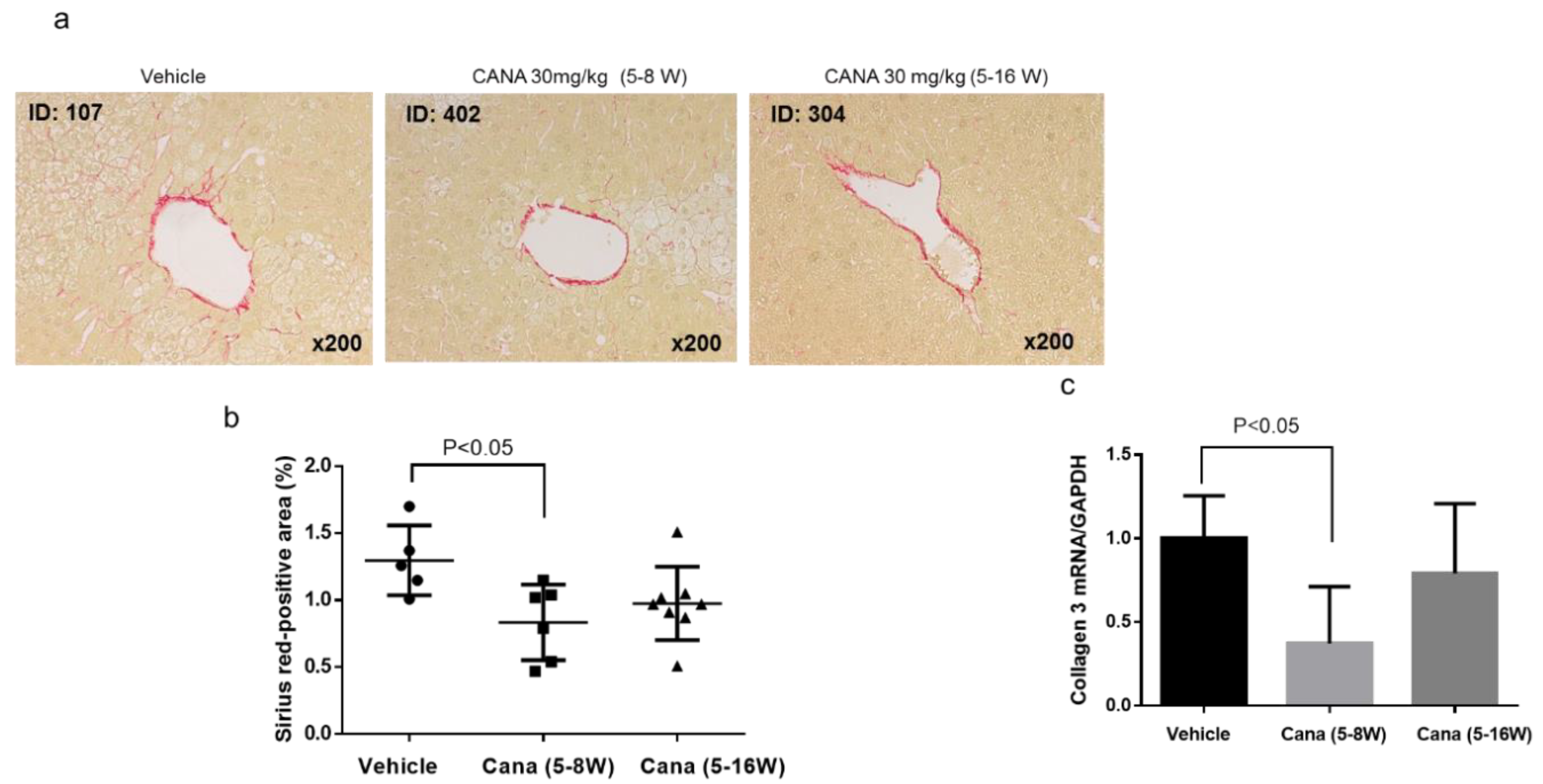
2.1. Effect of Canagliflozin on the NAFLD Histological Activity Score (NAS) and Hepatic Fibrosis
2.2. Effect of Canagliflozin on Body Weight, Liver Weight, and Biochemical Parameters
2.2.1. Effect of Early Canagliflozin Administration (5–9 Weeks) or Continuous Canagliflozin Administration (5–16 Weeks) on the NAS
2.2.2. Effect of Canagliflozin on Hepatic Fibrosis
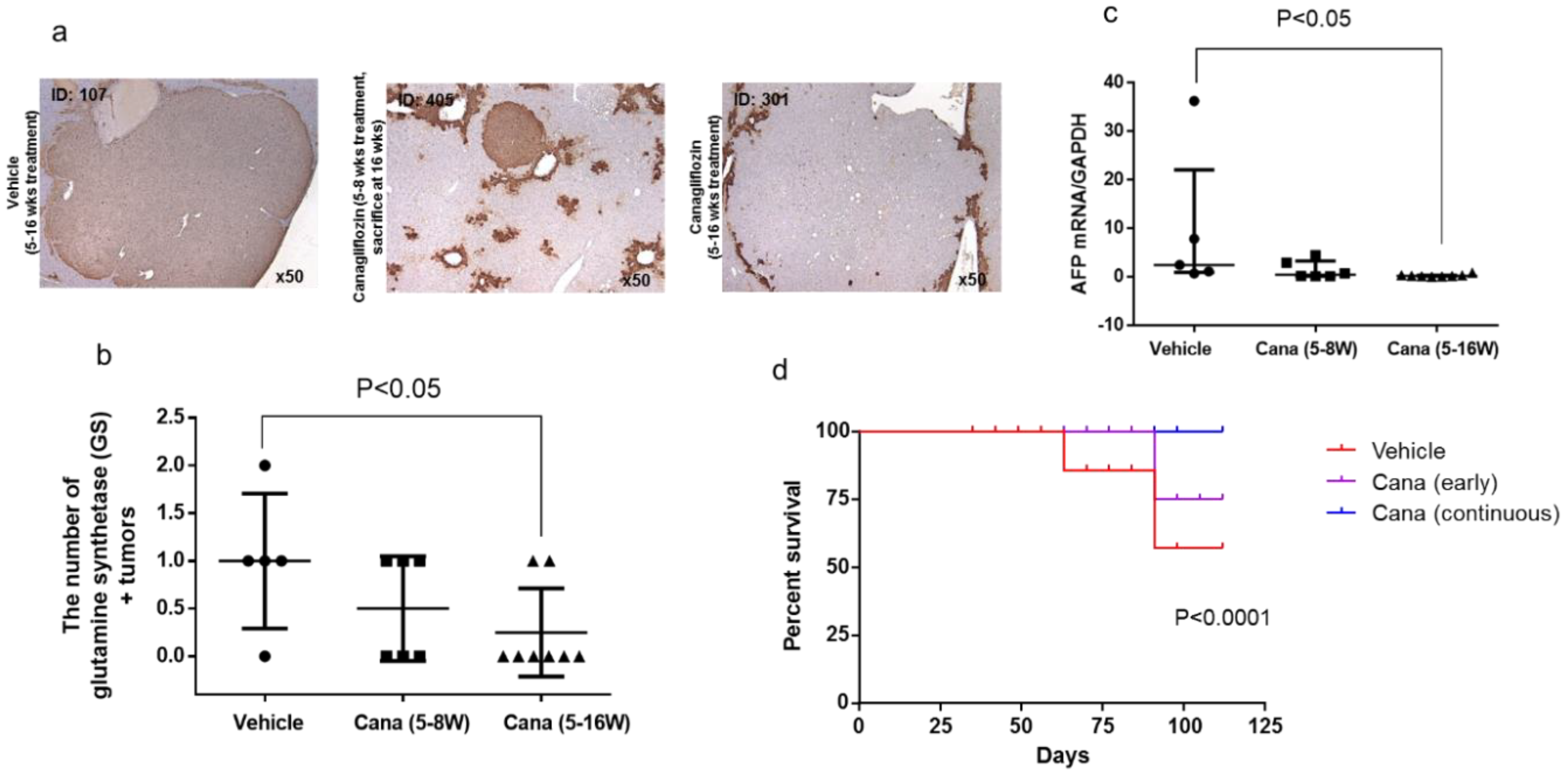
2.2.3. Canagliflozin Inhibits Hepatic Tumorigenesis
2.2.4. Canagliflozin Inhibits Progression of NASH to Hepatocarcinogenesis
2.2.5. Hepatic and Tumor SGLT1/SGLT2 Expression in the Vehicle Group at Eight and 16 Weeks
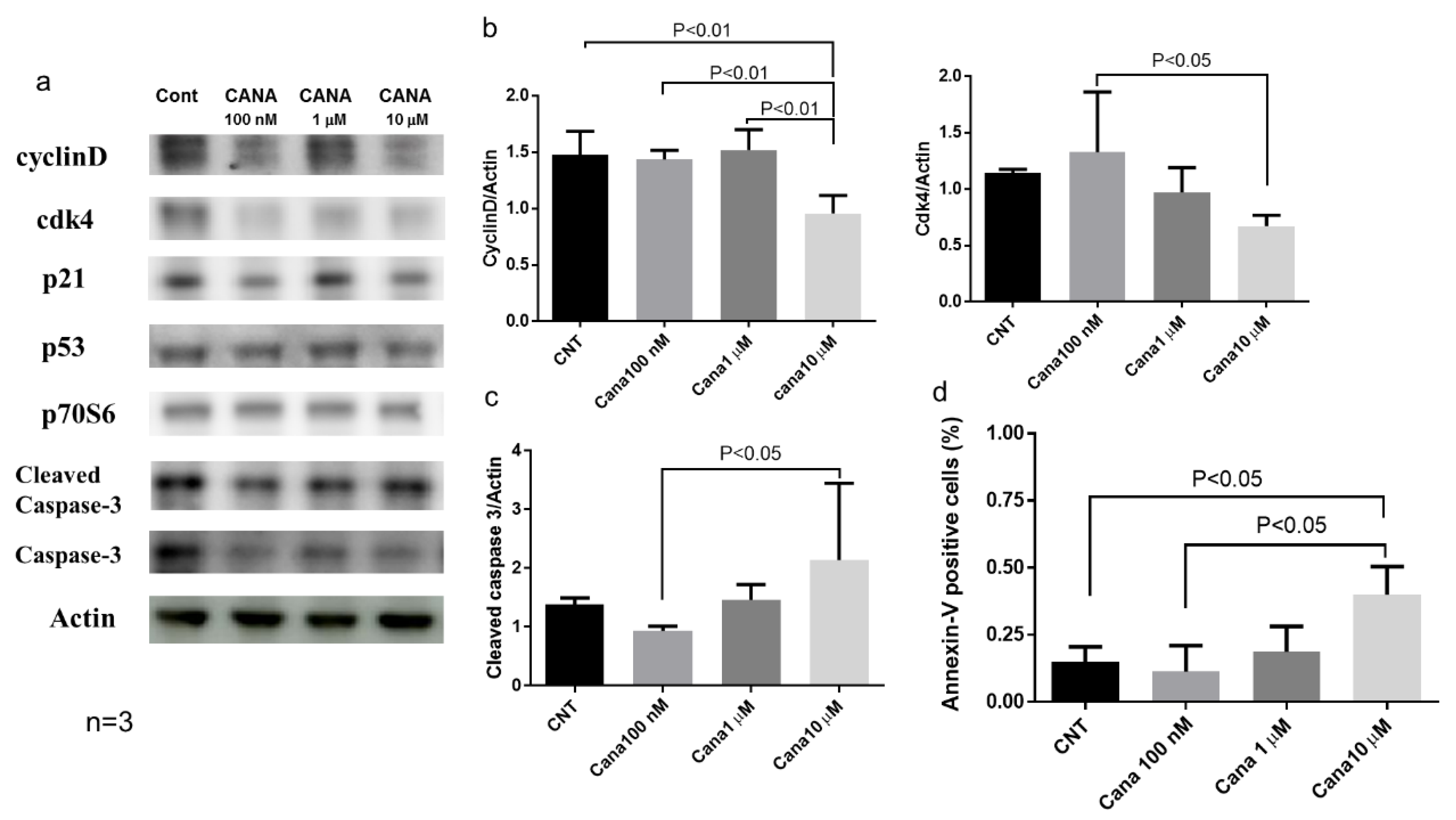
2.3. Effects of Canagliflozin on Real-Time Proliferation of HepG2 Cells Evaluated by the xCELLigence DP System
Effects of Canagliflozin on Cell Cycle-Related Proteins and Apoptosis
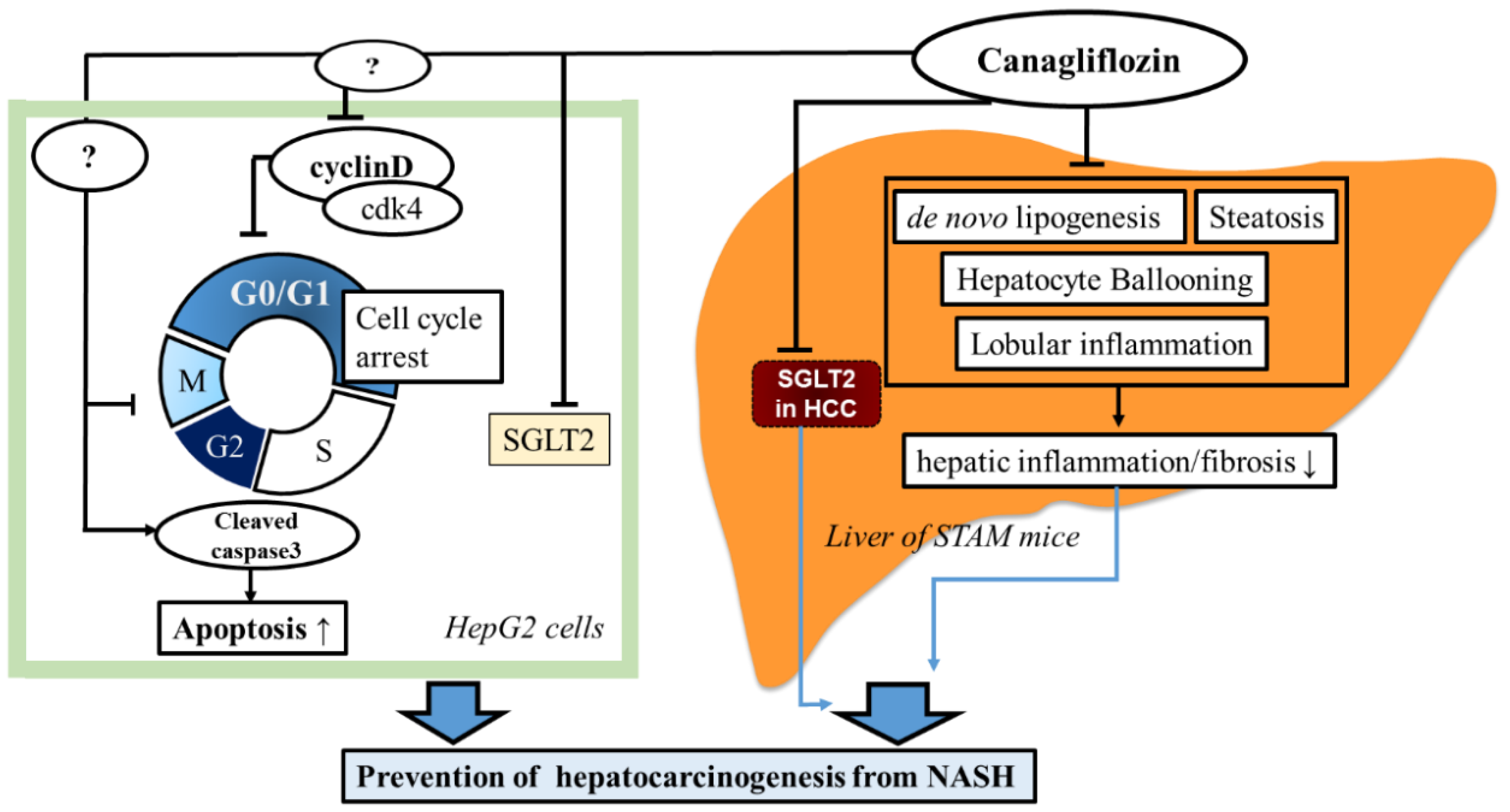
3. Discussion
4. Materials and Methods
4.1. Animal Model and Induction of NASH
4.1.1. Study 1
4.1.2. Study 2
4.2. Cell Culture and Reagents
4.3. Measurement of Cell Proliferation
4.4. Western Blotting
4.5. Quantitative Real-Time RT-PCR
4.6. Cell Cycle Analysis
4.7. Flow Cytometric Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forner, A.; Reiq, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ertle, J.; Dechêne, A.; Sowa, J.-P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.-K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; King, L.Y.; Chong, D.Q.; Nguyen, L.H.; Ma, Y.; VoPham, T.; Giovannucci, E.L.; Fuchs, C.S.; Meyerhardt, J.A.; Corey, K.E.; et al. Diabetes, metabolic comorbidities, and risk of hepatocellular carcinoma: Results from two prospective cohort studies. Hepatology 2018, 67, 1797–1806. [Google Scholar] [CrossRef]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef]
- Komiya, C.; Tsuchiya, K.; Shiba, K.; Miyachi, Y.; Furuke, S.; Shimazu, N.; Yamaguchi, S.; Kanno, K.; Ogawa, Y. Ipragliflozin Improves Hepatic Steatosis in Obese Mice and Liver Dysfunction in Type 2 Diabetic Patients Irrespective of Body Weight Reduction. PLoS ONE 2016, 11, e0151511. [Google Scholar] [CrossRef]
- Qiang, S.; Nakatsu, Y.; Seno, Y.; Fujishiro, M.; Sakoda, H.; Kushiyama, A.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Kamata, H.; et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol. Metab. Syndr. 2015, 7, 104. [Google Scholar] [CrossRef]
- Seko, Y.; Nishikawa, T.; Umemura, A.; Yamaguchi, K.; Moriguchi, M.; Yasui, K.; Kimura, M.; Iijima, H.; Hashimoto, T.; Sumida, Y.; et al. Efficacy and safety of canagliflozin in type 2 diabetes mellitus patients with biopsy-proven nonalcoholic steatohepatitis classified as stage 1–3 fibrosis. Diabetes Metab. Syndr. Obes. 2018, 11, 835–843. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Shimizu, M.; Suzuki, K.; Kato, K.; Jojima, T.; Iijima, T.; Murohisa, T.; Iijima, M.; Takekawa, H.; Usui, I.; Hiraishi, H.; et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2019, 21, 285–292. [Google Scholar] [CrossRef]
- Jakher, H.; Chang, T.I.; Tan, M.; Mahaffey, K.W. Canagliflozin review – safety and efficacy profile in patients with T2DM. Diabetes Metab. Syndr. Obes. 2019, 12, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Arakawa, K.; Ueta, K.; Matsushita, Y.; Kuriyama, C.; Martin, T.; Du, F.; Liu, Y.; Xu, J.; Conway, B.; et al. Effect of Canagliflozin on Renal Threshold for Glucose, Glycemia, and Body Weight in Normal and Diabetic Animal Models. PLoS ONE 2012, 7, e30555. [Google Scholar] [CrossRef] [PubMed]
- Akuta, N.; Kawamura, Y.; Watanabe, C.; Nishimura, A.; Okubo, M.; Mori, Y.; Fujiyama, S.; Sezaki, H.; Hosaka, T.; Kobayashi, M.; et al. Impact of sodium glucose cotransporter 2 inhibitor on histological features and glucose metabolism of non-alcoholic fatty liver disease complicated by diabetes mellitus. Hepatol. Res. 2019, 49, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Urstad, A.P.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: housekeeper or messenger? Biochim. Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.; Perdereau, D.; Foufelle, F.; Prip-Buus, C.; Ferré, P. Regulation of lipogenic enzyme gene expression by nutrients and hormones. FASEB J. 1994, 8, 36–42. [Google Scholar] [CrossRef]
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018, 8, 2362. [Google Scholar] [CrossRef]
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722. [Google Scholar] [CrossRef]
- Di Tommaso, L.; Franchi, G.; Park, Y.N.; Fiamengo, B.; Destro, A.; Morenghi, E.; Montorsi, M.; Torzilli, G.; Tommasini, M.; Terracciano, L.; et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 2007, 45, 725–734. [Google Scholar] [CrossRef]
- Osada, T.; Sakamoto, M.; Nagawa, H.; Yamamoto, J.; Matsuno, Y.; Iwamatsu, A.; Muto, T.; Hirohashi, S. Acquisition of glutamine synthetase expression in human hepatocarcinogenesis: relation to disease recurrence and possible regulation by ubiquitin-dependent proteolysis. Cancer 1999, 85, 819–831. [Google Scholar] [CrossRef]
- Abelev, G.; Eraiser, T. Cellular aspects of alpha-fetoprotein reexpression in tumors. Semin. Cancer Biol. 1999, 9, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Shirakami, Y.; Maruta, A.; Ideta, T.; Miyazaki, T.; Kochi, T.; Sakai, H.; Tanaka, T.; Seishima, M.; Shimizu, M. Preventive effects of the sodium glucose cotransporter 2 inhibitor tofogliflozin on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic mice. Oncotarget 2017, 8, 58353–58363. [Google Scholar]
- Devineni, D.; Curtin, C.R.; Polidori, D.; Gutierrez, M.J.; Murphy, J.; Rusch, S.; Rothenberg, P.L. Pharmacokinetics and Pharmacodynamics of Canagliflozin, a Sodium Glucose Co-Transporter 2 Inhibitor, in Subjects With Type 2 Diabetes Mellitus. J. Clin. Pharmacol. 2013, 53, 601–610. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Jojima, T.; Uchida, K.; Akimoto, K.; Tomotsune, T.; Yanagi, K.; Iijima, T.; Suzuki, K.; Kasai, K.; Aso, Y. Liraglutide, a GLP-1 receptor agonist, inhibits vascular smooth muscle cell proliferation by enhancing AMP-activated protein kinase and cell cycle regulation, and delays atherosclerosis in ApoE deficient mice. Atheroscler 2017, 261, 44–51. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle (n = 5) | Cana 30 mg/kg (5–8 weeks) (n = 6) | Cana 30 mg/kg (5–16 weeks) (n = 8) | |
|---|---|---|---|
| Body weight (g) Day 0 | 19.0 ± 1.0 | 18.7 ± 0.9 | 18.6 ± 1.0 |
| Body weight (g) Day 77 | 25.1 ± 1.7 | 21.5 ± 4.0 | 25.0 ± 3.5 |
| Liver-to-weight ratio (mg/g) | 7.84 ± 1.47 | 8.77 ± 1.72 | 6.08 ± 1.35 * |
| Plasma glucose (mg/dL) | 603 ± 91 | 542 ± 61 | 310 ± 81 † |
| ALT (U/L) | 74 (48, 82) | 86 (43, 226.5) | 34 (26.3, 37.8) ‡ |
| Triglyceride (mg/dL) | 687.6 ± 432.4 | 730.3 ± 392.3 | 514.9 ± 395.1 |
| Serum insulin (pg/mL) | 57.9 (39, 443.5) | 254 (99, 518.5) | 409 (198.8, 1187) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5237. https://doi.org/10.3390/ijms20205237
Jojima T, Wakamatsu S, Kase M, Iijima T, Maejima Y, Shimomura K, Kogai T, Tomaru T, Usui I, Aso Y. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2019; 20(20):5237. https://doi.org/10.3390/ijms20205237
Chicago/Turabian StyleJojima, Teruo, Sho Wakamatsu, Masato Kase, Toshie Iijima, Yuko Maejima, Kenju Shimomura, Takahiko Kogai, Takuya Tomaru, Isao Usui, and Yoshimasa Aso. 2019. "The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma" International Journal of Molecular Sciences 20, no. 20: 5237. https://doi.org/10.3390/ijms20205237
APA StyleJojima, T., Wakamatsu, S., Kase, M., Iijima, T., Maejima, Y., Shimomura, K., Kogai, T., Tomaru, T., Usui, I., & Aso, Y. (2019). The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 20(20), 5237. https://doi.org/10.3390/ijms20205237