Functional Prediction of Candidate MicroRNAs for CRC Management Using in Silico Approach




Abstract
:1. Introduction
2. Results
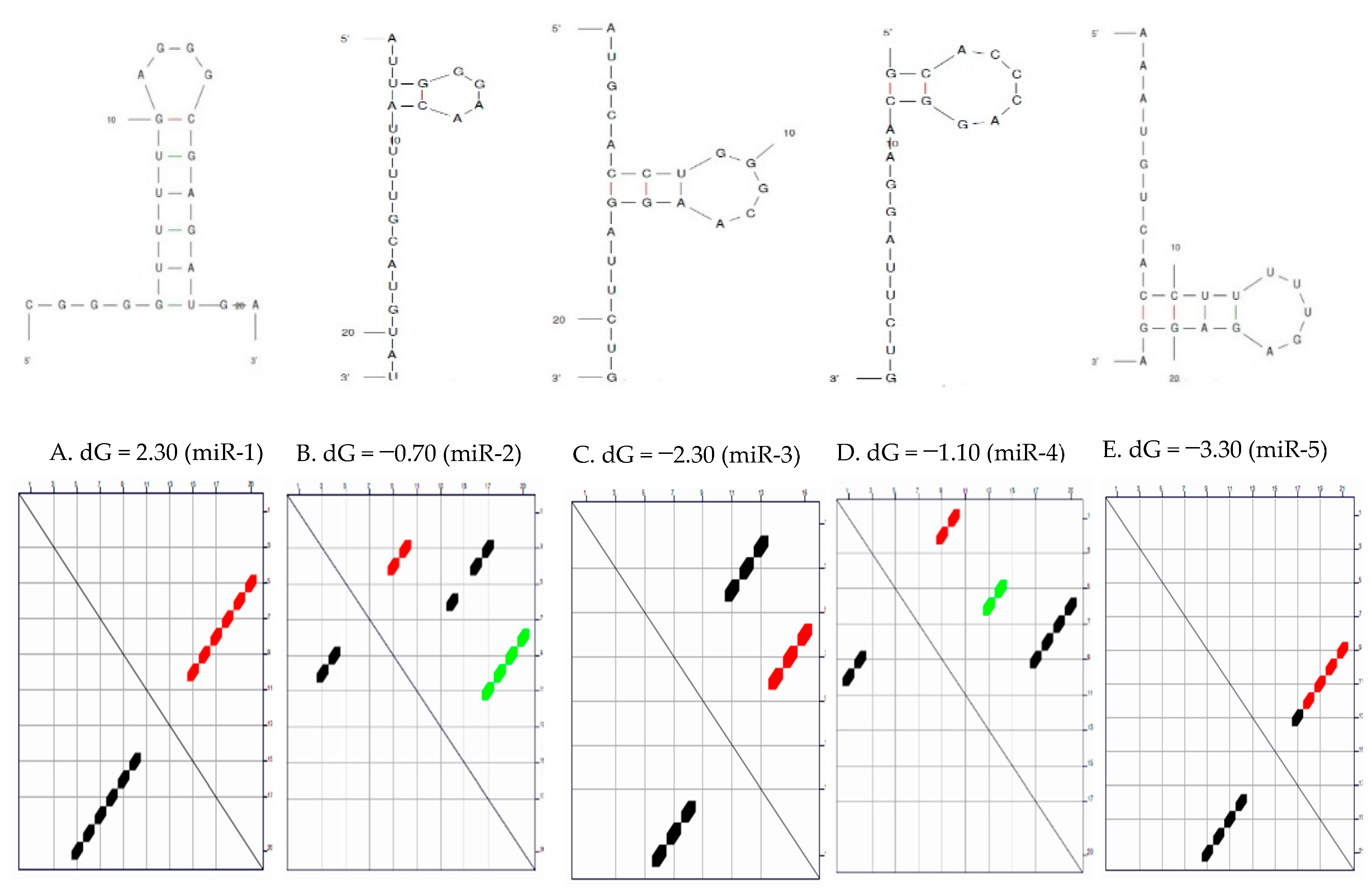
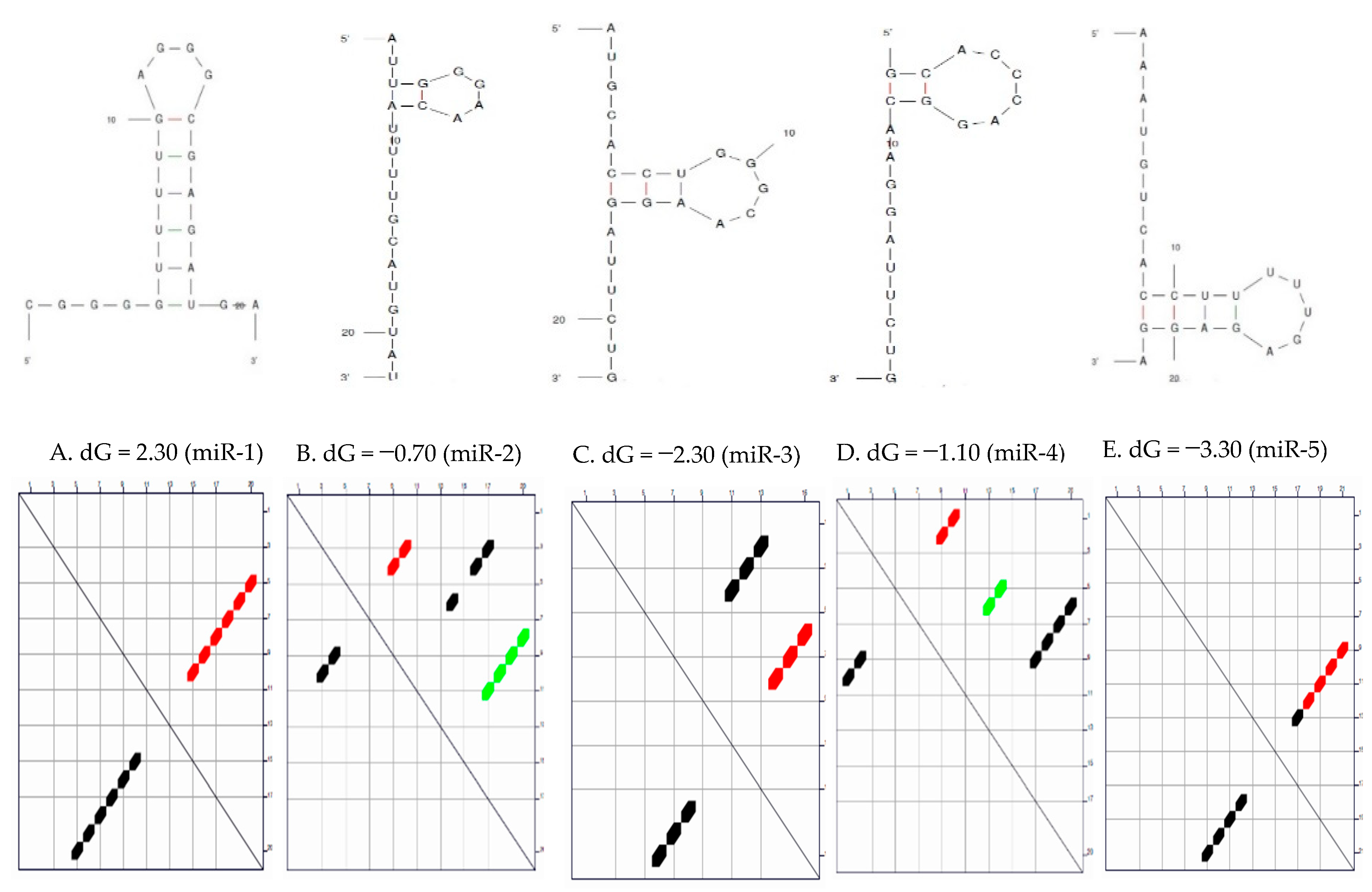
Secondary Structure of the Candidate MicroRNAs
3. Discussion
3.1. Secondary Structure and the Thermodynamic Energies of the Candidate MicroRNAs
3.2. CpG Island of the Promoter Sequences
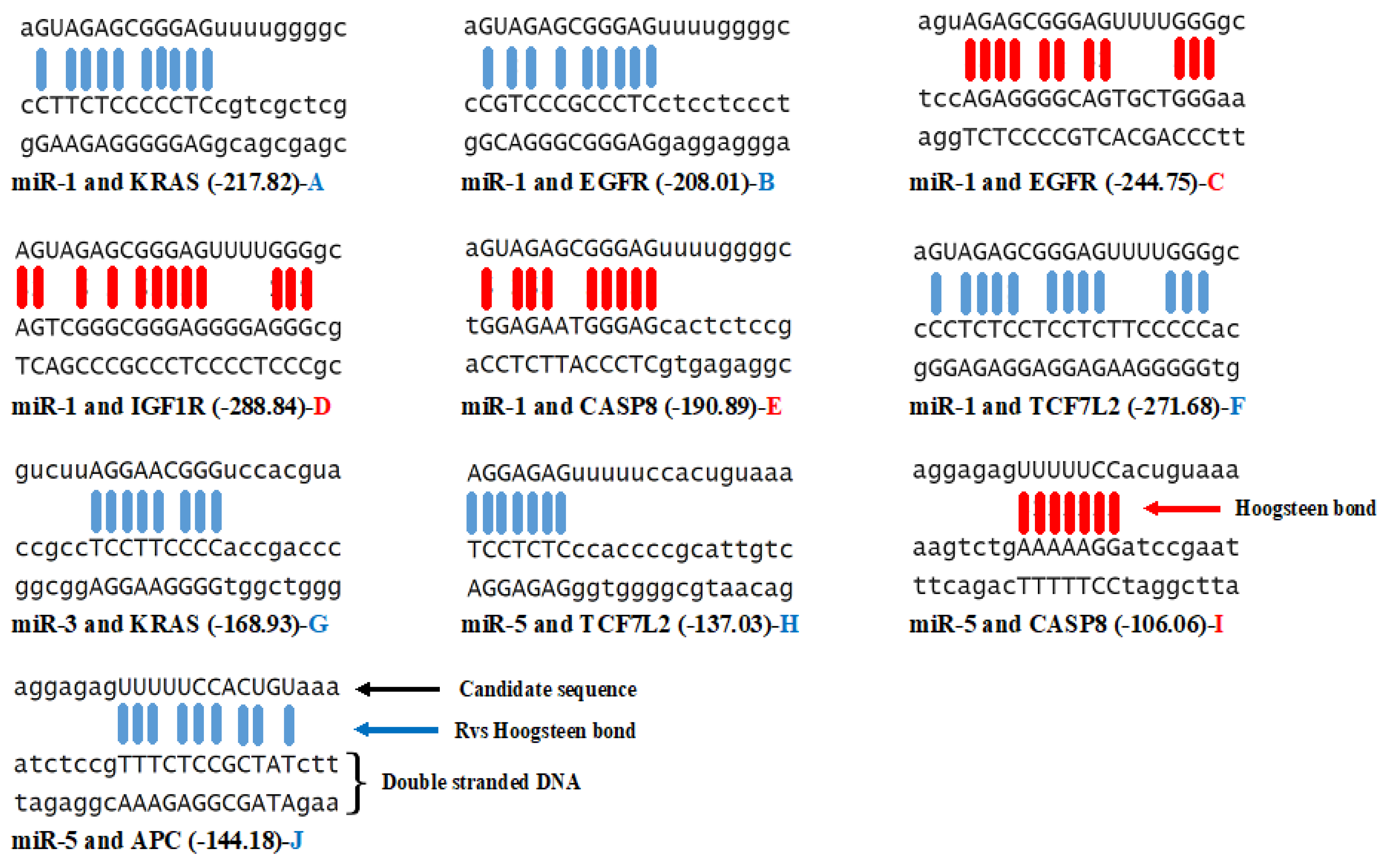
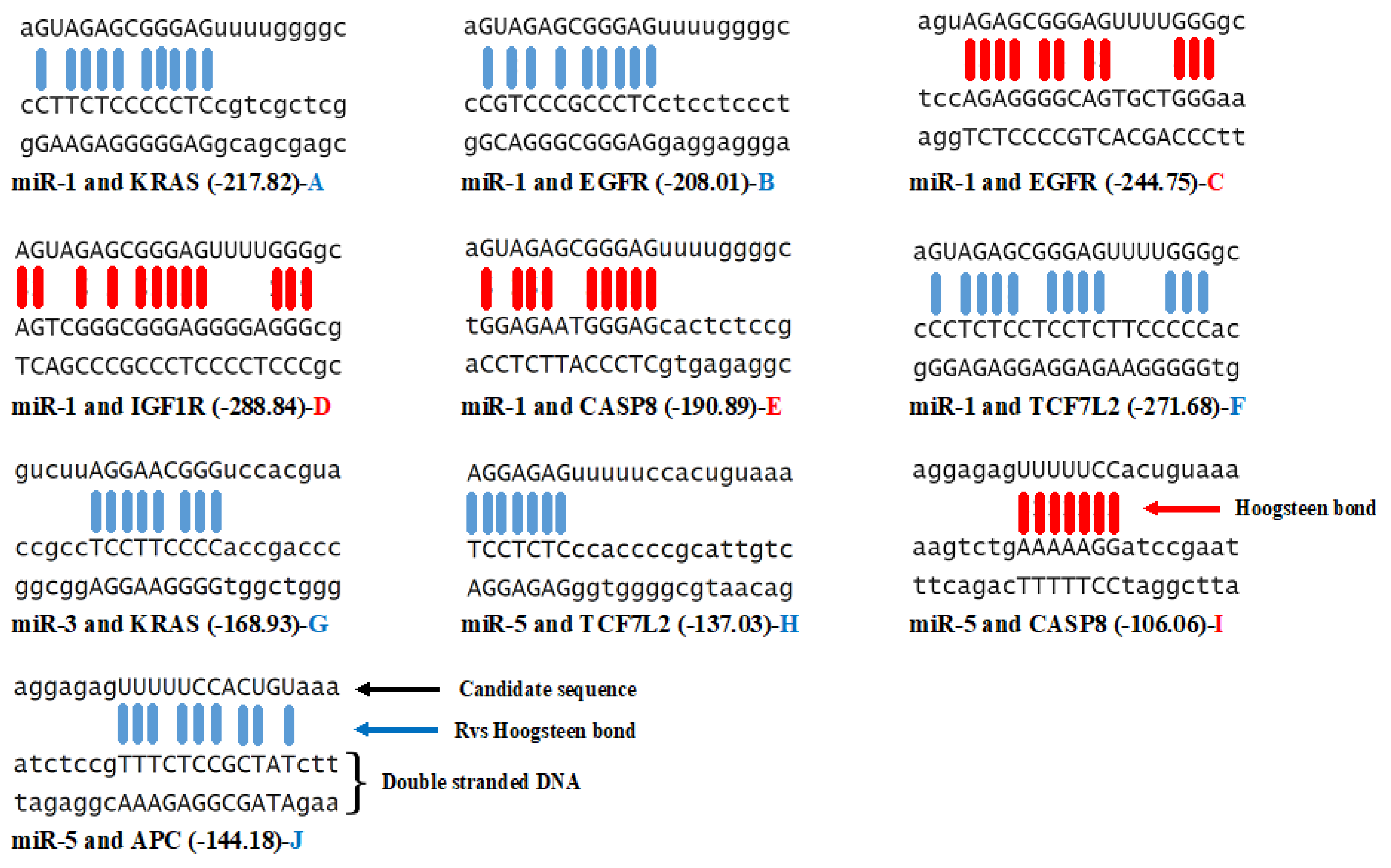
3.3. Triplex Binding Interaction of the MicroRNAs and Target Genes
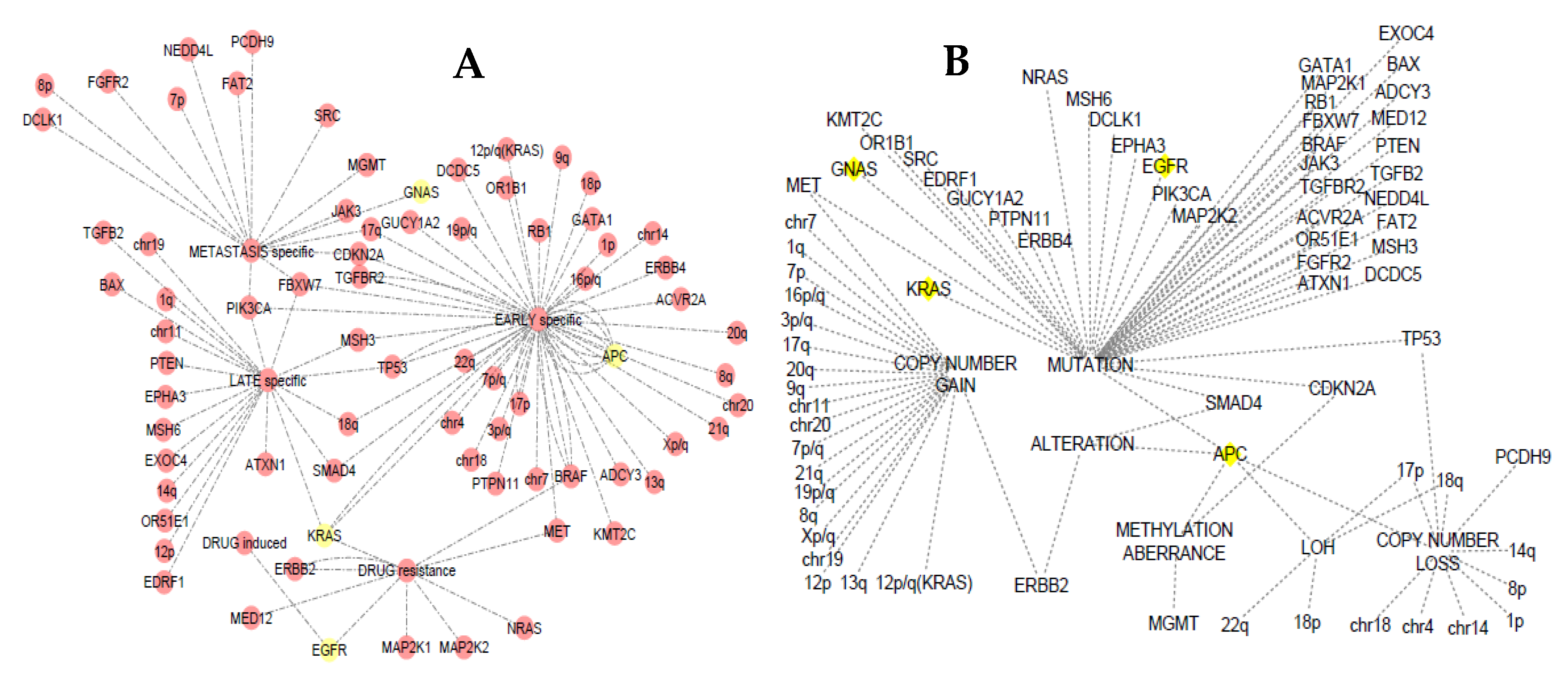
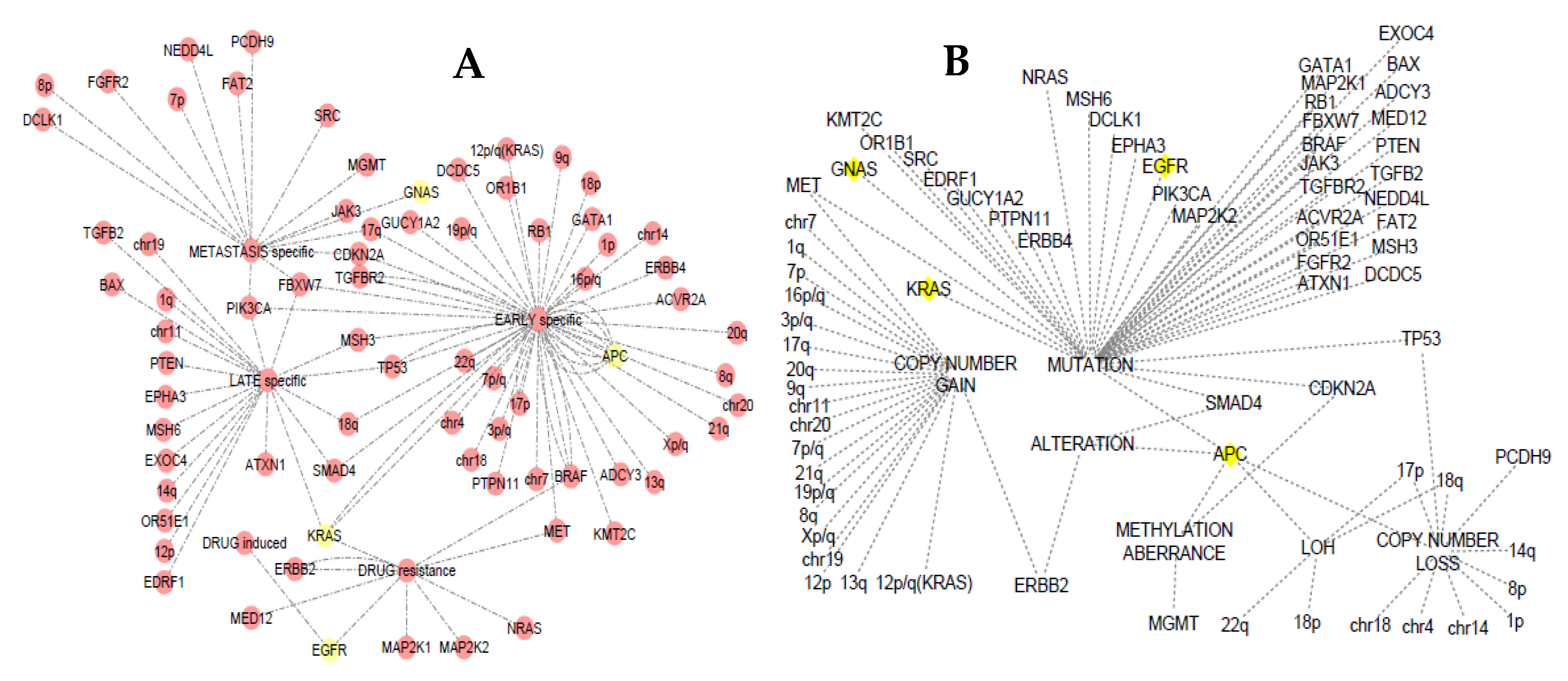
3.4. Somatic Event Evolution of the MicroRNA Target Genes
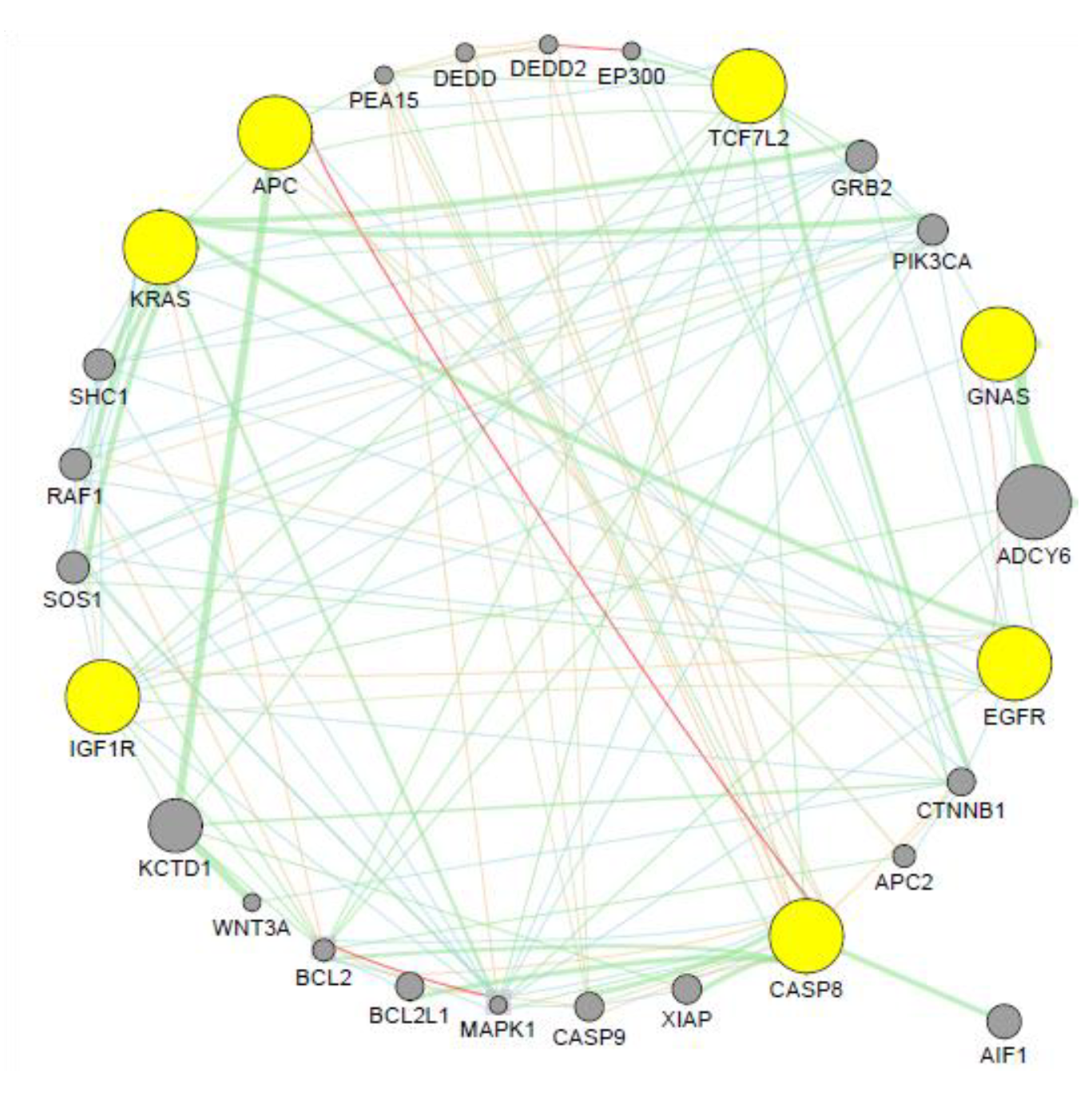
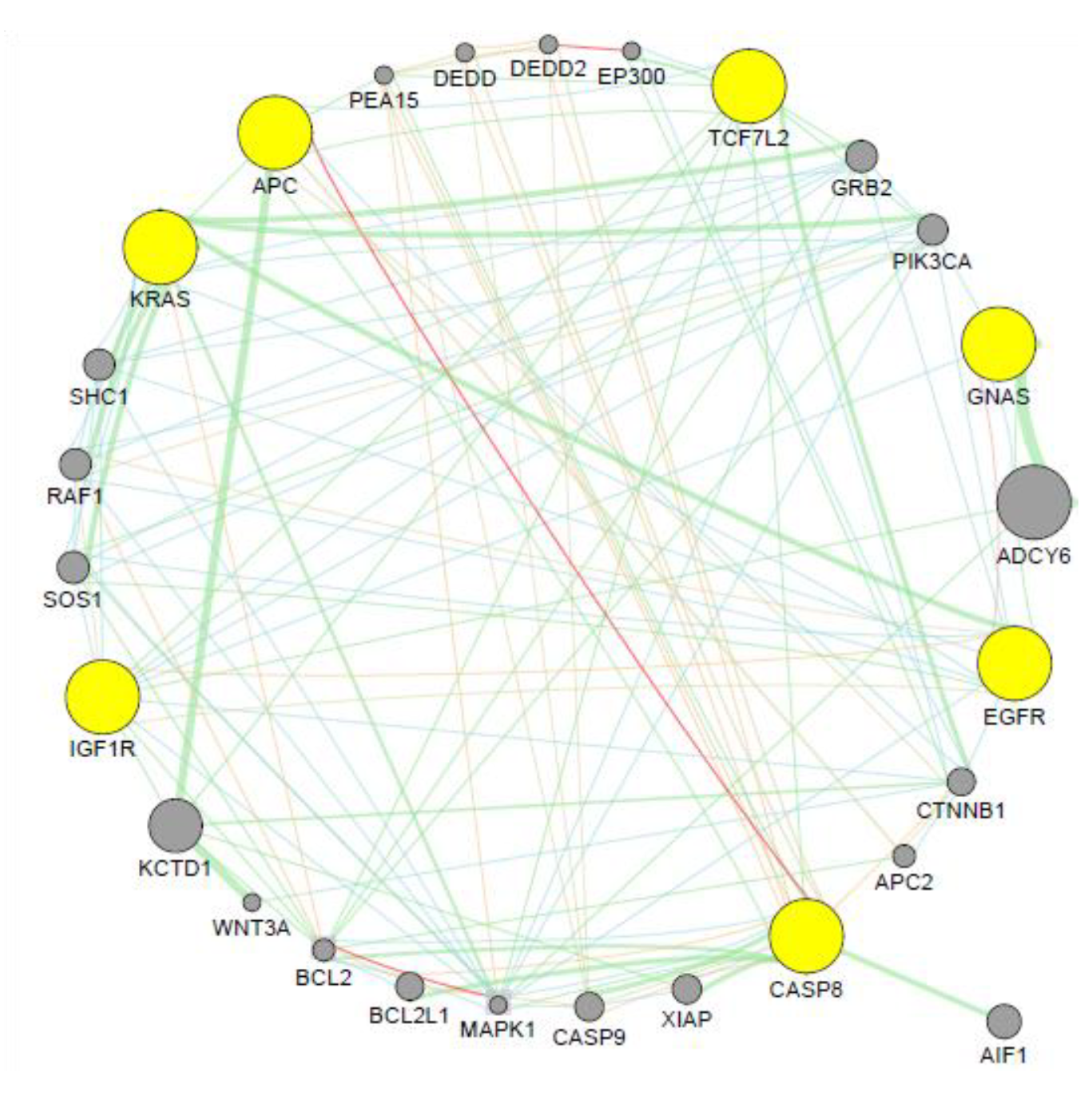
3.5. Co-expression Analysis
4. Materials and Methods
4.1. Datasets
4.2. Structural Determination of Candidate microRNA
4.3. Promoter Sequence Extraction
4.4. CpG Island Analysis
4.5. Triplex Binding Analysis
4.6. Staging Analysis
4.7. Co-expression Analysis
4.8. Statistical Analysis
4.9. Data Availability
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| CRC | Colorectal cancer |
| MRNA | Target genes |
| δG | Free enengy in plot profile |
| ΔG | Optinal energy of the secondery structure |
References
- Kuo, T.Y.; Hsi, E.; Yang, I.P.; Tsai, P.C.; Wang, J.Y.; Juo, S.H.H. Computational analysis of mRNA expression profiles identifies microRNA-29a/c as predictor of colorectal cancer early recurrence. PLoS ONE 2012, 7, e31587. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Meyerhardt, J.A.; Mamon, H.J.; Mayer, R.J. Adjuvant treatment of colorectal cancer. Cancer J. Clin. 2007, 57, 1681–1685. [Google Scholar] [CrossRef] [PubMed]
- Longo, W.E.; Johnson, F.E. The preoperative assessment and postoperative surveillance of patients with colon and rectal cancer. Surg. Clin. North Am. 2002, 82, 1091–1108. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Pretorius, A.; Klein, A. Biomarkers for Stratification in Colorectal Cancer: MicroRNAs. Cancer Control 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Schickel, R.; Boyerinas, B.; Park, S.; Peter, M. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959. [Google Scholar] [CrossRef]
- Hammond, S.M. MicroRNAs as tumor suppressors. Nat. Genet. 2007, 39, 582. [Google Scholar] [CrossRef]
- Liu, W.; Mao, S.Y.; Zhu, W.Y. Impact of tiny miRNAs on cancers. World J. Gastroenterol. 2007, 13, 497. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. MicroRNAs in cancer: Small molecules with a huge impact. J. Clin. Oncol. 2009, 27, 5848. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 2812–2897. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003, 113, 253–256. [Google Scholar] [CrossRef]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.O.; Ojo, B.A.; Adewale, O.B.; Esho, T.; Pretorius, A. Effect of dietary components on miRNA and colorectal carcinogenesis. Cancer Cell Int. 2018, 18, 130. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 199–227. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, K.; Inoue, H.; Takatsuno, Y.; Tanaka, F.; Mimori, K.; Uetake, H.; Sugihara, K.; Mori, M. Over-and under-expressed microRNAs in human colorectal cancer. Int. J. Oncol. 2009, 34, 1069–1075. [Google Scholar] [PubMed]
- Aslam, M.; Taylor, K.; Pringle, J.; Jameson, J. MicroRNAs are novel biomarkers of colorectal cancer. Br. J. Surg. 2009, 96, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Manne, U.; Shanmugam, C.; Bovell, L.; Katkoori, V.R.; Bumpers, H.L. miRNAs as biomarkers for management of patients with colorectal cancer. Biomark. Med. 2010, 4, 7617–7670. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Edwards, J.R.; Ju, J. Investigation of miRNA biology by bioinformatic tools and impact of miRNAs in colorectal cancer—Regulatory relationship of c-Myc and p53 with miRNAs. Cancer Inform. 2007, 3. [Google Scholar] [CrossRef]
- Ohler, U.; Niemann, H. Identification and analysis of eukaryotic promoters: Recent computational approaches. Trends Genet. 2001, 17, 56–60. [Google Scholar] [CrossRef]
- Moser, H.E.; Dervan, P.B. Sequence-specific cleavage of double helical DNA by triple helix formation. Science 1987, 238, 645–650. [Google Scholar] [CrossRef]
- Le Doan, T.; Perrouault, L.; Praseuth, D.; Habhoub, N.; Decout, J.L.; Thuong, N.T.; Lhomme, J.; Héène, C. Sequence-specific recognition, photocrosslinking and cleavage of the DNA double helix by an oligo-(α]-thymidylate covalently linked to an azidoproflavine derivative. Nucleic Acids Res. 1987, 15, 77497–77760. [Google Scholar] [CrossRef]
- Broitman, S.L.; Im, D.D.; Fresco, J.R. Formation of the triple-stranded polynucleotide helix, poly (AAU). Proc. Natl. Acad. Sci. USA 1987, 84, 5120–5124. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.; Collier, D.; Hanvey, J.; Shimizu, M.; Wohlrab, F. The chemistry and biology of unusual DNA structures adopted by oligopurine. oligopyrimidine sequences. FASEB J. 1988, 2, 29392–29949. [Google Scholar] [CrossRef]
- Buske, F.A.; Mattick, J.S.; Bailey, T.L. Potential in vivo roles of nucleic acid triple-helices. RNA Biol. 2011, 8, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalali, S.; Singh, A.; Maiti, S.; Scaria, V. Genome-wide computational analysis of potential long noncoding RNA mediated DNA: DNA: RNA triplexes in the human genome. J. Transl. Med. 2017, 15, 186. [Google Scholar] [CrossRef]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA triplex formation by long noncoding RNAs. Cell Chem. Biol. 2016, 23, 13251–13333. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Younger, S.T.; Hardy, D.B.; Ram, R.; Huffman, K.E.; Corey, D.R. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 2007, 3, 166. [Google Scholar] [CrossRef]
- Check, E. RNA interference: Hitting the on switch. Nature 2007, 448, 855–858. [Google Scholar] [CrossRef]
- Kanak, M.; Alseiari, M.; Balasubramanian, P.; Addanki, K.; Aggarwal, M.; Noorali, S.; Kalsum, A.; Mahalingam, K.; Pace, G.; Panasik, N. Triplex-forming MicroRNAs form stable complexes with HIV-1 provirus and inhibit its replication. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 5325–5345. [Google Scholar] [CrossRef]
- Schmitz, K.-M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [Green Version]
- Goñi, J.R.; De La Cruz, X.; Orozco, M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004, 32, 3543–3560. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Kuznetsov, V.A. In TTS mapping: Integrative WEB tool for analysis of triplex formation target DNA sequences, G-quadruplets and non-protein coding regulatory DNA elements in the human genome. BMC Genom. 2009, S9. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Chew, C.S.; Yong, T.P.; Choowongkomon, K.; Thammasorn, W.; Kuznetsov, V.A. The TTSMI database: A catalog of triplex target DNA sites associated with genes and regulatory elements in the human genome. Nucleic Acids Res. 2014, 43, D110–D116. [Google Scholar] [CrossRef] [PubMed]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplexator: Detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012, 22, 1372–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Gaddis, S.S.; MacLeod, M.C.; Walborg, E.F.; Thames, H.D.; DiGiovanni, J.; Vasquez, K.M. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol. Carcinog 2007, 46, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Bacani, J.; Begthel, H.; Jonkeer, S.; Gregorieff, A.; van de Born, M.; Malats, N.; Sancho, E.; Boon, E.; Pawson, T. EphB receptor activity suppresses colorectal cancer progression. Nature 2005, 435, 1126. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015, 161, 15391–15552. [Google Scholar] [CrossRef] [PubMed]
- Scholer-Dahirel, A.; Schlabach, M.R.; Loo, A.; Bagdasarian, L.; Meyer, R.; Guo, R.; Woolfenden, S.; Kristine, K.Y.; Markovits, J.; Killary, K. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 17135–17140. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532. [Google Scholar] [CrossRef]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Bass, A.J.; Lawrence, M.S.; Brace, L.E.; Ramos, A.H.; Drier, Y.; Cibulskis, K.; Sougnez, C.; Voet, D.; Saksena, G.; Sivachenko, A. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011, 43, 964. [Google Scholar] [CrossRef]
- Theodoropoulos, G.E.; Gazouli, M.; Vaiopoulou, A.; Leandrou, M.; Nikouli, S.; Vassou, E.; Kouraklis, G.; Nikiteas, N. Polymorphisms of caspase 8 and caspase 9 gene and colorectal cancer susceptibility and prognosis. Int. J. Colorectal Dis. 2011, 26, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Inno, A.; Di Salvatore, M.; Cenci, T.; Martini, M.; Orlandi, A.; Strippoli, A.; Ferrara, A.M.; Bagalà, C.; Cassano, A.; Larocca, L.M. Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin. Colorectal Cancer 2011, 10, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Idziaszczyk, S.; Wilson, C.; Smith, C.; Adams, D.; Cheadle, J. Analysis of the frequency of GNAS codon 201 mutations in advanced colorectal cancer. Cancer Genet. Cytogenetics 2010, 202, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Sudo, H.; Kawauchi, J.; Takizawa, S.; Kondou, S.; Nobumasa, H.; Ochiai, A. MicroRNA markers for the diagnosis of pancreatic and biliary-tract cancers. PLoS ONE 2015, 10, e0118220. [Google Scholar] [CrossRef]
- Plieskatt, J.L.; Rinaldi, G.; Feng, Y.; Peng, J.; Yonglitthipagon, P.; Easley, S.; Laha, T.; Pairojkul, C.; Bhudhisawasdi, V.; Sripa, B. Distinct miRNA signatures associate with subtypes of cholangiocarcinoma from infection with the tumourigenic liver fluke Opisthorchis viverrini. J. Hepatol. 2014, 61, 8508–8558. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Ng, S.-B.; Yan, J.; Huang, G.; Selvarajan, V.; Tay, J.L.-S.; Lin, B.; Bi, C.; Tan, J.; Kwong, Y.-L.; Shimizu, N. Dysregulated microRNAs affect pathways and targets of biologic relevance in nasal-type natural killer/T-cell lymphoma. Blood 2011, 118, 49194–49929. [Google Scholar] [CrossRef]
- Zhang, P.; Bill, K.; Liu, J.; Young, E.; Peng, T.; Bolshakov, S.; Hoffman, A.; Song, Y.; Demicco, E.G.; Terrada, D.L. MiR-155 is a liposarcoma oncogene that targets casein kinase-1α and enhances β-catenin signaling. Cancer Res. 2012, 72, 17511–17762. [Google Scholar] [CrossRef]
- Munding, J.B.; Adai, A.T.; Maghnouj, A.; Urbanik, A.; Zöllner, H.; Liffers, S.T.; Chromik, A.M.; Uhl, W.; Szafranska-Schwarzbach, A.E.; Tannapfel, A. Global microRNA expression profiling of microdissected tissues identifies miR-135b as a novel biomarker for pancreatic ductal adenocarcinoma. Int. J. Cancer 2012, 131, E86–E95. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, X.; Huo, Q.; Li, X.; Wang, H.; Schneider, P.; Hu, G.; Yang, Q. The oncogene metadherin modulates the apoptotic pathway based on the tumor necrosis factor superfamily member TRAIL (Tumor Necrosis Factor-related Apoptosis-inducing Ligand) in breast cancer. J. Biol. Chem. 2013, 288, 9396–9407. [Google Scholar] [CrossRef]
- Biagioni, F.; Ben-Moshe, N.B.; Fontemaggi, G.; Canu, V.; Mori, F.; Antoniani, B.; Di Benedetto, A.; Santoro, R.; Germoni, S.; De Angelis, F. miR-10b*, a master inhibitor of the cell cycle, is down-regulated in human breast tumours. EMBO Mol. Med. 2012, 4, 12141–12229. [Google Scholar] [CrossRef] [PubMed]
- Özata, D.M.; Caramuta, S.; Velázquez-Fernández, D.; Akçakaya, P.; Xie, H.; Höög, A.; Zedenius, J.; Bäckdahl, M.; Larsson, C.; Lui, W.-O. The role of microRNA deregulation in the pathogenesis of adrenocortical carcinoma. Endocr. Relat. Cancer 2011, 18, 6436–6455. [Google Scholar] [CrossRef] [PubMed]
- Namløs, H.M.; Meza-Zepeda, L.A.; Barøy, T.; Østensen, I.H.; Kresse, S.H.; Kuijjer, M.L.; Serra, M.; Bürger, H.; Cleton-Jansen, A.-M.; Myklebost, O. Modulation of the osteosarcoma expression phenotype by microRNAs. PLoS ONE 2012, 7, e48086. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.M.; Phillips, B.L.; Patel, R.S.; Cohen, D.M.; Jakymiw, A.; Kong, W.W.; Cheng, J.Q.; Chan, E.K. Keratinization-associated miR-7 and miR-21 regulate tumor suppressor reversion-inducing cysteine-rich protein with kazal motifs (RECK) in oral cancer. J. Biol. Chem. 2012, 287, 29261–29272. [Google Scholar] [CrossRef]
- Kretschmer-Kazemi Far, R.; Sczakiel, G. The activity of siRNA in mammalian cells is related to structural target accessibility: A comparison with antisense oligonucleotides. Nucleic Acids Res. 2003, 31, 44174–44424. [Google Scholar] [CrossRef]
- Overhoff, M.; Alken, M.; Far, R.K.-K.; Lemaitre, M.; Lebleu, B.; Sczakiel, G.; Robbins, I. Local RNA target structure influences siRNA efficacy: A systematic global analysis. J. Mol. Biol. 2005, 348, 8718–8781. [Google Scholar] [CrossRef]
- Mückstein, U.; Tafer, H.; Hackermüller, J.; Bernhart, S.H.; Stadler, P.F.; Hofacker, I.L. Thermodynamics of RNA–RNA binding. Bioinform. 2006, 22, 1177–1182. [Google Scholar] [CrossRef]
- Belter, A.; Gudanis, D.; Rolle, K.; Piwecka, M.; Gdaniec, Z.; Naskręt-Barciszewska, M.Z.; Barciszewski, J. Mature miRNAs form secondary structure, which suggests their function beyond RISC. PLoS ONE 2014, 9, e113848. [Google Scholar] [CrossRef]
- Maiti, M.; Nauwelaerts, K.; Lescrinier, E.; Schuit, F.C.; Herdewijn, P. Self-complementary sequence context in mature miRNAs. Biochem. Biophys. Res. Commun. 2010, 392, 5725–5776. [Google Scholar] [CrossRef]
- Belter, A.; Naskret-Barciszewska, M. The correlation of structural features of mature miRNAs with their biological function. Biotechnol. J. Biotechnol. Comput. Biol. Bionanotechnol. 2014, 95, 187–191. [Google Scholar]
- Ronchieri, E.; DAgostino, D.; Milanesi, L.; Merelli, I. microRNA-target interaction: A parallel approach for computing pairing energy. In Proceedings of the 2016 24th Euromicro International Conference on Parallel, Distributed, and Network-Based Processing (PDP), Heraklion, Greece, 17–19 February 2016; IEEE Press: Piscataway, NJ, USA, 2016; pp. 535–540. [Google Scholar]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical aspects of microRNA target prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278. [Google Scholar] [CrossRef]
- Arif, M.; Ochoa-Corona, F. Comparative assessment of 5′ A/T-rich overhang sequences with optimal and sub-optimal primers to increase PCR yields and sensitivity. Mol. Biotechnol. 2013, 55, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. New Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Oberley, M.J.; Farnham, P.J. Probing chromatin immunoprecipitates with CpG-island microarrays to identify genomic sites occupied by DNA-binding proteins. Methods Enzym. 2003, 371, 577–596. [Google Scholar]
- Curtin, K.; Slattery, M.L.; Samowitz, W.S. CpG island methylation in colorectal cancer: Past, present and future. Pathol. Res. Int. 2011. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. Gene expression: DNA methylation—How important in gene control? Nature 1984, 307, 503. [Google Scholar] [CrossRef]
- Bird, A.P. DNA methylation versus gene expression. Development 1984, 83, 31–40. [Google Scholar]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 37403–37745. [Google Scholar] [CrossRef]
- Gatter, K. Handbook of immunohistochemistry and in situ hybridisation of human carcinomas: Molecular genetics–lung and breast carcinomas. Br. J. Cancer 2005, 93, 1318. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.; Ladomery, M. Molecular Biology of RNA; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Pasquier, C.; Agnel, S.; Robichon, A. The Mapping of Predicted Triplex DNA: RNA in the Drosophila Genome Reveals a Prominent Location in Development-and Morphogenesis-Related Genes. Genes Genomes Genet. 2017, 7, 2295–2304. [Google Scholar]
- Park, C.W.; Zeng, Y.; Zhang, X.; Subramanian, S.; Steer, C.J. Mature microRNAs identified in highly purified nuclei from HCT116 colon cancer cells. RNA Biol. 2010, 7, 606–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, F.J.; Montoya, G. Transient DNA/RNA-protein interactions. FEBS J. 2011, 278, 16431–16650. [Google Scholar] [CrossRef] [PubMed]
- Semerad, C.L.; James Maher III, L. Exclusion of RNA strands from a purine motif triple helix. Nucleic Acids Res. 1994, 22, 53215–53325. [Google Scholar] [CrossRef]
- Roberts, R.W.; Crothers, D.M. Stability and properties of double and triple helices: Dramatic effects of RNA or DNA backbone composition. Science 1992, 258, 1463–1466. [Google Scholar] [CrossRef]
- Maine, I.P.; Kodadek, T. Efficient unwinding of triplex DNA by a DNA helicase. Biochem. Biophys. Res. Commun. 1994, 204, 1119–1124. [Google Scholar] [CrossRef]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M.; Waddell, M.B.; Ridout, G.; Naeve, D.; Leuze, M. MicroRNAs form triplexes with double stranded DNA at sequence-specific binding sites; a eukaryotic mechanism via which microRNAs could directly alter gene expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef]
- Umetani, N.; Sasaki, S.; Watanabe, T.; Shinozaki, M.; Matsuda, K.; Ishigami, H.; Ueda, E.; Muto, T. Genetic alterations in ulcerative colitis-associated neoplasia focusing on APC, K-ras gene and microsatellite instability. Jpn. J. Cancer Res. 1999, 90, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Matkowskyj, K.A.; Chen, Z.E.; Rao, M.S.; Yang, G.-Y. Dysplastic lesions in inflammatory bowel disease: Molecular pathogenesis to morphology. Arch. Pathol. Lab. Med. 2013, 137, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Redston, M.S.; Papadopoulos, N.; Caldas, C.; Kinzler, K.W.; Kern, S.E. Common occurrence of APC and K-ras gene mutations in the spectrum of colitis-associated neoplasias. Gastroenterology 1995, 108, 3833–3892. [Google Scholar] [CrossRef]
- Yashiro, M. Molecular alterations of colorectal cancer with inflammatory bowel disease. Dig. Dis. Sci. 2015, 60, 2251–2263. [Google Scholar] [CrossRef]
- Molinaro, A.; Terdiman, J.P.; Willenbucher, R.; Chang, F.; Baretton, G.B.; Waldman, F.; Aust, A.E.; Loehrs, U. The APC/beta-catenin pathway in ulcerative colitis-related colorectal carcinomas: A mutational analysis. Lancet 2002. [Google Scholar] [CrossRef]
- Segditsas, S.; Rowan, A.; Howarth, K.; Jones, A.; Leedham, S.; Wright, N.; Gorman, P.; Chambers, W.; Domingo, E.; Roylance, R. APC and the three-hit hypothesis. Oncogene 2009, 28, 146. [Google Scholar] [CrossRef]
- Kim, T.-M.; An, C.H.; Rhee, J.-K.; Jung, S.-H.; Lee, S.H.; Baek, I.-P.; Kim, M.S.; Lee, S.H.; Chung, Y.-J. Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget 2015, 6, 27725. [Google Scholar] [CrossRef]
- Sakai, E.; Fukuyo, M.; Matsusaka, K.; Ohata, K.; Doi, N.; Takane, K.; Matsuhashi, N.; Fukushima, J.; Nakajima, A.; Kaneda, A. TP 53 mutation at early stage of colorectal cancer progression from two types of laterally spreading tumors. Cancer Sci. 2016, 107, 8208–8227. [Google Scholar] [CrossRef]
- Fodde, R.; Kuipers, J.; Rosenberg, C.; Smits, R.; Kielman, M.; Gaspar, C.; van Es, J.H.; Breukel, C.; Wiegant, J.; Giles, R.H.; et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001, 3, 433–438. [Google Scholar] [CrossRef]
- Smolle, M.A.; Pichler, M.; Haybaeck, J.; Gerger, A. Genetic markers of recurrence in colorectal cancer. Pharmacogenomics 2015, 16, 1315–1328. [Google Scholar] [CrossRef]
- Burmer, G.C.; Rabinovitch, P.S.; Loeb, L.A. Analysis of c-Ki-ras mutations in human colon carcinoma by cell sorting, polymerase chain reaction, and DNA sequencing. Cancer Res. 1989, 49, 2141–2146. [Google Scholar] [PubMed]
- Youn, A.; Simon, R. Estimating the order of mutations during tumorigenesis from tumor genome sequencing data. Bioinformatics 2012, 28, 15551–15561. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.; Van Boxtel, R.; De Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, M.; Overman, M.; Dasari, A.; Kazmi, S.; Mazard, T.; Vilar, E.; Morris, V.; Lee, M.; Herron, D.; Eng, C. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 7317–7336. [Google Scholar] [CrossRef]
- Hong, S.H.; Goh, S.H.; Lee, S.J.; Hwang, J.A.; Lee, J.; Choi, I.J.; Seo, H.; Park, J.-H.; Suzuki, H.; Yamamoto, E. Upregulation of adenylate cyclase 3 (ADCY3) increases the tumorigenic potential of cells by activating the CREB pathway. Oncotarget 2013, 4, 1791. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, C.; Wang, F.; Huang, W.; Liang, Z.; Xiao, Y.; Wei, K.; Wan, Z.; Hu, X.; Xiang, S. KCTD1 suppresses canonical Wnt signaling pathway by enhancing β-catenin degradation. PLoS ONE 2014, 9, e94343. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhou, X.; Liu, Y.; Tang, Z.; Romeih, M. Wnt signaling in hepatocellular carcinoma: Analysis of mutation and expression of beta-catenin, T-cell factor-4 and glycogen synthase kinase 3-beta genes. J. Gastroenterol. Hepatol. 2003, 18, 2802–2887. [Google Scholar] [CrossRef]
- Ewan, K.B.; Dale, T.C. The potential for targeting oncogenic wnt/β-catenin signaling in therapy. Curr. Drug Targets 2008, 9, 5325–5347. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 2612–2682. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, S.; Zhang, X.; Liao, J.; Quan, F.; Zhao, E.; Zhou, C.; Yu, F.; Yin, W.; Zhang, Y. SEECancer: A resource for somatic events in evolution of cancer genome. Nucleic Acids Res. 2017, 46, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2017, 19, 5755–5792. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | MicroRNAs | Length | δG (kcal/mol) | Initial ΔG (kcal/mol) | StruC |
|---|---|---|---|---|---|
| 1 | miR-1 | 22 | 0.0 | −2.30 | 1 |
| 2 | miR-2 | 22 | 0.7 | −0.70 −0.40 0.00 | 3 |
| 3 | miR-3 | 22 | 0.7 | −2.80 −2.10 | 2 |
| 4 | miR-4 | 20 | 0.8 | −1.10 −0.80 −0.30 | 3 |
| 5 | miR-5 | 22 | 0.6 | −3.30 −2.70 | 2 |
| S/N | Gene_ID | Min. GC% | Max. GC% | Min. obs/exp | Max. obs/exp |
|---|---|---|---|---|---|
| 1 | APC | 51.00 | 57.50 | 0.61 | 0.67 |
| 2 | KRAS | 59.50 | 83.00 | 0.78 | 1.14 |
| 3 | TCF7L2 | 53.00 | 72.50 | 0.60 | 1.00 |
| 4 | EGFR | 56.00 | 57.00 | 0.62 | 0.90 |
| 5 | IGF1R | 53.00 | 81.50 | 0.61 | 1.22 |
| 6 | CASP8 | 67.50 | 70.50 | 0.60 | 0.74 |
| 7 | GNAS | 67.50 | 70.50 | 0.61 | 0.78 |
| MicroRNA/Gene | KRAS | TCF7L2 | APC | EGFR | CASP8 | IGF1R | GNAS |
|---|---|---|---|---|---|---|---|
| miR-1 | −4 | −9 | 0 | −3/+1 | +1 | +1 | 0 |
| miR-2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| miR-3 | −1 | 0 | 0 | 0 | 0 | 0 | 0 |
| miR-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| miR-5 | 0 | −2 | −1 | 0 | +1 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadaka, A.O.; Pretorius, A.; Klein, A. Functional Prediction of Candidate MicroRNAs for CRC Management Using in Silico Approach. Int. J. Mol. Sci. 2019, 20, 5190. https://doi.org/10.3390/ijms20205190
Fadaka AO, Pretorius A, Klein A. Functional Prediction of Candidate MicroRNAs for CRC Management Using in Silico Approach. International Journal of Molecular Sciences. 2019; 20(20):5190. https://doi.org/10.3390/ijms20205190
Chicago/Turabian StyleFadaka, Adewale Oluwaseun, Ashley Pretorius, and Ashwil Klein. 2019. "Functional Prediction of Candidate MicroRNAs for CRC Management Using in Silico Approach" International Journal of Molecular Sciences 20, no. 20: 5190. https://doi.org/10.3390/ijms20205190