Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence

 ,
,
Abstract
:1. Introduction
2. Mechanistic Rationales for Targeting PPARs in Various Human Diseases
3. Type 2 Diabetes Mellitus (T2DM)
4. Cardiovascular Diseases (CVDs)
5. Dyslipidemia
6. Metabolic Syndrome, Obesity, and Hypertension
6.1. Prediabetes and Metabolic Syndrome
6.2. Obesity
6.3. Hypertension
7. Liver Diseases
7.1. Non-Alcoholic Fatty Liver Disease (NAFLD)
7.2. Primary Biliary Cholangitis
7.3. Hepatitis C
8. Kidney Diseases
8.1. Chronic Kidney Disease (CKD)
8.2. Other Kidney Diseases
9. Neurodegenerative Diseases and Neurological Dysfunction
9.1. Alzheimer’s Disease and Parkinson’s Disease
9.2. Amyotrophic Lateral Sclerosis
9.3. Multiple Sclerosis
9.4. Other Neurological Disorders
10. Psychiatric Disorders
10.1. Addiction/Substance Dependency
10.2. Major Depressive Disorder and Bipolar Depression
10.3. Autism Spectrum Disorder
11. Autoimmune Diseases
11.1. Rheumatoid Arthritis
11.2. Systemic Lupus Erythematosus
11.3. Other Autoimmune Diseases
12. Inflammatory and Infectious Diseases
12.1. Malaria
12.2. Ulcerative Colitis
12.3. Asthma
12.4. Psoriasis
12.5. Endometriosis
12.6. Cystic Fibrosis
12.7. Other Inflammatory and Infectious Diseases
13. Malignancy
13.1. Head and Neck Squamous Cell Carcinoma (HNSCC)
13.2. Thyroid Cancer
13.3. Lung Cancer
13.4. Colorectal Cancer
13.5. Prostate Cancer
13.6. Blood Cancer (Leukemia, Lymphoma, Myeloma)
13.7. Skin Cancer
13.8. Liposarcoma
13.9. Breast Cancer
13.10. Brain Cancer
13.11. Recurrent/Progressive/Metastatic Cancer
14. Other Diseases
14.1. Polycystic Ovarian Syndrome
14.2. Muscular Disorders
14.3. Burn Injury
14.4. Miscellaneous Health Conditions
15. Clinical Prospects of PPAR Agonists and Antagonists
16. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, L.S.A.; Wells, R.A. Cross-talk between PPARs and the partners of RXR: A molecular perspective. PPAR Res. 2009, 2009, 925309. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural peroxisome proliferator-activated receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Vázquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARβ/δ. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; Ferranti, S.d.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management ofblood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [PubMed]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycaemia in type 2 diabetes, 2015: A patient-centred approach. Update to a Position Statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2015, 58, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Xu, P.; Zhai, Y. The opportunities and challenges of peroxisome proliferator-activated receptors ligands in clinical drug discovery and development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed]
- Bruckert, E.; Labreuche, J.; Deplanque, D.; Touboul, P.-J.; Amarenco, P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: A systematic review and meta-analysis. J. Cardiovasc. Pharmacol. 2011, 57, 267–272. [Google Scholar] [CrossRef]
- Abourbih, S.; Filion, K.B.; Joseph, L.; Schiffrin, E.L.; Rinfret, S.; Poirier, P.; Pilote, L.; Genest, J.; Eisenberg, M.J. Effect of fibrates on lipid profiles and cardiovascular outcomes: A systematic review. Am. J. Med. 2009, 122, 962.e1–962.e8. [Google Scholar] [CrossRef]
- Chiquette, E.; Ramirez, G.; DeFronzo, R. A meta-analysis comparing the effect of thiazolidinediones on cardiovascular risk factors. JAMA Intern. Med. 2004, 164, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Mavridis, D.; Nicolucci, A.; Johnson, D.W.; Tonelli, M.; Craig, J.C.; Maggo, J.; Gray, V.; De Berardis, G.; Ruospo, M.; et al. Comparison of clinical outcomes and adverse events associated with glucose-lowering drugs in patients with type 2 diabetes: A meta-analysis. JAMA 2016, 316, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J.; Schoonjans, K.; Fruchart, J.-C.; Staels, B. Regulation of triglyceride metabolism by PPARs: Fibrates and thiazolidinediones have distinct effects. J. Atheroscler. Thromb. 1996, 3, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Sugii, S.; Olson, P.; Sears, D.D.; Saberi, M.; Atkins, A.R.; Barish, G.D.; Hong, S.-H.; Castro, G.L.; Yin, Y.-Q.; Nelson, M.C.; et al. PPARγ activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. USA 2009, 106, 22504–22509. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.-X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ promotes running endurance by preserving glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Ravnskjaer, K.; Frigerio, F.; Boergesen, M.; Nielsen, T.; Maechler, P.; Mandrup, S. PPARδ is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J. Lipid Res. 2010, 51, 1370–1379. [Google Scholar] [CrossRef]
- Maccallini, C.; Mollica, A.; Amoroso, R. The positive regulation of eNOS signaling by PPAR agonists in cardiovascular diseases. Am. J. Cardiovasc. Drugs 2017, 17, 273–281. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Q.; Yan, J.T.; Zhao, C.; Cianflone, K.; Wang, D.W. Effects of bezafibrate on the expression of endothelial nitric oxide synthase gene and its mechanisms in cultured bovine endothelial cells. Atherosclerosis 2006, 187, 265–273. [Google Scholar] [CrossRef]
- Okayasu, T.; Tomizawa, A.; Suzuki, K.; Manaka, K.-I.; Hattori, Y. PPARα activators upregulate eNOS activity and inhibit cytokine-induced NF-κB activation through AMP-activated protein kinase activation. Life Sci. 2008, 82, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, H.; Wang, W.; Wang, X.; Huang, Y.; Huang, C.; Gao, F. Vascular insulin resistance in prehypertensive rats: Role of PI3-kinase/Akt/eNOS signaling. Eur. J. Pharmacol. 2010, 628, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Quintela, A.M.; Jiménez, R.; Piqueras, L.; Gómez-Guzmán, M.; Haro, J.; Zarzuelo, M.J.; Cogolludo, A.; Sanz, M.J.; Toral, M.; Romero, M.; et al. PPARβ activation restores the high glucose-induced impairment of insulin signalling in endothelial cells. Br. J. Pharmacol. 2014, 171, 3089–3102. [Google Scholar] [CrossRef] [PubMed]
- Polikandriotis, J.A.; Mazzella, L.J.; Rupnow, H.L.; Hart, C.M. Peroxisome proliferator-activated receptor γ ligands stimulate endothelial nitric oxide production through distinct peroxisome proliferator-activated receptor γ–dependent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Tian, X.Y.; Xu, A.; Yu, J.; Lau, C.W.; Hoo, R.L.; Wang, Y.; Lee, V.W.; Lam, K.S.; Vanhoutte, P.M.; et al. Adiponectin is required for PPARγ-mediated improvement of endothelial function in diabetic mice. Cell Metab. 2011, 14, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Wakino, S.; Hayashi, K.; Kanda, T.; Tatematsu, S.; Homma, K.; Yoshioka, K.; Takamatsu, I.; Saruta, T. Peroxisome proliferator-activated receptor γ ligands inhibit Rho/Rho kinase pathway by inducing protein tyrosine phosphatase SHP-2. Circ. Res. 2004, 95, e45–e55. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARα–leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Ibarra-Lara, M.D.L.L.; Sánchez-Aguilar, M.; Soria, E.; Torres-Narváez, J.C.; Del Valle-Mondragón, L.; Cervantes-Pérez, L.G.; Pérez-Severiano, F.; Ramírez-Ortega, M.D.C.; Pastelín-Hernández, G.; Oidor-Chan, V.H.; et al. Peroxisome proliferator-activated receptors (PPAR) downregulate the expression of pro-inflammatory molecules in an experimental model of myocardial infarction. Can. J. Physiol. Pharmacol. 2016, 94, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Paumelle, R.; Caron, S.; Hennuyer, N.; Mansouri, R.; Gervois, P.; Staels, B.; Haegeman, G.; De Bosscher, K. PPARα blocks glucocorticoid receptor α-mediated transactivation but cooperates with the activated glucocorticoid receptor α for transrepression on NF-κB. Proc. Natl. Acad. Sci. USA 2009, 106, 7397–7402. [Google Scholar] [CrossRef]
- Crisafulli, C.; Bruscoli, S.; Esposito, E.; Mazzon, E.; Di Paola, R.; Genovese, T.; Bramanti, P.; Migliorati, G.; Cuzzocrea, S. PPAR-α contributes to the anti-inflammatory activity of 17β-estradiol. J. Pharmacol. Exp. Ther. 2009, 331, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Sazatmari, I.; Rajnavolgyi, E.; Nagy, L. PPARγ, a lipid-activated transcription factor as a regulator of dendritic cell function. Ann. N. Y. Acad. Sci. 2006, 1088, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Kralj, D.; Jukić, L.V.; Stojsavljević, S.; Duvnjak, M.; Smolić, M.; Čurčić, I.B. Hepatitis C virus, insulin resistance, and steatosis. J. Clin. Transl. Hepatol. 2016, 4, 66–75. [Google Scholar] [PubMed]
- Kapadia, R.; Yi, J.-H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Nisbett, K.E.; Pinna, G. Emerging therapeutic role of PPAR–α in cognition and emotions. Front. Pharmacol. 2018, 9, 998. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Julie, D.; Renaud, J.; Olivier, C.; Pierre, T.; Regis, B. Therapeutic prospects of PPARs in psychiatric disorders: A comprehensive review. Curr. Drug Targets 2013, 14, 724–732. [Google Scholar]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-α-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Huang, J.-W.; Shiau, C.-W.; Yang, Y.-T.; Kulp, S.K.; Chen, K.-F.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.-S. Peroxisome proliferator-activated receptor γ-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol. Pharmacol. 2005, 67, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Brockman, J.A.; Sarraf, P.; Willson, T.M.; DuBois, R.N. Target genes of peroxisome proliferator-activated receptor γ in colorectal cancer cells. J. Biol. Chem. 2001, 276, 29681–29687. [Google Scholar] [CrossRef] [PubMed]
- Motomura, W.; Okumura, T.; Takahashi, N.; Obara, T.; Kohgo, Y. Activation of peroxisome proliferator-activated receptor γ by troglitazone inhibits cell growth through the increase of p27Kip1 in human pancreatic carcinoma cells. Cancer Res. 2000, 60, 5558–5564. [Google Scholar] [PubMed]
- Farrow, B.; Mark Evers, B. Activation of PPARγ increases PTEN expression in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003, 301, 50–53. [Google Scholar] [CrossRef]
- Bae, M.-A.; Song, B.J. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human hepG2 hepatoma cells. Mol. Pharmacol. 2003, 63, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Drori, S.; Girnun, G.D.; Tou, L.; Szwaya, J.D.; Mueller, E.; Kia, X.; Shivdasani, R.A.; Spiegelman, B.M. Hic-5 regulates an epithelial program mediated by PPARγ. Genes Dev. 2005, 19, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, X.; Kwong, J.S.W.; Li, L.; Li, Y.; Sun, X. Efficacy and safety of thiazolidinediones in diabetes patients with renal impairment: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 1717. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, X.; Zhang, M. Effect of thiazolidinediones on renal outcomes in diabetic patients with microalbuminuria or macroalbuminuria—A systematic review and meta-analysis. Diabetes 2018, 67, 532-P. [Google Scholar] [CrossRef]
- Alam, F.; Islam, M.A.; Mohamed, M.; Ahmad, I.; Kamal, M.A.; Donnelly, R.; Idris, I.; Gan, S.H. Efficacy and safety of pioglitazone monotherapy in type 2 diabetes mellitus: A systematic review and meta-analysis of randomised controlled trials. Sci. Rep. 2019, 9, 5389. [Google Scholar] [CrossRef]
- Rizos, C.V.; Elisaf, M.; Mikhailidis, D.P.; Liberopoulos, E.N. How safe is the use of thiazolidinediones in clinical practice? Expert Opin. Drug Saf. 2009, 8, 15–32. [Google Scholar] [CrossRef]
- Rubin, C.J.; De Pril, V.; Fiedorek, F.T. Coadministration of muraglitazar plus glyburide: Improvement of glycaemic and lipid profiles in patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2009, 6, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.J.; Viraswami-Appanna, K.; Fiedorek, F.T. Efficacy and safety of muraglitazar: A double-blind, 24-week, dose-ranging study in patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2009, 6, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.J.; Ledeine, J.-M.; Fiedorek, F.T. Improvement of glycaemic and lipid profiles with muraglitazar plus metformin in patients with type 2 diabetes: An active-control trial with glimepiride. Diabetes Vasc. Dis. Res. 2008, 5, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.; McElhattan, J.; Bryzinski, B.S. A double-blind, randomised trial of tesaglitazar versus pioglitazone in patients with type 2 diabetes mellitus. Diabetes Vasc. Dis. Res. 2007, 4, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, B.J.; Rosenstock, J.; Anzalone, D.; Tou, C.; Peter Öhman, K. Effect of tesaglitazar, a dual PPARα/γ agonist, on glucose and lipid abnormalities in patients with type 2 diabetes: A 12-week dose-ranging trial. Curr. Med. Res. Opin. 2006, 22, 2575–2590. [Google Scholar] [CrossRef]
- Göke, B.; Gause-Nilsson, I.; Persson, A. The effects of tesaglitazar as add-on treatment to metformin in patients with poorly controlled type 2 diabetes. Diabetes Vasc. Dis. Res. 2007, 4, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Ratner, R.E.; Parikh, S.; Tou, C. Efficacy, safety and tolerability of tesaglitazar when added to the therapeutic regimen of poorly controlled insulin-treated patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2007, 4, 214–221. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Gause-Nilsson, I.; Persson, A. Tesaglitazar, as add-on therapy to sulphonylurea, dose-dependently improves glucose and lipid abnormalities in patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2007, 4, 194–203. [Google Scholar] [CrossRef]
- Stirban, A.O.; Andjelkovic, M.; Heise, T.; Nosek, L.; Fischer, A.; Gastaldelli, A.; Herz, M. Aleglitazar, a dual peroxisome proliferator-activated receptor-α/γ agonist, improves insulin sensitivity, glucose control and lipid levels in people with type 2 diabetes: Findings from a randomized, double-blind trial. Diabetes Obes. Metab. 2016, 18, 711–715. [Google Scholar] [CrossRef]
- Henry, R.R.; Lincoff, A.M.; Mudaliar, S.; Rabbia, M.; Chognot, C.; Herz, M. Effect of the dual peroxisome proliferator-activated receptor-α/γ agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): A phase II, randomised, dose-ranging study. Lancet 2009, 374, 126–135. [Google Scholar] [CrossRef]
- Henry, R.R.; Buse, J.B.; Wu, H.; Durrwell, L.; Mingrino, R.; Jaekel, K.; El Azzouzi, B.; Andjelkovic, M.; Herz, M. Efficacy, safety and tolerability of aleglitazar in patients with type 2 diabetes: Pooled findings from three randomized phase III trials. Diabetes Obes. Metab. 2015, 17, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Kim, D.M.; Woo, J.-T.; Jang, H.C.; Chung, C.H.; Ko, K.S.; Park, J.H.; Park, Y.S.; Kim, S.J.; Choi, D.S. Efficacy and safety of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 24-weeks: A multicenter, randomized, double-blind, parallel-group, placebo controlled trial. PLoS ONE 2014, 9, e92843. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Park, C.Y.; Cho, Y.M.; Ku, B.J.; Ahn, C.W.; Cha, B.S.; Min, K.W.; Sung, Y.A.; Baik, S.H.; Lee, K.W.; et al. Lobeglitazone and pioglitazone as add-ons to metformin for patients with type 2 diabetes: A 24-week, multicentre, randomized, double-blind, parallel-group, active-controlled, phase III clinical trial with a 28-week extension. Diabetes Obes. Metab. 2015, 17, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Decochez, K.; Rippley, R.K.; Miller, J.L.; De Smet, M.; Yan, K.X.; Matthijs, Z.; Riffel, K.A.; Song, H.; Zhu, H.; Maynor, H.O.; et al. A dual PPAR α/γ agonist increases adiponectin and improves plasma lipid profiles in healthy subjects. Drugs R D 2006, 7, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Topol, E.J. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA 2005, 294, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Hamrén, B.; Öhman, K.P.; Svensson, M.K.; Karlsson, M.O. Pharmacokinetic-pharmacodynamic assessment of the interrelationships between tesaglitazar exposure and renal function in patients with type 2 diabetes mellitus. J. Clin. Pharmacol. 2012, 52, 1317–1327. [Google Scholar] [CrossRef]
- Herz, M.; Gaspari, F.; Perico, N.; Viberti, G.; Urbanowska, T.; Rabbia, M.; Kirk, D.W. Effects of high dose aleglitazar on renal function in patients with type 2 diabetes. Int. J. Cardiol. 2011, 151, 136–142. [Google Scholar] [CrossRef]
- Ruilope, L.; Hanefeld, M.; Lincoff, A.M.; Viberti, G.; Meyer-Reigner, S.; Mudie, N.; Wieczorek Kirk, D.; Malmberg, K.; Herz, M. Effects of the dual peroxisome proliferator-activated receptor-α/γ agonist aleglitazar on renal function in patients with stage 3 chronic kidney disease and type 2 diabetes: A Phase IIb, randomized study. BMC Nephrol. 2014, 15, 180. [Google Scholar] [CrossRef]
- Oleksiewicz, M.B.; Southgate, J.; Iversen, L.; Egerod, F.L. Rat urinary bladder carcinogenesis by dual-acting PPAR agonists. PPAR Res. 2008, 2008, 103167. [Google Scholar] [CrossRef]
- Lee, H.S.; Chang, M.; Lee, J.-E.; Kim, W.; Hwang, I.-C.; Kim, D.-H.; Park, H.-K.; Choi, H.-J.; Jo, W.; Cha, S.-W.; et al. Carcinogenicity study of CKD-501, a novel dual peroxisome proliferator-activated receptors α and γ agonist, following oral administration to Sprague Dawley rats for 94–101weeks. Regul. Toxicol. Pharmacol. 2014, 69, 207–216. [Google Scholar] [CrossRef]
- Moon, K.-S.; Lee, J.-E.; Lee, H.S.; Hwang, I.-C.; Kim, D.-H.; Park, H.-K.; Choi, H.-J.; Jo, W.; Son, W.-C.; Yun, H.-I. CKD-501, a novel selective PPARγ agonist, shows no carcinogenic potential in ICR mice following oral administration for 104 weeks. J. Appl. Toxicol. 2014, 34, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zaïr, Y.; Sauvinet, V.; Noël, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual Peroxisome Proliferator–Activated Receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Zaïr, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPARα/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Xu, Y.C.; Wang, Y.; Guo, L.; Cai, H.; Yang, T.; et al. 17-OR: Efficacy and safety of chiglitazar, a novel PPARα/γ/d pan-agonist, in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled phase 3 superiority trial (CMAP). Diabetes 2019, 68, 17-OR. [Google Scholar] [CrossRef]
- Jia, W.; Ma, J.; Miao, H.; Wang, C.; Wang, X.; Li, Q.; Lu, W.; Yang, J.; Zhang, L.; Yang, J.; et al. 18-OR: Efficacy and safety of chiglitazar vs. sitagliptin in patients with type 2 diabetes: A 24-week, randomized, double-blind, noninferiority phase 3 trial (CMAS). Diabetes 2019, 68, 18-OR. [Google Scholar] [CrossRef] [PubMed]
- The FIELD Study Investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Chew, E.Y.; Davis, M.D.; Danis, R.P.; Lovato, J.F.; Perdue, L.H.; Greven, C.; Genuth, S.; Goff, D.C.; Leiter, L.A.; Ismail-Beigi, F.; et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: The action to control cardiovascular risk in diabetes (ACCORD) Eye Study. Ophthalmology 2014, 121, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Tardif, J.-C.; Schwartz, G.G.; Nicholls, S.J.; Rydén, L.; Neal, B.; Malmberg, K.; Wedel, H.; Buse, J.B.; Henry, R.R.; et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: The AleCardio randomized clinical trial. JAMA 2014, 311, 1515–1525. [Google Scholar] [CrossRef]
- Erdmann, E.; Califf, R.; Gerstein, H.C.; Malmberg, K.; Ruilope, L.; Schwartz, G.G.; Wedel, H.; Volz, D.; Ditmarsch, M.; Svensson, A.; et al. Effects of the dual peroxisome proliferator–activated receptor activator aleglitazar in patients with type 2 diabetes mellitus or prediabetes. Am. Heart J. 2015, 170, 117–122. [Google Scholar] [CrossRef]
- Koomen, J.V.; Heerspink, H.J.L.; Schrieks, I.C.; Schwartz, G.G.; Lincoff, A.M.; Nicholls, S.J.; Svensson, A.; Wedel, H.; Weichert, A.; Grobbee, D.E.; et al. Exposure and response analysis of aleglitazar on cardiovascular risk markers and safety outcomes: An analysis of the AleCardio trial. Diabetes Obes. Metab. 2019. [Google Scholar] [CrossRef]
- Report from the Committee of Principal Investigators. A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. Br. Heart J. 1978, 40, 1069–1118. [Google Scholar] [CrossRef] [PubMed]
- Report of the Committee of Principal Investigators. WHO cooperative trial on primary prevention of ischaemic heart disease with clofibrate to lower serum cholesterol: Final mortality follow-up. Lancet 1984, 324, 600–604. [Google Scholar] [CrossRef]
- Meade, T.; Zuhrie, R.; Cook, C.; Cooper, J. Bezafibrate in men with lower extremity arterial disease: Randomised controlled trial. BMJ 2002, 325, 1139. [Google Scholar] [CrossRef] [PubMed]
- Arbel, Y.; Klempfner, R.; Erez, A.; Goldenberg, I.; Benzekry, S.; Shlomo, N.; Fisman, E.Z.; Tenenbaum, A. Bezafibrate for the treatment of dyslipidemia in patients with coronary artery disease: 20-year mortality follow-up of the BIP randomized control trial. Cardiovasc. Diabetol. 2016, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Elkeles, R.S.; Diamond, J.R.; Poulter, C.; Dhanjil, S.; Nicolaides, A.N.; Mahmood, S.; Richmond, W.; Mather, H.; Sharp, P.; Feher, M.D. Cardiovascular outcomes in type 2 diabetes: A double-blind placebo-controlled study of bezafibrate: The St. Mary’s, Ealing, Northwick Park Diabetes Cardiovascular Disease Prevention (SENDCAP) Study. Diabetes Care 1998, 21, 641–648. [Google Scholar] [CrossRef]
- Madrid-Miller, A.; Moreno-Ruiz, L.; Borrayo-Sánchez, G.; Almeida-Gutiérrez, E.; Martínez-Gómez, D.; Jáuregui-Aguilar, R. Impact of bezafibrate treatment in patients with hyperfibrinogenemia and ST-elevation acute myocardial infarction: A randomized clinical trial. Cir. Cir. 2010, 78, 229–237. [Google Scholar] [PubMed]
- d’Emden, M.C.; Jenkins, A.J.; Li, L.; Zannino, D.; Mann, K.P.; Best, J.D.; Stuckey, B.G.A.; Park, K.; Saltevo, J.; Keech, A.C. The FIELD Study Investigators, Favourable effects of fenofibrate on lipids and cardiovascular disease in women with type 2 diabetes: Results from the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetologia 2014, 57, 2296–2303. [Google Scholar] [CrossRef]
- Frick, M.H.; Elo, O.; Haapa, K.; Heinonen, O.P.; Heinsalmi, P.; Helo, P.; Huttunen, J.K.; Kaitaniemi, P.; Koskinen, P.; Manninen, V.; et al. Helsinki Heart Study: Primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. N. Engl. J. Med. 1987, 317, 1237–1245. [Google Scholar] [CrossRef]
- Michel, P.H. Impact of fenofibrate on type 2 diabetes patients with features of the metabolic syndrome: Subgroup analysis from FIELD. Curr. Cardiol. Rev. 2010, 6, 112–118. [Google Scholar]
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-González, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, CD009753. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.N. Peroxisome proliferator γ activated receptor gamma agonists for preventing recurrent stroke and other vascular events in people with stroke or transient ischaemic attack. Cochrane Database Syst. Rev. 2017, 12, CD010693. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.D.; Solis-Herrera, C.; Molina-Wilkins, M.; Martinez, S.; Merovci, A.; Cersosimo, E.; Chilton, R.J.; Iozzo, P.; Gastaldelli, A.; Abdul-Ghani, M.; et al. Pioglitazone improves left ventricular diastolic function in subjects with diabetes. Diabetes Care 2017, 40, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.; Bousser, M.-G.; Betteridge, D.J.; Schernthaner, G.; Pirags, V.; Kupfer, S.; Dormandy, J. Effects of pioglitazone in patients with type 2 diabetes with or without previous stroke. Stroke 2007, 38, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.; Kupfer, S.; Erdmann, E. Effects of pioglitazone on major adverse cardiovascular events in high-risk patients with type 2 diabetes: Results from PROspective pioglitAzone Clinical Trial In macro Vascular Events (PROactive 10). Am. Heart J. 2008, 155, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.A.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefèbvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after ischemic stroke or transient ischemic attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Choi, S.C.; Cho, J.Y.; Joo, H.J.; Park, J.H.; Yu, C.W.; Lim, D.-S. Pioglitazone increases circulating microRNA-24 with decrease in coronary neointimal hyperplasia in type 2 diabetic patients–optical coherence tomography analysis. Circ. J. 2015, 79, 880–888. [Google Scholar] [CrossRef]
- Kaneda, H.; Shiono, T.; Miyashita, Y.; Takahashi, S.; Taketani, Y.; Domae, H.; Matsumi, J.; Mizuno, S.; Minami, Y.; Sugitatsu, K.; et al. Efficacy and safety of pioglitazone in patients with ST elevation myocardial infarction treated with primary stent implantation. Heart 2009, 95, 1079–1084. [Google Scholar] [CrossRef]
- Takagi, T.; Okura, H.; Kobayashi, Y.; Kataoka, T.; Taguchi, H.; Toda, I.; Tamita, K.; Yamamuro, A.; Sakanoue, Y.; Ito, A.; et al. A prospective, multicenter, randomized trial to assess efficacy of pioglitazone on in-stent neointimal suppression in type 2 diabetes: POPPS (Prevention of In-Stent Neointimal Proliferation by Pioglitazone Study). JACC Cardiovasc. Interv. 2009, 2, 524–531. [Google Scholar] [CrossRef]
- Nishio, K.; Sakurai, M.; Kusuyama, T.; Shigemitsu, M.; Fukui, T.; Kawamura, K.; Itoh, S.; Konno, N.; Katagiri, T. A randomized comparison of pioglitazone to inhibit restenosis after coronary stenting in patients with type 2 diabetes. Diabetes Care 2006, 29, 101–106. [Google Scholar] [CrossRef]
- Mazzone, T.; Meyer, P.M.; Feinstein, S.B.; Davidson, M.H.; Kondos, G.T.; D’Agostino, R.B.; Perez, A.; Provost, J.-C.; Haffner, S.M. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 2006, 296, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochellière, R.; Staniloae, C.S.; Mavromatis, K.; et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Tahara, N.; Tahara, A.; Nitta, Y.; Kodama, N.; Oba, T.; Mawatari, K.; Yasukawa, H.; Kaida, H.; Ishibashi, M.; et al. Pioglitazone attenuates atherosclerotic plaque inflammation in patients with impaired glucose tolerance or diabetes: A prospective, randomized, comparator-controlled study using serial FDG PET/CT imaging study of carotid artery and ascending aorta. JACC Cardiovasc. Imaging 2011, 4, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Yamashiro, K.; Okuma, Y.; Shimura, H.; Nakamura, S.; Ueno, Y.; Tanaka, Y.; Miyamoto, N.; Tomizawa, Y.; Nakahara, T.; et al. Effects of pioglitazone for secondary stroke prevention in patients with impaired glucose tolerance and newly diagnosed diabetes: The J-SPIRIT Study. J. Atheroscler. Thromb. 2015, 22, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, S.; Ueno, M.; Tello-Montoliu, A.; Ferreiro, J.L.; Desai, B.; Rollini, F.; Box, L.C.; Zenni, M.M.; Guzman, L.A.; Bass, T.A.; et al. Effects of pioglitazone on platelet P2Y12-mediated signalling in clopidogrel-treated patients with type 2 diabetes mellitus. Thromb. Haemost. 2012, 108, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Giles, T.D.; Elkayam, U.; Bhattacharya, M.; Perez, A.; Miller, A.B. Comparison of pioglitazone vs glyburide in early heart failure: Insights from a randomized controlled study of patients with type 2 diabetes and mild cardiac disease. Congest. Heart Fail. 2010, 16, 111–117. [Google Scholar] [CrossRef]
- Vaccaro, O.; Masulli, M.; Nicolucci, A.; Bonora, E.; Del Prato, S.; Maggioni, A.P.; Rivellese, A.A.; Squatrito, S.; Giorda, C.B.; Sesti, G.; et al. Effects on the incidence of cardiovascular events of the addition of pioglitazone versus sulfonylureas in patients with type 2 diabetes inadequately controlled with metformin (TOSCA.IT): A randomised, multicentre trial. Lancet Diabetes Endocrinol. 2017, 5, 887–897. [Google Scholar] [CrossRef]
- de Jong, M.; van der Worp, H.B.; van der Graaf, Y.; Visseren, F.L.J.; Westerink, J. Pioglitazone and the secondary prevention of cardiovascular disease. A meta-analysis of randomized-controlled trials. Cardiovasc. Diabetol. 2017, 16, 134. [Google Scholar] [CrossRef]
- Bertrand, O.F.; Poirier, P.; Rodés-Cabau, J.; Rinfret, S.; Title, L.M.; Dzavik, V.; Natarajan, M.; Angel, J.; Batalla, N.; Alméras, N.; et al. Cardiometabolic effects of rosiglitazone in patients with type 2 diabetes and coronary artery bypass grafts: A randomized placebo-controlled clinical trial. Atherosclerosis 2010, 211, 565–573. [Google Scholar] [CrossRef]
- Lonn, E.M.; Gerstein, H.C.; Sheridan, P.; Smith, S.; Diaz, R.; Mohan, V.; Bosch, J.; Yusuf, S.; Dagenais, G.R. Effect of ramipril and of rosiglitazone on carotid intima-media thickness in people with impaired glucose tolerance or impaired fasting glucose: STARR (STudy of Atherosclerosis with Ramipril and Rosiglitazone). J. Am. Coll. Cardiol. 2009, 53, 2028–2035. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. JAMA Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Gao, H.; Li, W. Long-term risk of rosiglitazone on cardiovascular events—A systematic review and meta-analysis. Endokrynol. Pol. 2018, 69, 381–394. [Google Scholar] [PubMed]
- Loomba, R.S.; Arora, R. Prevention of cardiovascular disease utilizing fibrates—A pooled meta-analysis. Am. J. Ther. 2010, 17, e182–e188. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Saver, J.L.; Towfighi, A.; Chow, J.; Ovbiagele, B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: A meta-analysis. Atherosclerosis 2011, 217, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z. Fibrates are an essential part of modern anti-dyslipidemic arsenal: Spotlight on atherogenic dyslipidemia and residual risk reduction. Cardiovasc. Diabetol. 2012, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.-C. Pemafibrate (K-877), a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc. Diabetol. 2017, 16, 124. [Google Scholar] [CrossRef]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; K-877 Study Group. Efficacy and safety of Pemafibrate versus fenofibrate in patients with high triglyceride and low HDL cholesterol levels: A multicenter, placebo-controlled, double-blind, randomized trial. J. Atheroscler. Thromb. 2018, 25, 521–538. [Google Scholar] [CrossRef]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of Pemafibrate, a novel selective PPARα modulator, on lipid and glucose metabolism in patients with type 2 diabetes and hypertriglyceridemia: A randomized, double-blind, placebo- controlled, phase 3 trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S. Long-term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor-α modulator (SPPARMα), in dyslipidemic patients with renal impairment. Int. J. Mol. Sci. 2019, 20, 706. [Google Scholar] [CrossRef] [PubMed]
- Slim, A.; Castillo-Rojas, L.; Hulten, E.; Slim, J.N.; Pearce Moore, D.; Villines, T.C. Rosiglitazone and fenofibrate additive effects on lipids. Cholesterol 2011, 2011, 286875. [Google Scholar] [CrossRef]
- Sprecher Dennis, L.; Massien, C.; Pearce, G.; Billin Andrew, N.; Perlstein, I.; Willson Timothy, M.; Hassall David, G.; Ancellin, N.; Patterson Scott, D.; Lobe David, C.; et al. Triglyceride:High-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor δ agonist. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Risérus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator–activated receptor (PPAR)δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Olson Eric, J.; Pearce Gregory, L.; Jones Nigel, P.; Sprecher Dennis, L. Lipid effects of peroxisome proliferator-activated receptor-δ agonist GW501516 in subjects with low high-density lipoprotein cholesterol. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Ooi, E.M.M.; Watts, G.F.; Sprecher, D.L.; Chan, D.C.; Barrett, P.H.R. Mechanism of action of a peroxisome proliferator-activated receptor (PPAR)-δ agonist on lipoprotein metabolism in dyslipidemic subjects with central obesity. J. Clin. Endocrinol. Metab. 2011, 96, E1568–E1576. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., III; Kerzner, B.; Krauss, R.M.; Naim, S.; Wang, X.; Choi, Y.-J.; Roberts, B.K.; Karpf, D.B. MBX-8025, a novel peroxisome proliferator receptor-δ agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Roberts, B.K.; Wang, X.; Geaney, J.C.; Naim, S.; Wojnoonski, K.; Karpf, D.B.; Krauss, R.M. Effects of the PPAR-δ agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis 2012, 220, 470–476. [Google Scholar] [CrossRef]
- Pai, V.; Paneerselvam, A.; Mukhopadhyay, S.; Bhansali, A.; Kamath, D.; Shankar, V.; Gambhire, D.; Jani, R.H.; Joshi, S.; Patel, P. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared to pioglitazone 45 mg in diabetic dyslipidemia (PRESS V). J. Diabetes Sci. Technol. 2014, 8, 132–141. [Google Scholar] [CrossRef]
- Jani, R.H.; Pai, V.; Jha, P.; Mukhopadhyay, G.J.S.; Bhansali, A.; Joshi, S. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar] [PubMed]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist—Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Munigoti, S.; Harinarayan, C. Role of Glitazars in atherogenic dyslipidemia and diabetes: Two birds with one stone? Indian J. Endocrinol. Metab. 2014, 18, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Brisson, D.; Méthot, J.; Tremblay, K.; Tremblay, M.; Perron, P.; Gaudet, D. Comparison of the efficacy of fibrates on hypertriglyceridemic phenotypes with different genetic and clinical characteristics. Pharm. Genom. 2010, 20, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, M.-A.; Kobayashi, J.; Nohara, A.; Noguchi, T.; Tada, H.; Nakanishi, C.; Inazu, A.; Mabuchi, H.; Yamagishi, M. Impact of bezafibrate and atorvastatin on lipoprotein subclass in patients with type III hyperlipoproteinemia: Result from a crossover study. Clin. Chim. Acta 2011, 412, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Koopal, C.; Marais, A.D.; Westerink, J.; van der Graaf, Y.; Visseren, F.L.J. Effect of adding bezafibrate to standard lipid-lowering therapy on post-fat load lipid levels in patients with familial dysbetalipoproteinemia. A randomized placebo-controlled crossover trial. J. Lipid Res. 2017, 58, 2180–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelen, M.; Tran, L.; Ofman, R.; Brennecke, J.; Moser, A.B.; Dijkstra, I.M.E.; Wanders, R.J.A.; Poll-The, B.T.; Kemp, S. Bezafibrate for X-linked adrenoleukodystrophy. PLoS ONE 2012, 7, e41013. [Google Scholar] [CrossRef]
- Ørngreen, M.C.; Madsen, K.L.; Preisler, N.; Andersen, G.; Vissing, J.; Laforêt, P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: A randomized clinical trial. Neurology 2014, 82, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, D.; Saheb, S.; Bruckert, E.; de Graaf, J.; Langslet, G.; Tardif, J.C.; Bergheanu, S.; Steinberg, A.; Choi, Y.J.; Martin, R.; et al. A pilot study of MBX-8025 in the treatment of homozygous familial hypercholesterolemia (HoFH). Atherosclerosis 2016, 252, e253. [Google Scholar] [CrossRef]
- Naoumova, R.P.; Kindler, H.; Leccisotti, L.; Mongillo, M.; Khan, M.T.; Neuwirth, C.; Seed, M.; Holvoet, P.; Betteridge, J.; Camici, P.G. Pioglitazone improves myocardial blood flow and glucose utilization in nondiabetic patients with combined hyperlipidemia: A randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2007, 50, 2051–2058. [Google Scholar] [CrossRef]
- Thomas, E.L.; Potter, E.; Tosi, I.; Fitzpatrick, J.; Hamilton, G.; Amber, V.; Hughes, R.; North, C.; Holvoet, P.; Seed, M.; et al. Pioglitazone added to conventional lipid-lowering treatment in familial combined hyperlipidaemia improves parameters of metabolic control: Relation to liver, muscle and regional body fat content. Atherosclerosis 2007, 195, e181–e190. [Google Scholar] [CrossRef] [PubMed]
- Viale, P.; Manfredi, R.; Colangeli, V.; Calza, L.; Bon, I.; Re, M.C. Clinical management of dyslipidaemia associated with combination antiretroviral therapy in HIV-infected patients. J. Antimicrob. Chemother. 2016, 71, 1451–1465. [Google Scholar] [Green Version]
- Gerber, J.G.; Kitch, D.W.; Fichtenbaum, C.J.; Zackin, R.A.; Charles, S.; Hogg, E.; Acosta, E.P.; Connick, E.; Wohl, D.; Kojic, E.M.; et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: Results of ACTG A5186. JAIDS J. Acquir. Immune Defic. Syndr. 2008, 47, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; D’Amico, S.; Balasubramanyam, A.; Maldonado, M. Fenofibrate is effective in treating hypertriglyceridemia associated with HIV lipodystrophy. Am. J. Med. Sci. 2004, 327, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Badiou, S.; Merle De Boever, C.; Dupuy, A.-M.; Baillat, V.; Cristol, J.-P.; Reynes, J. Fenofibrate improves the atherogenic lipid profile and enhances LDL resistance to oxidation in HIV-positive adults. Atherosclerosis 2004, 172, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Dubé, M.P.; Komarow, L.; Fichtenbaum, C.J.; Cadden, J.J.; Overton, E.T.; Hodis, H.N.; Currier, J.S.; Stein, J.H. Extended-release niacin versus fenofibrate in HIV-infected participants with low high-density lipoprotein cholesterol: Effects on endothelial function, lipoproteins, and inflammation. Clin. Infect. Dis. 2015, 61, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, A.; Coraza, I.; Smith, E.O.B.; Scott, L.W.; Patel, P.; Iyer, D.; Taylor, A.A.; Giordano, T.P.; Sekhar, R.V.; Clark, P.; et al. Combination of niacin and fenofibrate with lifestyle changes improves dyslipidemia and hypoadiponectinemia in HIV patients on antiretroviral therapy: Results of “Heart Positive,” a randomized, controlled trial. J. Clin. Endocrinol. Metab. 2011, 96, 2236–2247. [Google Scholar] [CrossRef] [PubMed]
- Aberg, J.A.; Zackin, R.A.; Brobst, S.W.; Evans, S.R.; Alston, B.L.; Henry, W.K.; Glesby, M.J.; Torriani, F.J.; Yang, Y.; Owens, S.I.; et al. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res. Hum. Retrovir. 2005, 21, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, F.; Balestre, E.; Thiébaut, R.; Mercié, P.; Dupon, M.; Morlat, P.; Dabis, F.; Groupe d’Epidémiologie Clinique du SIDA en Aquitaine (GECSA). Fibrates or statins and lipid plasma levels in 245 patients treated with highly active antiretroviral therapy. Aquitaine Cohort, France, 1999–2001. HIV Med. 2004, 5, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Calza, L.; Manfredi, R.; Chiodo, F. Statins and fibrates for the treatment of hyperlipidaemia in HIV-infected patients receiving HAART. AIDS 2003, 17, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Calza, L.; Manfredi, R.; Colangeli, V.; Tampellini, L.; Sebastiani, T.; Pocaterra, D.; Chiodo, F. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005, 19, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Gavrila, A.; Martin, L.; Mantzoros, C.S.; Moses, A.C.; Hsu, W.; Karchmer, A.W.; Doweiko, J.; Tsiodras, S.; Gautam, S. Improvement in highly active antiretroviral therapy—Induced metabolic syndrome by treatment with pioglitazone but not with fenofibrate: A 2 × 2 factorial, randomized, double-blinded, placebo-controlled trial. Clin. Infect. Dis. 2005, 40, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Slama, L.; Lanoy, E.; Valantin, M.-A.; Bastard, J.-P.; Chermak, A.; Boutekatjirt, A.; William-Faltaos, D.; Billaud, E.; Molina, J.-M.; Capeau, J.; et al. Effect of pioglitazone on HIV-1-related lipodystrophy: A randomized double-blind placebo-controlled trial (ANRS 113). Antiral Ther. 2008, 13, 67–76. [Google Scholar]
- Okada, S.; Konishi, M.; Ishii, H. Pioglitazone therapy for HIV/HAART-associated lipodystrophy syndrome could increase subcutaneous fat mass in non-lipoatrophic but not in lipoatrophic regions. BMJ Case Rep. 2016, 2016, bcr2015213637. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Walmsley, S.; Kain, K.C.; Cheung, A.; Raboud, J.; Cavalcanti, R.B. A randomized, placebo-controlled trial of rosiglitazone for HIV-related lipoatrophy. J. Infect. Dis. 2007, 195, 1754–1761. [Google Scholar]
- van Wijk, J.P.H.; de Koning, E.J.P.; Cabezas, M.C.; op’t Roodt, J.; Joven, J.; Rabelink, T.J.; Hoepelman, A.I. Comparison of rosiglitazone and metformin for treating HIV lipodystrophy: A randomized trial. Ann. Intern. Med. 2005, 143, 337–346. [Google Scholar] [CrossRef]
- Tungsiripat, M.; El-Bejjani, D.; Rizk, N.; Dogra, V.; O’Riordan, M.A.; Ross, A.C.; Hileman, C.; Storer, N.; Harrill, D.; McComsey, G.A. Carotid intima media thickness, inflammatory markers, and endothelial activation markers in HIV patients with lipoatrophy increased at 48 weeks regardless of use of rosiglitazone or placebo. AIDS Res. Hum. Retrovir. 2011, 27, 295–302. [Google Scholar] [CrossRef]
- Sheth, S.H.; Larson, R.J. The efficacy and safety of insulin-sensitizing drugs in HIV-associated lipodystrophy syndrome: A meta-analysis of randomized trials. BMC Infect. Dis. 2010, 10, 183. [Google Scholar] [CrossRef]
- Fredriksen, J.; Ueland, T.; Dyrøy, E.; Halvorsen, B.; Melby, K.; Melbye, L.; Skalhegg, B.S.; Bohov, P.; Skorve, J.; Berge, R.K.; et al. Lipid-lowering and anti-inflammatory effects of tetradecylthioacetic acid in HIV-infected patients on highly active antiretroviral therapy. Eur. J. Clin. Investig. 2004, 34, 709–715. [Google Scholar] [CrossRef]
- Chilcott, J.; Tappenden, P.; Jones, M.L.; Wight, J.P. A systematic review of the clinical effectiveness of pioglitazone in the treatment of type 2 diabetes mellitus. Clin. Ther. 2001, 23, 1792–1823. [Google Scholar] [CrossRef]
- Ko, K.D.; Kim, K.K.; Lee, K.R. Does weight gain associated with thiazolidinedione use negatively affect cardiometabolic health? J. Obes. Metab. Syndr. 2017, 26, 102–106. [Google Scholar] [CrossRef]
- Berberoglu, Z.; Yazici, A.C.; Bayraktar, N.; Demirag, N.G. Rosiglitazone decreases fasting plasma peptide YY3–36 in type 2 diabetic women: A possible role in weight gain? Acta Diabetol. 2012, 49, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-M.; Li, Y.; Zhang, N.-N.; Xie, Y.-H.; Shi, Y.-Q. Combination therapy with metformin and fenofibrate for insulin resistance in obesity. J. Int. Med. Res. 2011, 39, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, T.D.; Kiortsis, D.N.; Liberopoulos, E.N.; Georgoula, M.; Mikhailidis, D.P.; Elisaf, M.S. Effect of orlistat, micronised fenofibrate and their combination on metabolic parameters in overweight and obese patients with the metabolic syndrome: The FenOrli study. Curr. Med. Res. Opin. 2005, 21, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.; Nian, H.; Yu, C.; Luther, J.M.; Brown, N.J. Fenofibrate lowers blood pressure in salt-sensitive but not salt-resistant hypertension. J. Hypertens. 2013, 31, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Chetty, V.T.; Sharma, A.M. Can PPARγ agonists have a role in the management of obesity-related hypertension? Vascul. Pharmacol. 2006, 45, 46–53. [Google Scholar] [CrossRef]
- Rivas, B.D.; Luque, M.; Martell, N.; Fernández, C.; Fernández-Cruz, A. Pioglitazone decreases ambulatory blood pressure in type 2 diabetics with difficult-to-control hypertension. J. Clin. Hypertens. 2007, 9, 530–537. [Google Scholar] [CrossRef]
- Qayyum, R.; Adomaityte, J. A meta-analysis of the effect of thiazolidinediones on blood pressure. J. Clin. Hypertens. 2006, 8, 19–28. [Google Scholar] [CrossRef]
- Pfützner, A.; Hanefeld, M.; Dekordi, L.A.; Müller, J.; Kleine, I.; Fuchs, W.; Forst, T. Effect of Pioglitazone and Ramipril on biomarkers of low-grade inflammation and vascular function in nondiabetic patients with increased cardiovascular risk and an activated inflammation: Results from the PIOace study. J. Diabetes Sci. Technol. 2011, 5, 989–998. [Google Scholar] [CrossRef]
- Straznicky, N.E.; Grima, M.T.; Sari, C.I.; Eikelis, N.; Lambert, G.W.; Nestel, P.J.; Richards, K.; Dixon, J.B.; Schlaich, M.P.; Lambert, E.A. Pioglitazone treatment enhances the sympathetic nervous system response to oral carbohydrate load in obese individuals with metabolic syndrome. Metabolism 2015, 64, 797–803. [Google Scholar] [CrossRef]
- Horio, T.; Suzuki, M.; Suzuki, K.; Takamisawa, I.; Hiuge, A.; Kamide, K.; Takiuchi, S.; Iwashima, Y.; Kihara, S.; Funahashi, T.; et al. Pioglitazone improves left ventricular diastolic function in patients with essential hypertension. Am. J. Hypertens. 2005, 18, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Raji, A.; Seely, E.W.; Bekins, S.A.; Williams, G.H.; Simonson, D.C. Rosiglitazone improves insulin sensitivity and lowers blood pressure in hypertensive patients. Diabetes Care 2003, 26, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Mittermayer, F.; Schaller, G.; Pleiner, J.; Krzyzanowska, K.; Kapiotis, S.; Roden, M.; Wolzt, M. Rosiglitazone prevents free fatty acid-induced vascular endothelial dysfunction. J. Clin. Endocrinol. Metab. 2007, 92, 2574–2580. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, A.; Uruno, A.; Kudo, M.; Matsuda, K.; Yang, C.W.; Ito, S. PPARγ agonist beyond glucose lowering effect. Korean J. Intern. Med. 2011, 26, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ningning, W.; Symons, J.D.; Hui, Z.; Zhanjun, J.; Gonzalez, F.J.; Tianxin, Y. Distinct functions of vascular endothelial and smooth muscle PPARγ in regulation of blood pressure and vascular tone. Toxicol. Pathol. 2009, 37, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sayiner, M.; Koenig, A.; Henry, L.; Younossi, Z.M. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin. Liver Dis. 2016, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef]
- Kim, S.; Ko, K.; Park, S.; Lee, D.R.; Lee, J. Effect of fenofibrate medication on renal function. Korean J. Fam. Med. 2017, 38, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H.; Armani, A.; McKenney, J.M.; Jacobson, T.A. Safety considerations with fibrate therapy. Am. J. Cardiol. 2007, 99, S3–S18. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A. Myopathy with statin–fibrate combination therapy: Clinical considerations. Nat. Rev. Endocrinol. 2009, 5, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Promrat, K.; Lutchman, G.; Uwaifo, G.I.; Freedman, R.J.; Soza, A.; Heller, T.; Doo, E.; Ghany, M.; Premkumar, A.; Park, Y.; et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004, 39, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: A randomized trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Hespenheide, E.E.; Redick, J.A.; Iezzoni, J.C.; Battle, E.H.; Sheppard, B.L. A pilot study of a thiazolidinedione, troglitazone, in nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Brunt, E.M.; Wehmeier, K.R.; Oliver, D.; Bacon, B.R. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-γ ligand rosiglitazone. Hepatology 2003, 38, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann–Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; LeNaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; Group, L.S. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Idilman, R.; Mizrak, D.; Corapcioglu, D.; Bektas, M.; Doganay, B.; Sayki, M.; Coban, S.; Erden, E.; Soykan, I.; Emral, R.; et al. Clinical trial: Insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2008, 28, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Akyüz, F.; Demir, K.; Özdil, S.; Aksoy, N.; Poturoğlu, Ş.; İbrişim, D.; Kaymakoğlu, S.; Beşışık, F.; Boztaş, G.; Çakaloğlu, Y.; et al. The effects of rosiglitazone, metformin, and diet with exercise in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2007, 52, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator−activated receptor−α and−δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.-J.; Cho, Y.M.; Lee, B.-W. Lobeglitazone, a novel thiazolidinedione, improves non-alcoholic fatty liver disease in type 2 diabetes: Its efficacy and predictive factors related to responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, G.; Luccarini, J.-M.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrêne, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Gershwin, M.E.; Ansari, A.A.; Mackay, I.R.; Nakanuma, Y.; Nishio, A.; Rowley, M.J.; Coppel, R.L. Primary biliary cirrhosis: An orchestrated immune response against epithelial cells. Immunol. Rev. 2000, 174, 210–225. [Google Scholar] [CrossRef]
- Lindor, K.D.; Gershwin, M.E.; Poupon, R.; Kaplan, M.; Bergasa, N.V.; Heathcote, E.J. Primary biliary cirrhosis. Hepatology 2009, 50, 291–308. [Google Scholar] [CrossRef]
- Corpechot, C.; Carrat, F.; Bahr, A.; Chrétien, Y.; Poupon, R.-E.; Poupon, R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology 2005, 128, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Itakura, J.; Izumi, N.; Nishimura, Y.; Inoue, K.; Ueda, K.; Nakanishi, H.; Tsuchiya, K.; Hamano, K.; Asahina, Y.; Kurosaki, M.; et al. Prospective randomized crossover trial of combination therapy with bezafibrate and UDCA for primary biliary cirrhosis. Hepatol. Res. 2004, 29, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Ohira, H.; Nishiguchi, S.; Zeniya, M.; Kaneko, S.; Onji, M.; Ishibashi, H.; Sakaida, I.; Kuriyama, S.; Ichida, T.; et al. The efficacy of ursodeoxycholic acid and bezafibrate combination therapy for primary biliary cirrhosis: A prospective, multicenter study. Hepatol. Res. 2008, 38, 557–564. [Google Scholar] [CrossRef]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M.; Namisaki, T.; Hashimoto, N.; Kawata, K.; Takahashi, A.; et al. Bezafibrate improves GLOBE and UK-PBC scores and long-term outcomes in patients with primary biliary cholangitis. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Peter, J.A.; Nelson, D.R.; Keach, J.; Petz, J.; Cabrera, R.; Clark, V.; Firpi, R.J.; Morelli, G.; Soldevila-Pico, C.; et al. Pilot study: Fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment. Pharmacol. Ther. 2011, 33, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, A.Y.; Mardini, H.E.; Corpechot, C.; Poupon, R.; Levy, C. Fenofibrate is effective adjunctive therapy in the treatment of primary biliary cirrhosis: A meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 296–306. [Google Scholar] [CrossRef]
- Han, X.F.; Wang, Q.X.; Liu, Y.; You, Z.R.; Bian, Z.L.; Qiu, D.K.; Ma, X. Efficacy of fenofibrate in Chinese patients with primary biliary cirrhosis partially responding to ursodeoxycholic acid therapy. J. Dig. Dis. 2012, 13, 219–224. [Google Scholar] [CrossRef]
- Cheung, A.C.; Lapointe-Shaw, L.; Kowgier, M.; Meza-Cardona, J.; Hirschfield, G.M.; Janssen, H.L.A.; Feld, J.J. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment. Pharmacol. Ther. 2016, 43, 283–293. [Google Scholar] [CrossRef]
- Liberopoulos, E.N.; Florentin, M.; Elisaf, M.S.; Mikhailidis, D.P.; Tsianos, E. Fenofibrate in primary biliary cirrhosis: A pilot study. Open Cardiovasc. Med. J. 2010, 4, 120–126. [Google Scholar] [CrossRef]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef]
- McHutchison, J.; Goodman, Z.; Patel, K.; Makhlouf, H.; Rodriguez-Torres, M.; Shiffman, M.; Rockey, D.; Husa, P.; Chuang, W.L.; Levine, R.; et al. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology 2010, 138, 1365–1373. [Google Scholar] [CrossRef]
- Overbeck, K.; Genné, D.; Golay, A.; Negro, F. Pioglitazone in chronic hepatitis C not responding to pegylated interferon-α and ribavirin. J. Hepatol. 2008, 49, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Chojkier, M.; Elkhayat, H.; Sabry, D.; Donohue, M.; Buck, M. Pioglitazone decreases hepatitis C viral load in overweight, treatment naïve, genotype 4 infected-patients: A pilot study. PLoS ONE 2012, 7, e31516. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-S.; Hsu, S.-J.; Lin, H.H.; Tseng, T.-C.; Wang, C.-C.; Chen, D.-S.; Kao, J.-H. A pilot study of add-on oral hypoglycemic agents in treatment-naïve genotype-1 chronic hepatitis C patients receiving peginterferon alfa-2b plus ribavirin. J. Formos. Med. Assoc. 2014, 113, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Vachon, M.-L.; Dieterich, D.T. The era of direct-acting antivirals has begun: The beginning of the end for HCV? Semin. Liver Dis. 2011, 31, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Bolier, R.; de Vries, E.S.; Parés, A.; Helder, J.; Kemper, E.M.; Zwinderman, K.; Elferink, R.P.O.; Beuers, U.; van Buuren, H.R.; Drenth, J.P.; et al. Fibrates for the treatment of cholestatic itch (FITCH): Study protocol for a randomized controlled trial. Trials 2017, 18, 230. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef]
- Satirapoj, B.; Watanakijthavonkul, K.; Supasyndh, O. Safety and efficacy of low dose pioglitazone compared with standard dose pioglitazone in type 2 diabetes with chronic kidney disease: A randomized controlled trial. PLoS ONE 2018, 13, e0206722. [Google Scholar] [CrossRef] [PubMed]
- Zanchi, A.; Tappy, L.; Lê, K.-A.; Bortolotti, M.; Theumann, N.; Halabi, G.; Gauthier, T.; Mathieu, C.; Tremblay, S.; Bertrand, P.C.; et al. Pioglitazone improves fat distribution, the adipokine profile and hepatic insulin sensitivity in non-diabetic end-stage renal disease subjects on maintenance dialysis: A randomized cross-over pilot study. PLoS ONE 2014, 9, e109134. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.T.; Watts, G.F.; Irish, A.B.; Dogra, G.K. Rosiglitazone does not improve vascular function in subjects with chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 3543–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Ferrannini, E.; DeFronzo, R.; Schernthaner, G.; Yates, J.; Erdmann, E. Effect of pioglitazone on cardiovascular outcome in diabetes and chronic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Werzowa, J.; Hecking, M.; Haidinger, M.; Lechner, F.; Döller, D.; Pacini, G.; Stemer, G.; Pleiner, J.; Frantal, S.; Säemann, M.D. Vildagliptin and pioglitazone in patients with impaired glucose tolerance after kidney transplantation: A randomized, placebo-controlled clinical trial. Transplantation 2013, 95, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Hur, K.Y.; Kim, Y.S.; Kang, E.S.; Kim, S.I.; Kim, M.S.; Kwak, J.Y.; Kim, D.J.; Choi, S.H.; Cha, B.S.; et al. Effects of pioglitazone on subclinical atherosclerosis and insulin resistance in nondiabetic renal allograft recipients. Nephrol. Dial. Transplant. 2009, 25, 976–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maalouf, N.M.; Poindexter, J.R.; Adams-Huet, B.; Moe, O.W.; Sakhaee, K. Increased production and reduced urinary buffering of acid in uric acid stone formers is ameliorated by pioglitazone. Kidney Int. 2019, 95, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Tzimopoulou, S.; Cunningham, V.J.; Nichols, T.E.; Searle, G.; Brid, N.P.; Mistry, P.; Dixon, I.J.; Hallett, W.A.; Whitcher, B.; Brown, A.P.; et al. A multi-center randomized proof-of-concept clinical trial applying [18F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 1241–1256. [Google Scholar] [CrossRef]
- Risner, M.E.; Saunders, A.M.; Altman, J.F.B.; Ormandy, G.C.; Craft, S.; Foley, I.M.; Zvartau-Hind, M.E.; Hosford, D.A.; Roses, A.D.; Group for the Rosiglitazone in Alzheimer’s Disease Study. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharm. J. 2006, 6, 246–254. [Google Scholar] [CrossRef]
- Gold, M.; Alderton, C.; Zvartau-Hind, M.; Egginton, S.; Saunders, A.M.; Irizarry, M.; Craft, S.; Landreth, G.; Linnamägi, Ü.; Sawchak, S. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: Results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord. 2010, 30, 131–146. [Google Scholar] [CrossRef]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.; Saunders, A.M.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.; van Dyck, C.; et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimers disease: Two phase 3 studies. Curr. Alzheimer Res. 2011, 8, 592–606. [Google Scholar] [CrossRef]
- Hildreth, K.L.; Van Pelt, R.E.; Moreau, K.L.; Grigsby, J.; Hoth, K.F.; Pelak, V.; Anderson, C.A.; Parnes, B.; Kittelson, J.; Wolfe, P.; et al. Effects of pioglitazone or exercise in older adults with mild cognitive impairment and insulin resistance: A pilot study. Dement. Geriatr. Cogn. Disord. Extra 2015, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. JAMA Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar] [CrossRef]
- Dutta, K.; Patel, P.; Julien, J.-P. Protective effects of Withania somnifera extract in SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2018, 309, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Schütz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schürmann, B.; Zimmer, A.; Heneka, M.T. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef] [PubMed]
- Joardar, A.; Menzl, J.; Podolsky, T.C.; Manzo, E.; Estes, P.S.; Ashford, S.; Zarnescu, D.C. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum. Mol. Genet. 2014, 24, 1741–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Dengler, R.; Heneka, M.T.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; Steiner, F.; Lindauer, E.; Otto, M.; et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e37885. [Google Scholar] [CrossRef]
- Levine, T.D.; Bowser, R.; Hank, N.C.; Gately, S.; Stephan, D.; Saperstein, D.S.; Van Keuren-Jensen, K. A pilot trial of pioglitazone HCl and tretinoin in ALS: Cerebrospinal fluid biomarkers to monitor drug efficacy and predict rate of disease progression. Neurol. Res. Int. 2012, 2012, 582075. [Google Scholar] [CrossRef]
- Höftberger, R.; Leisser, M.; Bauer, J.; Lassmann, H. Autoimmune encephalitis in humans: How closely does it reflect multiple sclerosis ? Acta Neuropathol. Commun. 2015, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.C.; Shukla, D.K.; Stebbins, G.T.; Skias, D.D.; Jeffery, D.R.; Stefoski, D.; Katsamakis, G.; Feinstein, D.L. A pilot test of pioglitazone as an add-on in patients with relapsing remitting multiple sclerosis. J. Neuroimmunol. 2009, 211, 124–130. [Google Scholar] [CrossRef]
- Weinstein, D.; Boyko, A.; Pugliese, L.; Tang, H.; Lanfear, D.; Zivadinov, R.; Finck, B. CHS-131, A novel once daily oral treatment, decreased lesion burden of patients with relapsing-remitting course of multiple sclerosis (RRMS) in a randomized, double-blind, phase 2b, multicenter study (S50.002). Neurology 2017, 88, S50.002. [Google Scholar]
- Puligheddu, M.; Melis, M.; Pillolla, G.; Milioli, G.; Parrino, L.; Terzano, G.M.; Aroni, S.; Sagheddu, C.; Marrosu, F.; Pistis, M.; et al. Rationale for an adjunctive therapy with fenofibrate in pharmacoresistant nocturnal frontal lobe epilepsy. Epilepsia 2017, 58, 1762–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Brassington, R.; Pandolfo, M. Pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2016, 2016, CD007791. [Google Scholar] [CrossRef] [PubMed]
- Foll, B.L.; Ciano, P.D.; Leigh, V.P.; Steven, R.G.; Roberto, C. Peroxisome proliferator-activated receptor (PPAR) agonists as promising new medications for drug addiction: Preclinical evidence. Curr. Drug Targets 2013, 14, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Gendy, M.N.S.; Di Ciano, P.; Kowalczyk, W.J.; Barrett, S.P.; George, T.P.; Heishman, S.; Le Foll, B. Testing the PPAR hypothesis of tobacco use disorder in humans: A randomized trial of the impact of gemfibrozil (a partial PPARα agonist) in smokers. PLoS ONE 2018, 13, e0201512. [Google Scholar] [CrossRef] [PubMed]
- Perkins, K.A.; Karelitz, J.L.; Michael, V.C.; Fromuth, M.; Conklin, C.A.; Chengappa, K.N.R.; Hope, C.; Lerman, C. Initial evaluation of fenofibrate for efficacy in aiding smoking abstinence. Nicotine Tob. Res. 2015, 18, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Comer, S.D.; Metz, V.E.; Manubay, J.M.; Mogali, S.; Ciccocioppo, R.; Martinez, S.; Mumtaz, M.; Bisaga, A. Pioglitazone, a PPARγ agonist, reduces nicotine craving in humans, with marginal effects on abuse potential. Pharmacol. Biochem. Behav. 2017, 163, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.M.; Green, C.E.; Hasan, K.M.; Vincent, J.; Suchting, R.; Weaver, M.F.; Moeller, F.G.; Narayana, P.A.; Cunningham, K.A.; Dineley, K.T.; et al. PPAR-gamma agonist pioglitazone modifies craving intensity and brain white matter integrity in patients with primary cocaine use disorder: A double-blind randomized controlled pilot trial. Addiction 2017, 112, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Sullivan, M.A.; Manubay, J.M.; Mogali, S.; Metz, V.E.; Ciccocioppo, R.; Comer, S.D. The effects of pioglitazone, a PPARγ receptor agonist, on the abuse liability of oxycodone among nondependent opioid users. Physiol. Behav. 2016, 159, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.R.; Phillips, K.A.; Epstein, D.H.; Jobes, M.L.; Furnari, M.A.; Kennedy, A.P.; Heilig, M.; Preston, K.L. Assessment of pioglitazone and proinflammatory cytokines during buprenorphine taper in patients with opioid use disorder. Psychopharmacology (Berl) 2018, 235, 2957–2966. [Google Scholar] [CrossRef]
- Kemp, D.E.; Schinagle, M.; Gao, K.; Conroy, C.; Ganocy, S.J.; Ismail-Beigi, F.; Calabrese, J.R. PPAR-γ agonism as a modulator of mood: Proof-of-concept for pioglitazone in bipolar depression. CNS Drugs 2014, 28, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Rasgon, N.L.; Kenna, H.A.; Williams, K.E.; Powers, B.; Wroolie, T.; Schatzberg, A.F. Rosiglitazone add-on in treatment of depressed patients with insulin resistance: A pilot study. Sci. World J. 2010, 10, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Sepanjnia, K.; Modabbernia, A.; Ashrafi, M.; Modabbernia, M.-J.; Akhondzadeh, S. Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: Randomized double-blind placebo-controlled trial. Neuropsychopharmacology 2012, 37, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.W.; Wroolie, T.E.; Robakis, T.; Rasgon, N.L. Adjuvant pioglitazone for unremitted depression: Clinical correlates of treatment response. Psychiatry Res. 2015, 230, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.C.; Jin, H.; Li, C.; Bark, N.; Shekhar, A.; Dwivedi, S.; Mortiere, C.; Lohr, J.; Hu, Q.; Davis, J.M. Effects of pioglitazone on metabolic abnormalities, psychopathology, and cognitive function in schizophrenic patients treated with antipsychotic medication: A randomized double-blind study. Schizophr. Res. 2013, 143, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Aftab, A.; Kemp, D.E.; Ganocy, S.J.; Schinagle, M.; Conroy, C.; Brownrigg, B.; D’Arcangelo, N.; Goto, T.; Woods, N.; Serrano, M.B.; et al. Double-blind, placebo-controlled trial of pioglitazone for bipolar depression. J. Affect. Disord. 2019, 245, 957–964. [Google Scholar] [CrossRef]
- Boris, M.; Kaiser, C.C.; Goldblatt, A.; Elice, M.W.; Edelson, S.M.; Adams, J.B.; Feinstein, D.L. Effect of pioglitazone treatment on behavioral symptoms in autistic children. J. Neuroinflamm. 2007, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Ghaleiha, A.; Rasa, S.M.; Nikoo, M.; Farokhnia, M.; Mohammadi, M.-R.; Akhondzadeh, S. A pilot double-blind placebo-controlled trial of pioglitazone as adjunctive treatment to risperidone: Effects on aberrant behavior in children with autism. Psychiatry Res. 2015, 229, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Capano, L.; Dupuis, A.; Brian, J.; Mankad, D.; Genore, L.; Hastie Adams, R.; Smile, S.; Lui, T.; Odrobina, D.; Foster, J.A.; et al. A pilot dose finding study of pioglitazone in autistic children. Mol. Autism 2018, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, M.J.; Oeser, A.M.; Cunningham, A.; Bian, A.; Shintani, A.; Solus, J.; Tanner, S.B.; Stein, C.M. Peroxisome proliferator-activated receptor γ agonist effect on rheumatoid arthritis: A randomized controlled trial. Arthrit. Res. Ther. 2013, 15, R110. [Google Scholar] [CrossRef]
- Marder, W.; Khalatbari, S.; Myles, J.D.; Hench, R.; Lustig, S.; Yalavarthi, S.; Parameswaran, A.; Brook, R.D.; Kaplan, M.J. The peroxisome proliferator activated receptor-γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J. Am. Heart Assoc. 2013, 2, e000441. [Google Scholar] [CrossRef] [PubMed]
- Shahin, D.; Toraby, E.E.; Abdel-Malek, H.; Boshra, V.; Elsamanoudy, A.Z.; Shaheen, D. Effect of peroxisome proliferator-activated receptor gamma agonist (pioglitazone) and methotrexate on disease activity in rheumatoid arthritis (experimental and clinical study). Clin. Med. Insights Arthrit. Musculoskelet. Disord. 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Kaplan, M.J. Cardiovascular disease in lupus: Insights and updates. Curr. Opin. Rheumatol. 2013, 25, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Rojas, J.; Medina-Urrutia, A.; Jorge-Galarza, E.; Caracas-Portilla, N.; Posadas-Sánchez, R.; Cardoso-Saldaña, G.; Goycochea-Robles, M.; Silveira, L.; Lino-Pérez, L.; Mas-Oliva, J.; et al. Pioglitazone improves the cardiovascular profile in patients with uncomplicated systemic lupus erythematosus: A double-blind randomized clinical trial. Lupus 2012, 21, 27–35. [Google Scholar] [CrossRef]
- Boggild, A.K.; Krudsood, S.; Patel, S.N.; Serghides, L.; Tangpukdee, N.; Katz, K.; Wilairatana, P.; Liles, W.C.; Looareesuwan, S.; Kain, K.C. Use of peroxisome proliferator-activated receptor γ agonists as adjunctive treatment for Plasmodium falciparum malaria: A randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 2009, 49, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Varo, R.; Crowley, V.M.; Sitoe, A.; Madrid, L.; Serghides, L.; Bila, R.; Mucavele, H.; Mayor, A.; Bassat, Q.; Kain, K.C. Safety and tolerability of adjunctive rosiglitazone treatment for children with uncomplicated malaria. Malar. J. 2017, 16, 215. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef]
- Pedersen, G.; Brynskov, J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-γ activity. Am. J. Gastroenterol. 2010, 105, 1595–1603. [Google Scholar] [CrossRef]
- Benayoun, L.; Letuve, S.; Druilhe, A.; Boczkowski, J.; Dombret, M.-C.; Mechighel, P.; Megret, J.; Leseche, G.; Aubier, M.; Pretolani, M. Regulation of peroxisome proliferator-activated receptor γ expression in human asthmatic airways. Am. J. Respir. Crit. Care Med. 2001, 164, 1487–1494. [Google Scholar] [CrossRef]
- Richards, D.B.; Bareille, P.; Lindo, E.L.; Quinn, D.; Farrow, S.N. Treatment with a peroxisomal proliferator activated receptor gamma agonist has a modest effect in the allergen challenge model in asthma: A randomised controlled trial. Respir. Med. 2010, 104, 668–674. [Google Scholar] [CrossRef]
- Anderson, J.R.; Mortimer, K.; Pang, L.; Smith, K.M.; Bailey, H.; Hodgson, D.B.; Shaw, D.E.; Knox, A.J.; Harrison, T.W. Evaluation of the PPAR-γ agonist pioglitazone in mild asthma: A double-blind randomized controlled trial. PLoS ONE 2016, 11, e0160257. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.E.; Subramanian, M.; DeSarno, M.; Black, K.; Lane, L.; Holguin, F. A pilot randomized controlled trial of pioglitazone for the treatment of poorly controlled asthma in obesity. Respir. Res. 2015, 16, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaler, M.; Barochia, A.V.; Weir, N.A.; Cuento, R.A.; Stylianou, M.; Roth, M.J.; Filie, A.C.; Vaughey, E.C.; Nathan, S.D.; Levine, S.J. A randomized, placebo-controlled, double-blinded, crossover trial of pioglitazone for severe asthma. J. Allergy Clin. Immunol. 2017, 140, 1716–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, A.; Shafiq, N.; Rajagopalan, S.; Dogra, S.; Malhotra, S. Thiazolidinediones for plaque psoriasis: A systematic review and meta-analysis. Evid. Based Med. 2012, 17, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Malhotra, S.; Pandhi, P.; Kaur, I.; Dogra, S. Efficacy and safety of combination acitretin and pioglitazone therapy in patients with moderate to severe chronic plaque-type psoriasis: A randomized, double-blind, placebo-controlled clinical trial. JAMA Dermatol. 2009, 145, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Moravek, M.B.; Ward, E.A.; Lebovic, D.I. Thiazolidinediones as therapy for endometriosis: A case series. Gynecol. Obstet. Investig. 2009, 68, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.; Hilliard, K.; Bucur, C.; Xue, W.; Schluchter, M.; Divoky, E.; JE, K.; Hilliard, J. Effect of pioglitazone on sputum markers of inflammation in cystic fibrosis [abstract]. Pediatr. Pulmonol. 2008, 43, 310. [Google Scholar]
- Kaplan, J.M.; Zingarelli, B.; Krallman, K.; Girdwood, S.T.; Lagory, D.; Mizuno, T.; Fei, L.; Wong, H.R.; Vinks, A.A. Phase 1 safety and pharmacokinetic study on the use of pioglitazone in critically ill patients with sepsis: A randomized clinical trial. Intensive Care Med. 2018, 44, 2006–2008. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Huang, H.J.; Byers, D.E.; Shifren, A.; Belikoff, B.; Engle, J.T.; Arentson, E.; Kemp, D.; Phillips, S.; Scherrer, D.E.; et al. The peroxisome proliferator-activated receptor agonist pioglitazone and 5-lipoxygenase inhibitor zileuton have no effect on lung inflammation in healthy volunteers by positron emission tomography in a single-blind placebo-controlled cohort study. PLoS ONE 2018, 13, e0191783. [Google Scholar] [CrossRef] [PubMed]
- Migliavacca, M.; Assanelli, A.; Ferrua, F.; Cicalese, M.P.; Biffi, A.; Frittoli, M.; Silvani, P.; Chidini, G.; Calderini, E.; Mandelli, A.; et al. Pioglitazone as a novel therapeutic approach in chronic granulomatous disease. J. Allergy Clin. Immunol. 2016, 137, 1913–1915.e2. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, C.K.; Ferguson, J.F.; Tabita-Martinez, J.; Kong, S.; Shah, R.Y.; Patel, P.N.; Master, S.R.; Usman, M.H.U.; Propert, K.J.; Shah, R.; et al. Peroxisome proliferator–activated receptor-α agonism with fenofibrate does not suppress inflammatory responses to evoked endotoxemia. J. Am. Heart Assoc. 2012, 1, e002923. [Google Scholar] [CrossRef]
- Rhodus, N.; Rohrer, M.; Pambuccian, S.; Keel, S.; Bliss, R.L.; Szabo, E.; Ondrey, F. Phase IIa chemoprevention clinical trial of pioglitazone for oral leukoplakia. In Proceedings of the 89th General Session & Exhibition of the International Association for Dental Research, San Diego, CA, USA, 16–19 March 2011. [Google Scholar]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an oral PPAR-γ agonist, in combination with paclitaxel in anaplastic thyroid cancer: Results of a multicenter phase 1 trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J.; Haugen, B.R.; Sherman, S.I.; Shah, M.H.; Caoili, E.M.; Koenig, R.J. Pioglitazone therapy of PAX8-PPARγ fusion protein thyroid carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Lindsay, S.; Clark, O.H.; Woeber, K.A.; Hawkins, R.; Greenspan, F.S. Results of rosiglitazone therapy in patients with thyroglobulin-positive and radioiodine-negative advanced differentiated thyroid cancer. Thyroid 2009, 19, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Ahn, M.J.; Ahn, J.S.; Lee, J.; Sun, J.M.; Yi, J.H.; Cho, S.H.; Lee, J.Y.; Park, S.; Hwang, D.W. Phase 1b Study of Oral PPAR-gamma Agonist Efatutazone (CS-7017) in Combination With Carboplatin and Paclitaxel in Chemotherapy-Naive Korean Patients With Metastatic or Unresectable Locally Advanced Non-Small Cell Lung Cancer. Eur. J. Cancer 2011, 47, S609. [Google Scholar] [CrossRef]
- Kim, S.-W.; Choi, C.-M.; Lee, D.H.; Lee, J.C.; Suh, C.; Lee, J.S. Phase Ib study of oral PPARγ agonist efatutazone (CS-7017) in combination with erlotinib in Korean patients with metastatic or unresectable locally advanced non-small cell lung cancer (NSCLC) who progressed after first-line therapy. J. Clin. Oncol. 2012, 30, e18052. [Google Scholar]
- Wigle, D.A.; Mandrekar, S.J.; Allen-Ziegler, K.; Gesthalter, Y.; Holland, P.; Aubry, M.-C.; Limburg, P.J.; Avi, S.; Szabo, E. Pioglitazone as a candidate chemoprevention agent for lung cancer: A pilot window trial in early stage NSCLC. J. Clin. Oncol. 2014, 32, 1581. [Google Scholar] [CrossRef]
- Keith, R.L.; Blatchford, P.J.; Merrick, D.T.; Bunn, P.A.; Bagwell, B.; Dwyer-Nield, L.D.; Jackson, M.K.; Geraci, M.; Miller, Y.E. A randomized phase II trial of pioglitazone for lung cancer chemoprevention in high risk current and former smokers. Cancer Prev. Res. 2019, 12, 721–730. [Google Scholar] [CrossRef]
- Marshall, J.; Shuster, D.E.; Goldberg, T.R.; Copigneaux, C.; Chen, S.; Zahir, H.; Dutta, D.; Saleh, M.N.; Pishvaian, M.J.; Varela, M.S.; et al. A randomized, open-label phase II study of efatutazone in combination with FOLFIRI as second-line therapy for metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2014, 32, 535. [Google Scholar] [CrossRef]
- Smith, M.R.; Manola, J.; Kaufman, D.S.; George, D.; Oh, W.K.; Mueller, E.; Slovin, S.; Spiegelman, B.; Small, E.; Kantoff, P.W. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer 2004, 101, 1569–1574. [Google Scholar] [CrossRef]
- Mueller, E.; Smith, M.; Sarraf, P.; Kroll, T.; Aiyer, A.; Kaufman, D.S.; Oh, W.; Demetri, G.; Figg, W.D.; Zhou, X.-P.; et al. Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 10990–10995. [Google Scholar] [CrossRef] [PubMed]
- Vogelhuber, M.; Feyerabend, S.; Stenzl, A.; Suedhoff, T.; Schulze, M.; Huebner, J.; Oberneder, R.; Wieland, W.; Mueller, S.; Eichhorn, F.; et al. Biomodulatory treatment of patients with castration-resistant prostate cancer: A phase II study of imatinib with pioglitazone, etoricoxib, dexamethasone and low-dose treosulfan. Cancer Microenviron. 2015, 8, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.A.; Khanim, F.L.; Hayden, R.E.; Craddock, C.F.; Holyoake, T.L.; Jackson, N.; Lumley, M.; Bunce, C.M.; Drayson, M.T. Combined bezafibrate and medroxyprogesterone acetate have efficacy without haematological toxicity in elderly and relapsed acute myeloid leukaemia (AML). Br. J. Haematol. 2010, 149, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Pratt, G.; Jacob, A.; Clark, F.; Blundred, R.; Fox, S.; Bishop, R.; Wheatley, K.; Khanim, F.; Bunce, C.; et al. Single arm phase II trial assessing the safety, compliance with and activity of Bezafibrate and medroxyProgesterone acetate (BaP) therapy against myeloid and lymphoid cancers. Contemp. Clin. Trials Commun. 2019, 14, 100361. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Sepmeyer, J.A.; Greer, J.P.; Koyama, T.; Zic, J.A. Open-label pilot study of combination therapy with rosiglitazone and bexarotene in the treatment of cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 2007, 56, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, E.; Merrick, B.; Khanim, F.L.; Banda, K.; Dunn, J.A.; Iqbal, G.; Bunce, C.M.; Drayson, M.T. Bezafibrate and medroxyprogesterone acetate in resistant and relapsed endemic Burkitt lymphoma in Malawi; an open-label, single-arm, phase 2 study (ISRCTN34303497). Br. J. Haematol. 2014, 164, 888–890. [Google Scholar] [CrossRef]
- Hart, C.; Vogelhuber, M.; Hafner, C.; Landthaler, M.; Berneburg, M.; Haferkamp, S.; Herr, W.; Reichle, A. Biomodulatory metronomic therapy in stage IV melanoma is well-tolerated and may induce prolonged progression-free survival, a phase I trial. J. Eur. Acad. Dermatol. Venereol. 2016, 30, e119–e121. [Google Scholar] [CrossRef] [PubMed]
- Reichle, A.; Bross, K.; Vogt, T.; Bataille, F.; Wild, P.; Berand, A.; Krause, S.W.; Andreesen, R. Pioglitazone and rofecoxib combined with angiostatically scheduled trofosfamide in the treatment of far-advanced melanoma and soft tissue sarcoma. Cancer 2004, 101, 2247–2256. [Google Scholar] [CrossRef]
- Tontonoz, P.; Singer, S.; Forman, B.M.; Sarraf, P.; Fletcher, J.A.; Fletcher, C.D.M.; Brun, R.P.; Mueller, E.; Altiok, S.; Oppenheim, H.; et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 237–241. [Google Scholar] [CrossRef]
- Demetri, G.D.; Fletcher, C.D.M.; Mueller, E.; Sarraf, P.; Naujoks, R.; Campbell, N.; Spiegelman, B.M.; Singer, S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-γ ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. USA 1999, 96, 3951–3956. [Google Scholar] [CrossRef] [PubMed]
- Debrock, G.; Vanhentenrijk, V.; Sciot, R.; Debiec-Rychter, M.; Oyen, R.; Van Oosterom, A. A phase II trial with rosiglitazone in liposarcoma patients. Br. J. Cancer 2003, 89, 1409–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, H.J.; Demetri, G.D.; Mueller, E.; Sarraf, P.; Spiegelman, B.M.; Winer, E.P. Use of the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Res. Treat. 2003, 79, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.D.; Williams, N.; Wen, P.; Young, D.C.; Lester, J.; Johnson, M.V.; Farrar, W.B.; Walker, M.J.; Povoski, S.P.; Suster, S.; et al. Pilot study of rosiglitazone therapy in women with breast cancer: Effects of short-term therapy on tumor tissue and serum markers. Clin. Cancer Res. 2007, 13, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Bogazzi, F.; Rossi, G.; Lombardi, M.; Raggi, F.; Urbani, C.; Sardella, C.; Cosci, C.; Martino, E. Effect of rosiglitazone on serum IGF-I concentrations in uncontrolled acromegalic patients under conventional medical therapy: Results from a pilot phase 2 study. J. Endocrinol. Investig. 2011, 34, e43–e51. [Google Scholar] [CrossRef] [PubMed]
- Robison, N.J.; Campigotto, F.; Chi, S.N.; Manley, P.E.; Turner, C.D.; Zimmerman, M.A.; Chordas, C.A.; Werger, A.M.; Allen, J.C.; Goldman, S.; et al. A phase II trial of a multi-agent oral antiangiogenic (metronomic) regimen in children with recurrent or progressive cancer. Pediatr. Blood Cancer 2014, 61, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Cotarla, I.; Wagner, A.J.; Deeken, J.F.; He, A.R.; Hwang, J.J.; Demetri, G.D.; Halim, A.; Copigneaux, C.; Marshall, J. Final reporting of a phase I clinical trial of the oral PPAR-gamma agonist, CS-7017, in patients with advanced malignancies. J. Clin. Oncol. 2010, 28, 2526. [Google Scholar] [CrossRef]
- Du, Q.; Yang, S.; Wang, Y.-J.; Wu, B.; Zhao, Y.-Y.; Fan, B. Effects of thiazolidinediones on polycystic ovary syndrome: A meta-analysis of randomized placebo-controlled trials. Adv. Ther. 2012, 29, 763–774. [Google Scholar] [CrossRef]
- Gupta, A.; Jakubowicz, D.; Nestler, J.E. Pioglitazone therapy increases insulin-stimulated release of d-Chiro-Inositol-containing Inositolphosphoglycan mediator in women with polycystic ovary syndrome. Metab. Syndr. Relat. Disord. 2016, 14, 391–396. [Google Scholar] [CrossRef]
- Tfayli, H.; Ulnach, J.W.; Lee, S.; Sutton-Tyrrell, K.; Arslanian, S. Drospirenone/Ethinyl estradiol versus rosiglitazone treatment in overweight adolescents with polycystic ovary syndrome: Comparison of metabolic, hormonal, and cardiovascular risk factors. J. Clin. Endocrinol. Metab. 2011, 96, 1311–1319. [Google Scholar] [CrossRef]
- Glintborg, D.; Højlund, K.; Andersen, N.R.; Hansen, B.F.; Beck-Nielsen, H.; Wojtaszewski, J.F.P. Impaired insulin activation and dephosphorylation of glycogen synthase in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. J. Clin. Endocrinol. Metab. 2008, 93, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Glintborg, D.; Hermann, A.P.; Rasmussen, L.M.; Andersen, M. Plasma osteoprotegerin is associated with testosterone levels but unaffected by pioglitazone treatment in patients with polycystic ovary syndrome. J. Endocrinol. Investig. 2013, 36, 460–465. [Google Scholar]
- Ortega-González, C.; Luna, S.; Hernández, L.; Crespo, G.; Aguayo, P.; Arteaga-Troncoso, G.; Parra, A. Responses of serum androgen and insulin resistance to metformin and pioglitazone in obese, insulin-resistant women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.W.; Kilpatrick, E.S.; Keevil, B.G.; Coady, A.M.; Atkin, S.L. Effect of metformin, orlistat and pioglitazone treatment on mean insulin resistance and its biological variability in polycystic ovary syndrome. Clin. Endocrinol. (Oxf.) 2009, 70, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-J.; Yu, Y.-X.; Liu, C.-Q.; Zhang, W.; Zhang, H.-J.; Yan, B.; Wang, L.-Y.; Yang, S.-Y.; Zhang, S.-H. Metformin vs thiazolidinediones for treatment of clinical, hormonal and metabolic characteristics of polycystic ovary syndrome: A meta-analysis. Clin. Endocrinol. (Oxf.) 2011, 74, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, L.; Diaz, M.; Sebastiani, G.; Sánchez-Infantes, D.; Salvador, C.; Lopez-Bermejo, A.; de Zegher, F. Treatment of androgen excess in adolescent girls: Ethinylestradiol-cyproteroneacetate versus low-dose pioglitazone-flutamide-metformin. J. Clin. Endocrinol. Metab. 2011, 96, 3361–3366. [Google Scholar] [CrossRef] [PubMed]
- El-khayat, W.; Abdel Moety, G.; Al Mohammady, M.; Hamed, D. A randomized controlled trial of clomifene citrate, metformin, and pioglitazone versus letrozole, metformin, and pioglitazone for clomifene-citrate-resistant polycystic ovary syndrome. Int. J. Gynecol. Obstet. 2016, 132, 206–209. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, T.; Havekes, B.; Bilet, L.; Hoeks, J.; Sparks, L.; Bosma, M.; Paglialunga, S.; Jorgensen, J.; Janssen Mirian, C.H.; Schaart, G.; et al. Effects of bezafibrate treatment in a patient and a carrier with mutations in the PNPLA2 gene, causing neutral lipid storage disease with myopathy. Circ. Res. 2013, 112, e51–e54. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Wahli, W. Nuclear hormone receptors and epidermal differentiation. In Lipids and Skin Health; Pappas, A., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 91–106. [Google Scholar]
- Elijah, I.E.; Børsheim, E.; Maybauer, D.M.; Finnerty, C.C.; Herndon, D.N.; Maybauer, M.O. Role of the PPAR-α agonist fenofibrate in severe pediatric burn. Burns 2012, 38, 481–486. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Kraft, R.; Emdad, F.; Kulp, G.A.; Williams, F.N.; Herndon, D.N. Glucose control in severely thermally injured pediatric patients: What glucose range should be the target? Ann. Surg. 2010, 252, 521–528. [Google Scholar] [CrossRef]
- Cree, M.G.; Zwetsloot, J.J.; Herndon, D.N.; Qian, T.; Morio, B.; Fram, R.; Sanford, A.P.; Aarsland, A.; Wolfe, R.R. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann. Surg. 2007, 245, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Bruckert, E.; Duchêne, E.; Bonnefont-Rousselot, D.; Hansel, B.; Ansquer, J.-C.; Dubois, A.; Gaymard, B. Proof of concept study: Does fenofibrate have a role in sleep apnoea syndrome? Curr. Med. Res. Opin. 2010, 26, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Kim, S.H.; Ariel, D.; Abbasi, F.; Lamendola, C.; Cardell, J.; Xu, S.; Patel, S.; Tomasso, V.; Mojaddidi, H.; et al. Does enhanced insulin sensitivity improve sleep measures in patients with obstructive sleep apnea: A randomized, placebo-controlled pilot study. Sleep Med. 2016, 22, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Richir, M.C.; Ellger, B.; Teerlink, T.; Siroen, M.P.C.; Visser, M.; Spreeuwenberg, M.; Girbes, A.R.J.; van der Hoven, B.; van den Berghe, G.; Wilhelm, A.J.; et al. The effect of rosiglitazone on asymmetric dimethylarginine (ADMA) in critically ill patients. Pharmacol. Res. 2009, 60, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Doggrell, S. Do peroxisome proliferation receptor-γ antagonists have clinical potential as combined antiobesity and antidiabetic drugs? Expert Opin. Investig. Drugs 2003, 12, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.S.; Lee, J.X.T.; Wahli, W.; Tan, N.S. Exploiting vulnerabilities of cancer by targeting nuclear receptors of stromal cells in tumor microenvironment. Mol. Cancer 2019, 18, 51. [Google Scholar] [CrossRef]
- Tan, E.H.P.; Sng, M.K.; How, I.S.B.; Chan, J.S.K.; Chen, J.; Tan, C.K.; Wahli, W.; Tan, N.S. ROS release by PPARβ/δ-null fibroblasts reduces tumor load through epithelial antioxidant response. Oncogene 2018, 37, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Wahli, W.; Tan, N.S. Nuclear receptor peroxisome proliferator activated receptor (PPAR) β/δ in skin wound healing and cancer. Eur. J. Dermatol. 2015, 25, 4–11. [Google Scholar]
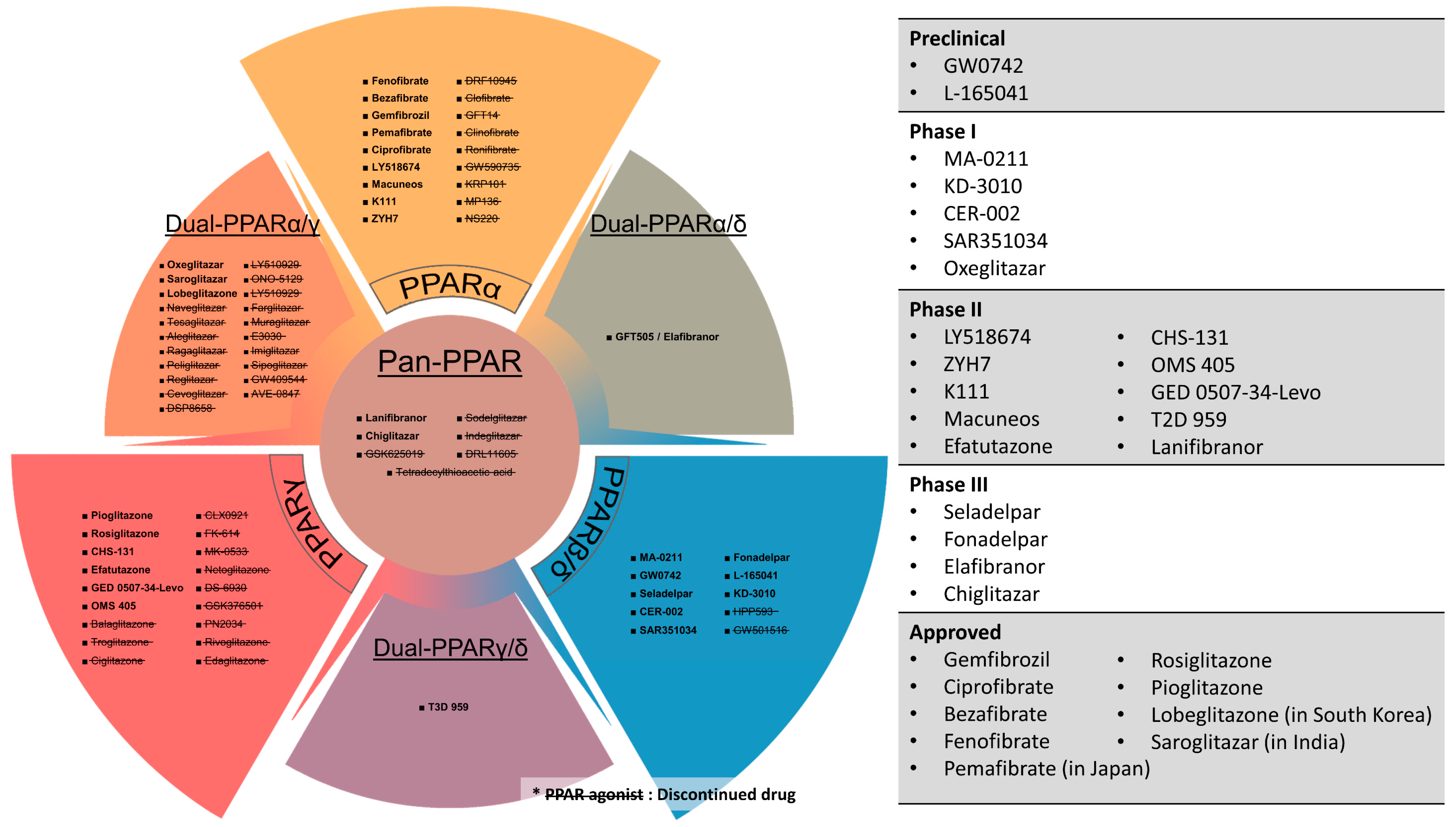
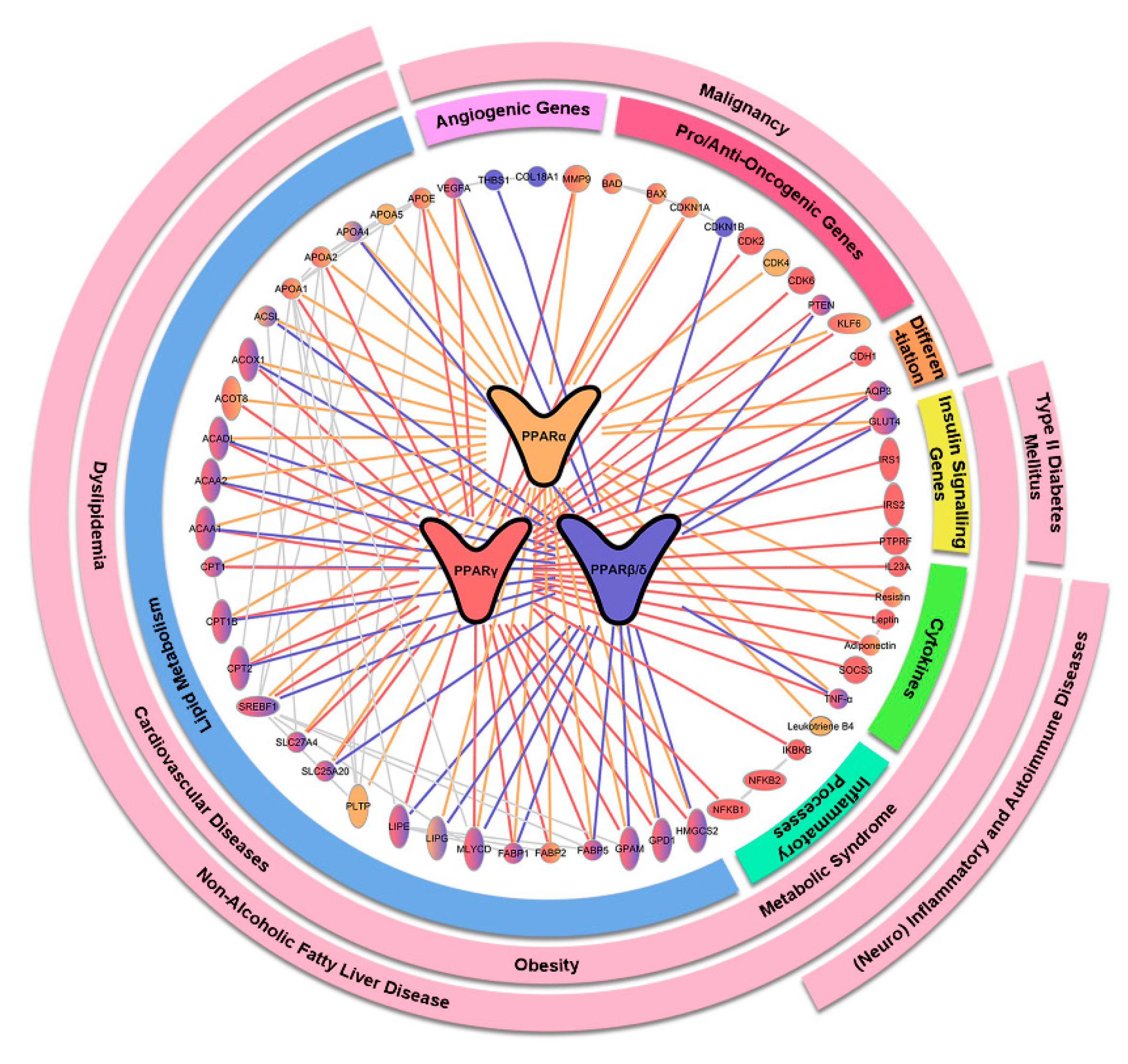

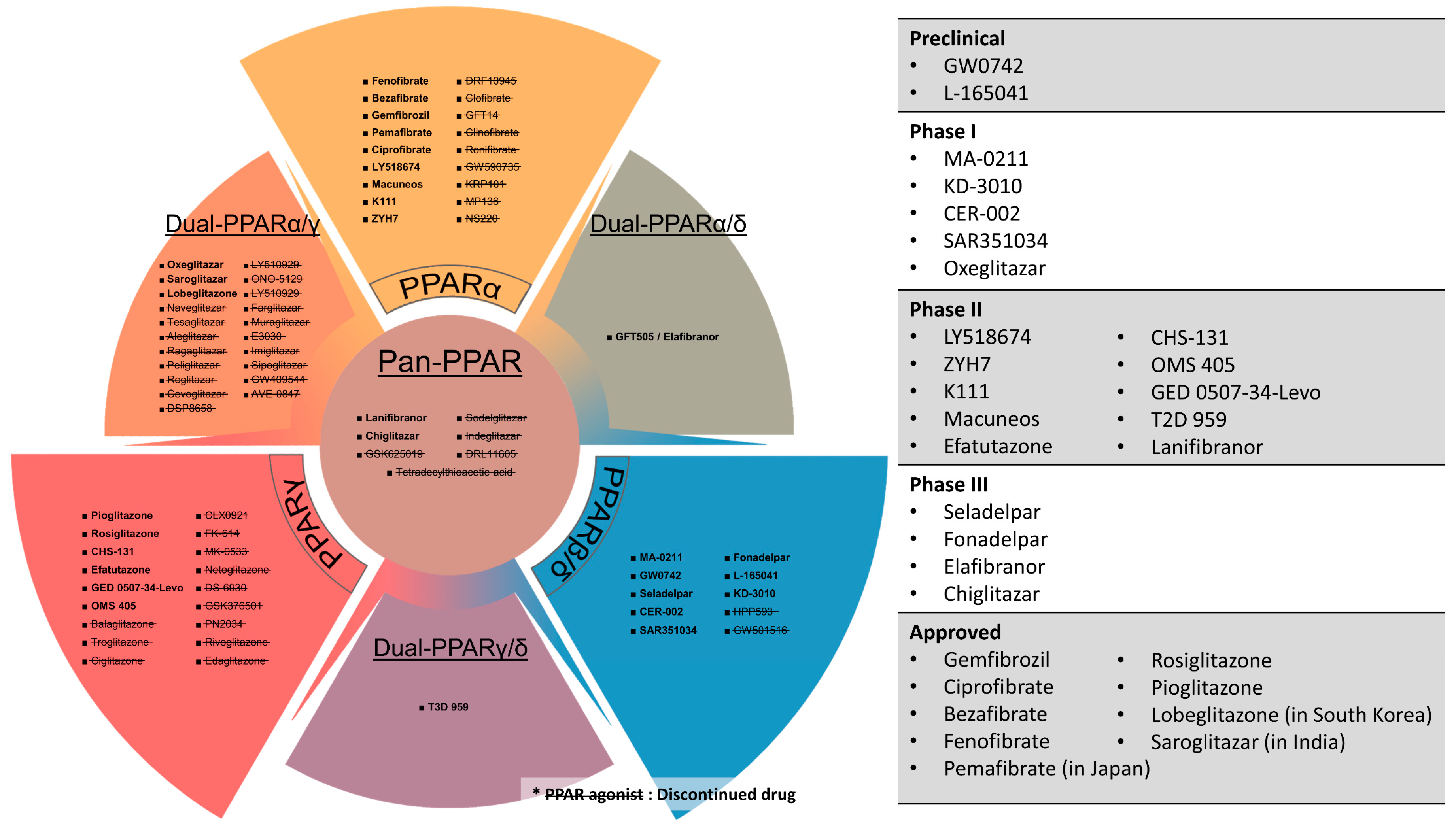{kind=link}
{kind=link}
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| T2DM | Dual PPARα/γ | Muraglitazar | II & III (1477) |
| [52] |
| II & III (3725) |
| [65] | |||
| Muraglitazar + sulphonylurea | III (583) |
| [51] | ||
| Muraglitazar + metformin | III (1805) |
| [53] | ||
| Tesaglitazar | II (500) |
| [55] | ||
| III-terminated (1707) |
| [54] | |||
| Tesaglitazar + metformin | III-terminated (555) |
| [56] | ||
| Tesaglitazar + insulin | III-terminated (392) |
| [57] | ||
| Tesaglitazar + sulphonylurea | III-terminated (568) |
| [58] | ||
| Aleglitazar | II (40) |
| [59] | ||
| II (332) |
| [60] | |||
| III-terminated (591) |
| [61] | |||
| Lobeglitazone | III (173) |
| [62] | ||
| III (253) |
| [63] | |||
| MK-0767 | NA (8) |
| [64] | ||
| III-terminated (382) |
| NCT00543361 | |||
| III-terminated (247) |
| NCT00543010 | |||
| III-terminated (129) | NCT00543491 | ||||
| III-terminated (99) | NCT00543517 | ||||
| III-terminated (114) | NCT00543738 | ||||
| III-terminated (610) | NCT00543751 | ||||
| III-terminated (111) | NCT00543816 | ||||
| III-terminated (100) | NCT00547274 | ||||
| Naveglitazar | II-completed |
| NCT00065312 | ||
| ON-5129 | II-completed (81) |
| NCT00335712 | ||
| II-completed (105) |
| NCT00212641 | |||
| DSP-8658 | I-completed (40) |
| NCT01042106 | ||
| Lobeglitazone | IV-ongoing (78) |
| NCT02338921 | ||
| IV-ongoing (174) |
| NCT03770052 | |||
| Dual-PPAR α/δ | Elafibranor | II (22) |
| [72] | |
| II (47) |
| [73] | |||
| Pan-PPAR | Indelglitazar | II-completed (108) |
| EUCTR 2005-004227-19 | |
| II-completed (500) |
| NCT00425919 | |||
| Tetradecylthioacetic acid | II-completed (16) |
| NCT00605787 | ||
| Chiglitazar | III (535) |
| [74] | ||
| III (739) |
| [75] | |||
| Lanifibranor | II-ongoing (84) |
| NCT03459079 | ||
| PPARβ/δ | GW677954 | II-terminated (1) |
| NCT00437164 | |
| II-completed (448) |
| NCT00196989 | |||
| Diabetic retinopathy | PPARα | Fenofibrate | III (3472) |
| [77] |
| IV-ongoing (1060) |
| NCT03439345 | |||
| Pemafibrate | III-terminated (15) |
| NCT03345901 |
| Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|
| Dual PPARα/γ | Aleglitazar | III-terminated (7226) |
| [78] |
| III-terminated (1999) |
| [79] | ||
| II (332) |
| [60] | ||
| PPARα | Bezafibrate | NA (1568) |
| [83] |
| NA (3090) |
| [84] | ||
| NA (164) |
| [85] | ||
| NA (50) |
| [86] | ||
| Fenofibrate | NA (9795) |
| [87] | |
| Gemfibrozil | NA (4081) |
| [88] | |
| Pemafibrate | III (10000) |
| NCT03071692 | |
| PPARγ | Pioglitazone | IV (24) |
| [92] |
| III (5238) |
| [93] | ||
| III (5238) |
| [94] | ||
| III (5238) |
| [95] | ||
| III (3876) |
| [96] | ||
| NA (72) |
| [97] | ||
| NA (96) |
| [98] | ||
| NA (97) |
| [99] | ||
| NA (28) |
| [100] | ||
| III (462) |
| [101] | ||
| III (543) |
| [102] | ||
| NA (56) |
| [103] | ||
| NA (120) |
| [104] | ||
| NA (15) |
| [105] | ||
| III (300) |
| [106] | ||
| Pioglitazone + metformin | IV (3028) |
| [107] | |
| Rosiglitazone | III (193) |
| [109] | |
| III (1425) |
| [110] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Dyslipidemia | PPARα | Pemafibrate | III (225) |
| [118] |
| III (526) |
| [119] | |||
| III (166) |
| [120] | |||
| III (189) |
| [121] | |||
| PPARα, PPARγ | Rosiglitazone and/or fenofibrate | NA (41) |
| [122] | |
| PPARβ/δ | GW501516 | II (268) |
| [125] | |
| IV (13) |
| [126] | |||
| Seladelpar | II (181) |
| [128] | ||
| Seladelpar and/or statins | II (166) |
| [129] | ||
| Dual PPARα/γ | Muraglitazar | II & III-Completed (330) |
| NCT00245388 | |
| Dual PPAR α/δ | Elafibranor | II (94) |
| [73] | |
| Diabetic dyslipidemia | Dual PPARα/γ | Saroglitazar | III (109) |
| [130] |
| III (302) |
| [131] | |||
| Familial dysbetalipoproteinemia | PPARα | Fenofibrate or gemfibrozil | NA (146) |
| [134] |
| Bezafibrate | NA (14) |
| [135] | ||
| Bezafibrate + Statins | NA (15) |
| [136] | ||
| X-linked adrenoleukodystrophy | PPARα | Bezafibrate | NA (10) |
| [137] |
| CPT II and VLCAD deficiencies. | PPARα | Bezafibrate | NA (10) |
| [138] |
| Familial hypercholesterolemia | PPARβ/δ | Seladelpar | II (13) |
| [139] |
| Familial combined hyperlipidemia | PPARγ | Pioglitazone + lipid-lowering drugs | NA (26) |
| [140] |
| NA (22) |
| [141] | |||
| HIV-associated dyslipidemia and lipodystrophy syndrome | PPARα | Fenofibrate + fish oil | II (100) |
| [143] |
| Fenofibrate | NA (55) |
| [144] | ||
| NA (36) |
| [145] | |||
| II (99) |
| [146] | |||
| NA (191) |
| [147] | |||
| Fenofibrate + Pravastatin | III (174) |
| [148] | ||
| Fibrate | NA (245) |
| [149] | ||
| NA (656) |
| [150] | |||
| Bezafibrate | NA (130) |
| [151] | ||
| PPARα, PPARγ | Fenofibrate, pioglitazone | NA (14) |
| [152] | |
| PPARγ | Pioglitazone | III (130) |
| [153] | |
| Rosigltazone | NA (96) |
| [155] | ||
| NA (39) |
| [156] | |||
| II (71) |
| [157] | |||
| Pan-PPAR | Tetradecylthioacetic acid | NA (10) |
| [159] | |
| Dyslipidemia due to spinal cord injury | PPARα | Fenofibrate | II and III -Completed (23) |
| NCT02455336 |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Prediabetes and MetS | Dual PPAR α/δ | Elafibranor | II (47) |
| [73] |
| II (22) |
| [72] | |||
| Obesity | PPARβ/δ | GW501516 | IV (13) |
| [126] |
| PPARγ | Rosiglitazone | IV (28) |
| [162] | |
| PPARα | Fenofibrate + metformin | NA (87) |
| [163] | |
| Fenofibrate | NA (89) |
| [164] | ||
| Hypertension | PPARγ | Pioglitazone | NA (27) |
| [167] |
| NA (149) |
| [169] | |||
| III (42) |
| [170] | |||
| NA (30) |
| [171] | |||
| Rosiglitazone | NA (24) |
| [172] | ||
| NA (16) |
| [173] | |||
| PPARα | Fenofibrate | NA (31) |
| [165] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| NAFLD | PPARα | Gemfibrozil | NA (46) |
| [178] |
| Fenofibrate | NA (16) |
| [179] | ||
| NA (27) |
| [180] | |||
| Pemafibrate | II-ongoing (100) |
| NCT03350165 | ||
| PPARγ | Pioglitazone | NA (18) |
| [185] | |
| IV (55) |
| [186] | |||
| NA (74) |
| [187] | |||
| III (247) |
| [188] | |||
| IV (101) |
| [189] | |||
| NA (10) |
| [190] | |||
| Rosiglitazone | NA (30) |
| [191] | ||
| II (63) |
| [192] | |||
| II (44) |
| [193] | |||
| NA (74) |
| [194] | |||
| NA (47) |
| [195] | |||
| dual PPAR α/δ | Elafibranor | II (276) |
| [196] | |
| III-ongoing (2000) |
| NCT02704403 | |||
| Dual-PPAR α/γ | Saroglitazar | III-unknown status (100) |
| NCT02265276 | |
| II-ongoing (106) |
| NCT03061721 | |||
| II-ongoing (15) |
| NCT03863574 | |||
| II-ongoing (60) |
| NCT03617263 | |||
| II-ongoing (15) |
| NCT03639623 | |||
| Lobeglitazone | IV (38) |
| [197] | ||
| Pan-PPAR | Lanifibranor | II-ongoing (225) |
| NCT03008070 | |
| II-ongoing (84) |
| NCT03459079 | |||
| PBC | PPARα | Bezafibrate | III (100) |
| [202] |
| NA (16) |
| [203] | |||
| NA (66) |
| [204] | |||
| III-ongoing (34) |
| NCT02937012 | |||
| III-ongoing (84) |
| [217] | |||
| Fenofibrate | II (20) |
| [206] | ||
| NA (22) |
| [208] | |||
| NA (120) |
| [209] | |||
| II (10) |
| [210] | |||
| PPARβ/δ | Seladelpar | III-ongoing (240) |
| NCT03602560 | |
| II & III-ongoing (356) |
| NCT03301506 | |||
| II-ongoing (116) |
| NCT02955602 | |||
| II (41) |
| [211] | |||
| Dual PPAR α/δ | Elafibranor | II (45) |
| NCT03124108 | |
| Dual PPAR α/γ | Saroglitazar | II-ongoing (36) |
| NCT03112681 | |
| Hepatitis C | PPARγ | Farglitazar | II (265) |
| [212] |
| Pioglitazone | II (5) |
| [213] | ||
| II (40) |
| [214] | |||
| IV (80) |
| [215] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| CKD | PPARγ | Pioglitazone | IV (16) |
| [220] |
| NA-completed (95) |
| NCT01301027 | |||
| NA (75) |
| [219] | |||
| NA-terminated (36) |
| NCT00586261 | |||
| IV-ongoing (28) |
| NCT03471117 | |||
| Rosiglitazone | NA (70) |
| [221] | ||
| Renal transplant complication | PPARγ | Pioglitazone | NA (48) |
| [223] |
| NA (83) |
| [224] | |||
| Kidney stone | PPARγ | Pioglitazone | NA (36) |
| [225] |
| resistant focal segmental glomerulosclerosis | PPARγ | Pioglitazone | I-completed (21) |
| NCT00193648 |
| Polycystic kidney disease | PPARγ | Pioglitazone | II-ongoing (18) |
| NCT02697617 |
| Disease | Target | Drug Name | Clinical Phase (Sample size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Alzheimer′s Disease | PPARγ | Rosiglitazone | II (30) |
| [226] |
| II (687) |
| [228] | |||
| II-completed (337) |
| EUCTR2004-000985-12 | |||
| II (80) |
| [227] | |||
| II-completed (40) |
| NCT00381238 | |||
| III (639) |
| [229] | |||
| III (1496; 1485; 1461) |
| [230] | |||
| Pioglitazone | III-terminated (3494) |
| NCT01931566 | ||
| II (78) |
| [231] | |||
| II (25) |
| [232] | |||
| PPARα | Gemfibrozil | I-ongoing (72) |
| NCT02045056 | |
| Amyotrophic Lateral Sclerosis | PPARγ | Pioglitazone | II (219) |
| [237] |
| II (27) |
| [238] | |||
| Multiple Sclerosis | PPARγ | Pioglitazone | I (24) |
| [240] |
| CHS-131 | II (227) |
| [241] | ||
| Drug-resistant Nocturnal Frontal Lobe Epilepsy | PPARα | Fenofibrate | II (12) |
| [242] |
| Postherpetic Neuralgia | PPARγ | ATx08-001/FK614 | II (61) |
| NCT01318226 |
| Friedreich′s Ataxia | PPARγ | Pioglitazone | III (40) |
| [243] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Alcoholism | PPARα | Fenofibrate | II-completed (50) |
| NCT02158273 |
| Gemfibrozil | II-terminated (3) |
| NCT03539432 | ||
| PPARγ | Pioglitazone | II-terminated (16) |
| NCT01631630 | |
| Nicotine dependence/Smoking | PPARα | Gemfibrozil | II (27) |
| [245] |
| II-completed (16) |
| NCT02638597 | |||
| Fenofibrate | II (38) |
| [246] | ||
| PPARγ | Pioglitazone | I/II (42) |
| [247] | |
| Cocaine dependence | PPARγ | Pioglitazone | I/II (30) |
| [248] |
| Opioid dependence | PPARγ | Pioglitazone | II (32) |
| [249] |
| I (40) |
| [250] | |||
| Bipolar disorder | PPARγ | Pioglitazone | IV (34) |
| [251] |
| IV (38) |
| [256] | |||
| III-ongoing (60) |
| EUCTR 2014-003803-31 | |||
| Rosiglitazone | NA-completed (12) |
| [252] | ||
| PPARα | Bezafibrate | II-ongoing (30) |
| NCT02481245 | |
| Major depressive disorder | PPARγ | Pioglitazone | II-completed (23) |
| NCT00671515 |
| II/III (50) |
| [253] | |||
| IV (37) |
| [254] | |||
| Schizophrenia | PPARγ | Pioglitazone | IV (56) |
| [255] |
| Autism spectrum disorder | PPARγ | Pioglitazone | NA (25) |
| [257] |
| II (44) |
| [258] | |||
| II (25) |
| [259] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Rheumatoid arthritis | PPARγ | Pioglitazone | NA (34) |
| [260] |
| III (143) |
| [261] | |||
| NA (49) |
| [262] | |||
| Systemic Lupus Erythematosus | PPARγ | Pioglitazone | IV (30) |
| [264] |
| I/II-ongoing (88) |
| NCT02338999 | |||
| Autoimmune pulmonary alveolar proteinosis | PPARγ | Pioglitazone | I-ongoing (3) |
| NCT03231033 |
| Diffuse cutaneous systemic sclerosis | Pan-PPAR | Lanifibranor | III-completed (145) |
| NCT02503644 |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Malaria | PPARγ | Rosiglitazone | I/II (140) |
| [265] |
| II (30) |
| [266] | |||
| Ulcerative colitis | PPARγ | Rosiglitazone | II (105) |
| [267] |
| NA (14) |
| [268] | |||
| Asthma | PPARγ | Rosiglitazone | I (34) |
| [270] |
| Pioglitazone | IV (68) |
| [271] | ||
| II (23) |
| [272] | |||
| NA (34) |
| [273] | |||
| Plaque psoriasis | PPARγ | Pioglitazone + acitretin | II (41) |
| [275] |
| Endometriosis | PPARγ | Rosiglitazone | II-terminated (3) |
| [276] |
| Pioglitazone | II and III-withdrawn (20) |
| NCT01184144 | ||
| Cystic fibrosis | PPARγ | Pioglitazone | NA (20) |
| [277] |
| I-completed (24) |
| NCT00719381 | |||
| NA (21) |
| NCT00322868 | |||
| Sepsis | PPARγ | Pioglitazone | I and II (12) |
| [278] |
| Lung inflammation | PPARγ | Pioglitazone | I (18) |
| [279] |
| Lung inflammation due to alcoholism | PPARγ | Pioglitazone | II-ongoing (36) |
| NCT03060772 |
| Chronic granulomatous | PPARγ | Pioglitazone | I and II-ongoing (100) |
| NCT03080480 |
| Gastric phlogosis due to H. pylori infection | PPARγ | Pioglitazone | III-ongoing (80) |
| EUCTR 2005-001218-42 |
| Induced endotoxemia | PPARα | Fenofibrate | NA (36) |
| [281] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| Cushing′s disease/ Pituitary tumors | PPARγ | Rosiglitazone Rosiglitazone | II-terminated (2) |
| NCT00612066 |
| II-terminated (1) |
| NCT00616642 | |||
| NA-ongoing (24) |
| NCT03309319 | |||
| Medullablastoma | PPARα | Fenofibrate | II-ongoing (40) |
| NCT01356290 |
| Oral leukoplakia | PPARγ | Pioglitazone | II (44) |
| [282] |
| II-terminated (52) |
| NCT00951379 | |||
| Rosiglitazone | II-completed (25) |
| NCT00369174 | ||
| Oral Cavity / Oropharngeal cancer | PPARγ | Pioglitazone | II-terminated (39) |
| NCT02917629 |
| Anaplastic thyroid carcinoma | PPARγ | Efatutazone/ CS-7017 | I (15) |
| [283] |
| II-ongoing (19) |
| NCT02152137 | |||
| PAX8-PPARγ anaplastic thyroid carcinoma | PPARγ | Pioglitazone | II (1) |
| [284] |
| Differentiated thyroid carcinoma | PPARγ | Rosiglitazone | II (20) |
| [285] |
| Non-small cell lung carcinoma | PPARγ | Efatutazone/ CS-7017 | I (16) |
| [286] |
| II-completed (111) |
| NCT00806286 | |||
| I (14) |
| [287] | |||
| II-completed (90) |
| NCT01101334 | |||
| Pioglitazone | II (6) |
| [288] | ||
| II (92) |
| [289] | |||
| II-ongoing (86) |
| NCT02852083 | |||
| Colorectal cancer | PPARγ | Efatutazone/ CS-7017 | II-completed (86) |
| NCT00986440 |
| II (100) |
| [290] | |||
| Prostate carcinoma | PPARγ | Rosiglitazone | III (105) |
| [291] |
| Troglitazone | II (41) |
| [292] | ||
| Castration-resistant prostate cancer | PPARγ | Pioglitazone | II (61) |
| [293] |
| PPARα & PPARγ | Fenofibrate, Pioglitazone, Rosiglitazone | II-terminated (49) |
| EUCTR 2006-001398-44 | |
| Acute myeloid leukemia | PPARα | Bezafibrate | II (20) |
| [294] |
| II (18) |
| [295] | |||
| PPARγ | Pioglitazone | II-ongoing (94) |
| NCT02942758 | |
| Chronic myeloid leukemia | PPARγ | Pioglitazone | I/II-ongoing (100) |
| NCT02767063 |
| II-ongoing (26) |
| NCT02889003 | |||
| II-ongoing (31) |
| NCT02852486 | |||
| II-terminated (9) |
| NCT02730195 | |||
| II-unknown status (20) |
| NCT02687425 | |||
| II (24) |
| [296] | |||
| Cutaneous T-cell lymphoma | PPARγ | Rosiglitazone | II (4) |
| [297] |
| Endemic Burkitt′s lymphoma | PPARα | Bezafibrate | II (95) |
| [298] |
| Multiple myeloma | PPARα | Fenofibrate | II-terminated (6) |
| NCT01965834 |
| PPARγ | Efatutazone | I-terminated (9) |
| NCT01504490 | |
| I/II-unknwon status (54) |
| NCT01010243 | |||
| Melanoma | PPARγ | Pioglitazone | I (6) |
| [299] |
| Melanoma/Soft Tissue Sarcoma | PPARγ | Pioglitazone | II (40) |
| [300] |
| Skin squamous cell carcinoma | PPARγ | Pioglitazone | II-ongoing (40) |
| NCT02347813 |
| Liposarcoma | PPARγ | Troglitazone | II (3) |
| [302] |
| II-completed (85) |
| NCT00003058 | |||
| Rosiglitazone | II (12) |
| [303] | ||
| Refractory breast cancer | PPARγ | Troglitazone | II (22) |
| [304] |
| Breast cancer | PPARγ | Pioglitazone | I/II (38) |
| [305] |
| Acromegaly (macroadenoma and microadenoma) | PPARγ | Rosiglitazone | II (5) |
| [306] |
| Progressive pediatric malignancies | PPARα | Fenofibrate | II (101) |
| [307] |
| Advanced or metastatic solid tumors | PPARγ | Pioglitazone | I-completed (28) |
| NCT02133625 |
| Efatutazone/ CS-7017 | I (32) |
| [308] |
| Disease | Target | Drug Name | Clinical Phase (Sample Size) | Main Findings/Primary Endpoint | Reference/Clinical Trial Identifier |
|---|---|---|---|---|---|
| PCOS | PPARγ | Pioglitazone | NA (52) |
| [314] |
| NA (30) |
| [315] | |||
| Pioglitazone + flutamide + metformin | NA (34) |
| [317] | ||
| Pioglitazone + metformin + letrozole | I (50) |
| [318] | ||
| Mitochondrial dysfunction and myopathy | PPARα | Bezafibrate | II-completed (6) |
| NCT02398201 |
| II-ongoing (24) |
| EUCTR 2012-002692 | |||
| NA (2) |
| [319] | |||
| Neutral Lipid Storage Disease With Myopathy | PPARα | Bezafibrate | IV-completed (6) |
| NCT01527318 |
| Sporadic inclusion body myositis | PPARγ | Pioglitazone | I -ongoing (15) |
| NCT03440034 |
| PPARβ/δ | HPP593 | I -terminated (24) |
| NCT01524406 | |
| Burn injury | PPARα | Fenofibrate | II (21) |
| [323] |
| II & III-ongoing (330) |
| NCT02452255 | |||
| Obstructive sleep apnea | PPARα | Fenofibrate | II (34) |
| [324] |
| PPARγ | Pioglitazone | NA (45) |
| [325] | |
| Sexual dysfunction | PPARα | Fenofibrate | IV-unknown (300) |
| NCT00923676 |
| Huntington′s disease | PPARα | Fenofibrate | II-ongoing (20) |
| NCT03515213 |
| Critically ill patients | PPARγ | Rosiglitazone | NA (12) |
| [326] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. https://doi.org/10.3390/ijms20205055
Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. International Journal of Molecular Sciences. 2019; 20(20):5055. https://doi.org/10.3390/ijms20205055
Chicago/Turabian StyleCheng, Hong Sheng, Wei Ren Tan, Zun Siong Low, Charlie Marvalim, Justin Yin Hao Lee, and Nguan Soon Tan. 2019. "Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence" International Journal of Molecular Sciences 20, no. 20: 5055. https://doi.org/10.3390/ijms20205055