RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia

{kind=link}
{kind=link}
Abstract
1. Introduction
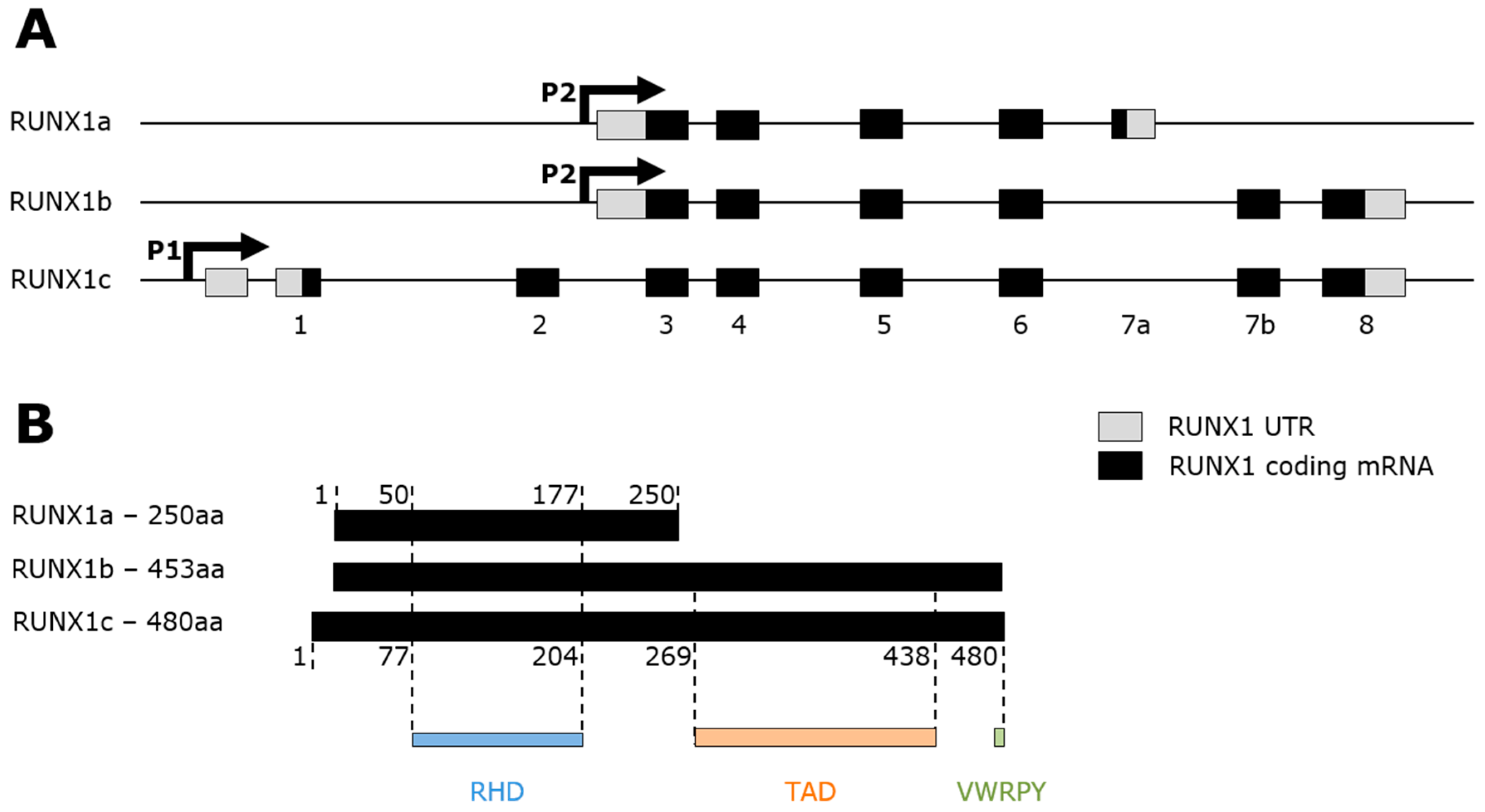
2. RUNX1 Transcription Factor
2.1. RUNX1 and Hematopoiesis
2.2. RUNX1 Structure
2.3. RUNX1 and the CBFβ Complex
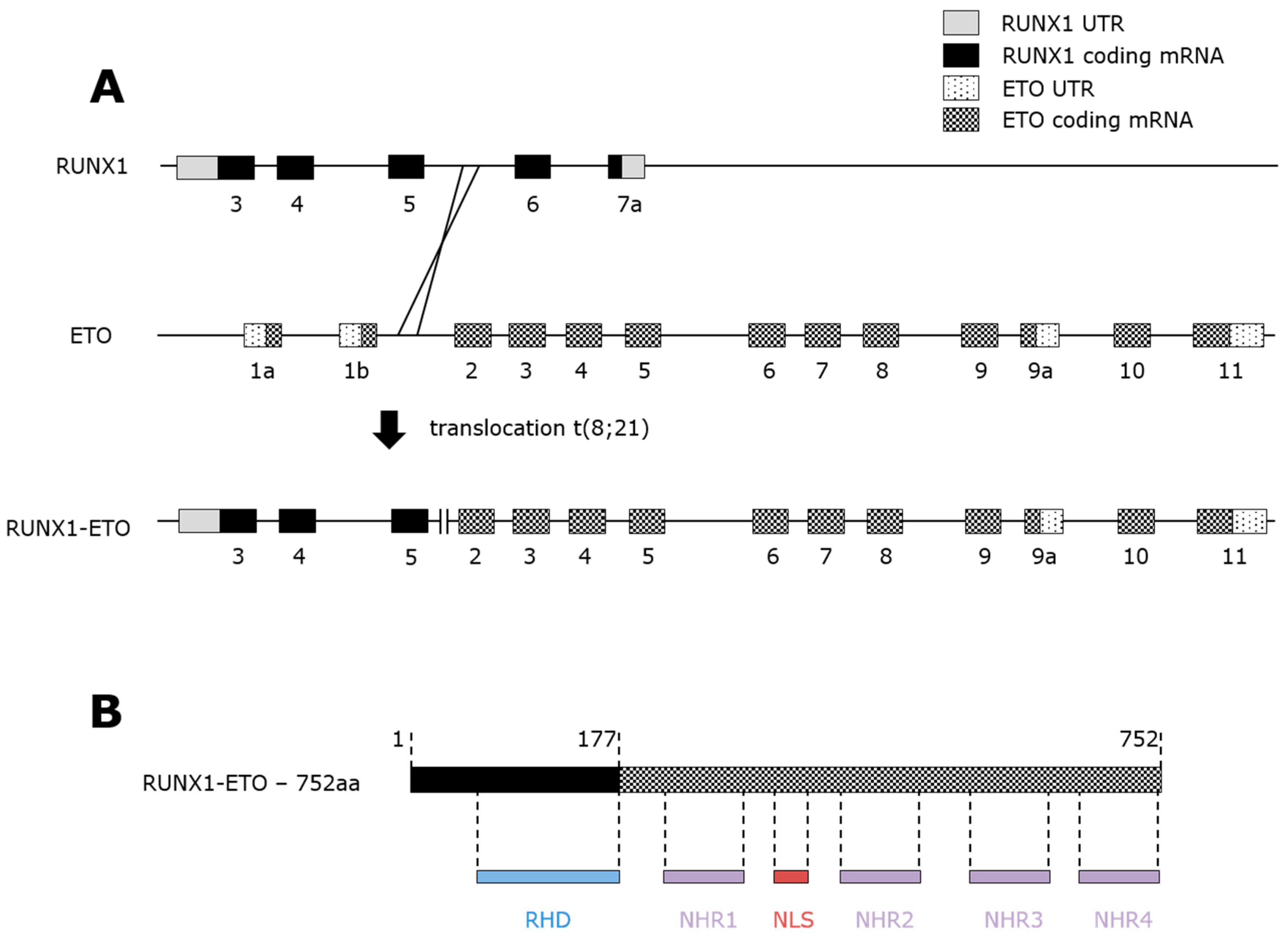
3. RUNX1-ETO Fusion Protein
3.1. t(8;21) AML
3.2. RUNX1-ETO Structure and Repressive Activity
3.3. RUNX1-ETO Cooperation with RUNX1
3.4. RUNX1-ETO Decreases DNA Repair Capabilities and Compromises Genomic Stability
4. RUNX1-ETO is Associated with Altered Methylation
5. RUNX1-ETO Recruits Histone Markers
6. Higher-Order Chromatin Structure Differentially Regulates the RUNX1/RUNX1-ETO Balance
7. Summary and Final Remarks
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann. Genet. 1973, 16, 109–112. [Google Scholar]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the united kingdom medical research council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Lin, S.; Mulloy, J.C.; Goyama, S. RUNX1-ETO leukemia. Adv. Exp. Med. Biol. 2017, 962, 151–173. [Google Scholar] [PubMed]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Biological implications of consistent chromosome rearrangements in leukemia and lymphoma. Cancer Res. 1984, 44, 3159–3168. [Google Scholar] [PubMed]
- Gergen, J.P.; Wieschaus, E.F. The localized requirements for a gene affecting segmentation in drosophila: Analysis of larvae mosaic for runt. Dev. Biol. 1985, 109, 321–335. [Google Scholar] [CrossRef]
- Van Wijnen, A.J.; Stein, G.S.; Gergen, J.P.; Groner, Y.; Hiebert, S.W.; Ito, Y.; Liu, P.; Neil, J.C.; Ohki, M.; Speck, N. Nomenclature for runt-related (RUNX) proteins. Oncogene 2004, 23, 4209–4210. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K.; et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403. [Google Scholar] [CrossRef]
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. T(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434. [Google Scholar] [CrossRef]
- Lacaud, G.; Kouskoff, V.; Trumble, A.; Schwantz, S.; Keller, G. Haploinsufficiency of RUNX1 results in the acceleration of mesodermal development and hemangioblast specification upon in vitro differentiation of es cells. Blood 2004, 103, 886–889. [Google Scholar] [CrossRef] [PubMed]
- North, T.; Gu, T.L.; Stacy, T.; Wang, Q.; Howard, L.; Binder, M.; Marin-Padilla, M.; Speck, N.A. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development 1999, 126, 2563–2575. [Google Scholar]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999, 126, 5073–5084. [Google Scholar]
- Ferkowicz, M.J.; Yoder, M.C. Blood island formation: Longstanding observations and modern interpretations. Exp. Hematol. 2005, 33, 1041–1047. [Google Scholar] [CrossRef]
- Kieusseian, A.; Brunet de la Grange, P.; Burlen-Defranoux, O.; Godin, I.; Cumano, A. Immature hematopoietic stem cells undergo maturation in the fetal liver. Development 2012, 139, 3521–3530. [Google Scholar] [CrossRef]
- Rybtsov, S.; Ivanovs, A.; Zhao, S.; Medvinsky, A. Concealed expansion of immature precursors underpins acute burst of adult hsc activity in foetal liver. Development 2016, 143, 1284–1289. [Google Scholar] [CrossRef]
- North, T.E.; Stacy, T.; Matheny, C.J.; Speck, N.A.; de Bruijn, M.F. RUNX1 is expressed in adult mouse hematopoietic stem cells and differentiating myeloid and lymphoid cells, but not in maturing erythroid cells. Stem Cells 2004, 22, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Lorsbach, R.B.; Moore, J.; Ang, S.O.; Sun, W.; Lenny, N.; Downing, J.R. Role of RUNX1 in adult hematopoiesis: Analysis of RUNX1-IRES-GFP knock-in mice reveals differential lineage expression. Blood 2004, 103, 2522–2529. [Google Scholar] [CrossRef]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef]
- Wang, Q.; Stacy, T.; Miller, J.D.; Lewis, A.F.; Gu, T.L.; Huang, X.; Bushweller, J.H.; Bories, J.C.; Alt, F.W.; Ryan, G.; et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell 1996, 87, 697–708. [Google Scholar] [CrossRef]
- Takakura, N.; Watanabe, T.; Suenobu, S.; Yamada, Y.; Noda, T.; Ito, Y.; Satake, M.; Suda, T. A role for hematopoietic stem cells in promoting angiogenesis. Cell 2000, 102, 199–209. [Google Scholar] [CrossRef]
- Wang, Q.; Stacy, T.; Binder, M.; Marin-Padilla, M.; Sharpe, A.H.; Speck, N.A. Disruption of the cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.M.; Medvinsky, A.; Strouboulis, J.; Grosveld, F.; Dzierzak, E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1994, 1, 291–301. [Google Scholar] [CrossRef]
- Cai, Z.; de Bruijn, M.; Ma, X.; Dortland, B.; Luteijn, T.; Downing, R.J.; Dzierzak, E. Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity 2000, 13, 423–431. [Google Scholar] [CrossRef]
- Goetz, T.L.; Gu, T.L.; Speck, N.A.; Graves, B.J. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor alpha2. Mol. Cell. Biol. 2000, 20, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Imperato, M.R.; Cauchy, P.; Obier, N.; Bonifer, C. The RUNX1-PU.1 axis in the control of hematopoiesis. Int. J. Hematol. 2015, 101, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Elagib, K.E.; Racke, F.K.; Mogass, M.; Khetawat, R.; Delehanty, L.L.; Goldfarb, A.N. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood 2003, 101, 4333–4341. [Google Scholar] [CrossRef]
- Takahashi, A.; Satake, M.; Yamaguchi-Iwai, Y.; Bae, S.C.; Lu, J.; Maruyama, M.; Zhang, Y.W.; Oka, H.; Arai, N.; Arai, K.; et al. Positive and negative regulation of granulocyte-macrophage colony-stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood 1995, 86, 607–616. [Google Scholar]
- Zhang, D.E.; Fujioka, K.; Hetherington, C.J.; Shapiro, L.H.; Chen, H.M.; Look, A.T.; Tenen, D.G. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1). Mol. Cell. Biol. 1994, 14, 8085–8095. [Google Scholar] [CrossRef]
- Zhang, Y.; Derynck, R. Transcriptional regulation of the transforming growth factor-beta -inducible mouse germ line Ig alpha constant region gene by functional cooperation of Smad, CREB, and AML family members. J. Biol. Chem. 2000, 275, 16979–16985. [Google Scholar] [CrossRef]
- Libermann, T.A.; Pan, Z.; Akbarali, Y.; Hetherington, C.J.; Boltax, J.; Yergeau, D.A.; Zhang, D.E. AML1 (CBFα2) cooperates with b cell-specific activating protein (BSAP/PAX5) in activation of the B cell-specific BLK gene promoter. J. Biol. Chem. 1999, 274, 24671–24676. [Google Scholar] [CrossRef]
- Sun, W.; Graves, B.J.; Speck, N.A. Transactivation of the moloney murine leukemia virus and T-cell receptor beta-chain enhancers by cbf and ets requires intact binding sites for both proteins. J. Virol. 1995, 69, 4941–4949. [Google Scholar] [PubMed]
- Puig-Kroger, A.; Sanchez-Elsner, T.; Ruiz, N.; Andreu, E.J.; Prosper, F.; Jensen, U.B.; Gil, J.; Erickson, P.; Drabkin, H.; Groner, Y.; et al. RUNX/AML and C/EBP factors regulate cd11a integrin expression in myeloid cells through overlapping regulatory elements. Blood 2003, 102, 3252–3261. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.J.; Ferec, C.; De Braekeleer, M. RUNX1 translocations and fusion genes in malignant hemopathies. Future Oncol. (London, England) 2011, 7, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Rock, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the aml study group. J. Clin. Oncol. 2011, 29, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 mutations are frequent in de novo aml with noncomplex karyotype and confer an unfavorable prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Hong, D.; Gupta, R.; Matsuo, K.; Seto, M.; Enver, T. Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med. 2007, 4, e172. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Shia, W.J.; Lo, M.C.; Fan, J.B.; Knorr, D.A.; Ferrell, P.I.; Ye, Z.; Yan, M.; Cheng, L.; Kaufman, D.S.; et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood 2013, 121, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Goodell, M.A. RUNX1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp. Hematol. 2010, 38, 403–416. [Google Scholar] [CrossRef]
- Draper, J.E.; Sroczynska, P.; Tsoulaki, O.; Leong, H.S.; Fadlullah, M.Z.; Miller, C.; Kouskoff, V.; Lacaud, G. RUNX1b expression is highly heterogeneous and distinguishes megakaryocytic and erythroid lineage fate in adult mouse hematopoiesis. PLoS Genet. 2016, 12, e1005814. [Google Scholar]
- Draper, J.E.; Sroczynska, P.; Leong, H.S.; Fadlullah, M.Z.H.; Miller, C.; Kouskoff, V.; Lacaud, G. Mouse RUNX1c regulates premegakaryocytic/erythroid output and maintains survival of megakaryocyte progenitors. Blood 2017, 130, 271–284. [Google Scholar] [CrossRef]
- Imai, Y.; Kurokawa, M.; Tanaka, K.; Friedman, A.D.; Ogawa, S.; Mitani, K.; Yazaki, Y.; Hirai, H. Tle, the human homolog of groucho, interacts with AML1 and acts as a repressor of AML1-induced transactivation. Biochem. Biophys. Res. Commun. 1998, 252, 582–589. [Google Scholar] [CrossRef]
- Meyers, S.; Downing, J.R.; Hiebert, S.W. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: The runt homology domain is required for DNA binding and protein-protein interactions. Mol. Cell. Biol. 1993, 13, 6336–6345. [Google Scholar] [CrossRef]
- Golling, G.; Li, L.; Pepling, M.; Stebbins, M.; Gergen, J.P. Drosophila homologs of the proto-oncogene product PEBP2/CBF beta regulate the DNA-binding properties of runt. Mol. Cell. Biol. 1996, 16, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Blake, T.; Adya, N.; Kim, C.H.; Oates, A.C.; Zon, L.; Chitnis, A.; Weinstein, B.M.; Liu, P.P. Zebrafish homolog of the leukemia gene CBFB: Its expression during embryogenesis and its relationship to scl and gata-1 in hematopoiesis. Blood 2000, 96, 4178–4184. [Google Scholar]
- Ogawa, E.; Maruyama, M.; Kagoshima, H.; Inuzuka, M.; Lu, J.; Satake, M.; Shigesada, K.; Ito, Y. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the drosophila runt gene and the human AML1 gene. Proc. Natl. Acad. Sci. USA 1993, 90, 6859–6863. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Q.; Crute, B.E.; Melnikova, I.N.; Keller, S.R.; Speck, N.A. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol. Cell. Biol. 1993, 13, 3324–3339. [Google Scholar] [CrossRef] [PubMed]
- Tahirov, T.H.; Bushweller, J. Structure and biophysics of CBFβ/RUNX and its translocation products. Adv. Exp. Med. Biol. 2017, 962, 21–31. [Google Scholar]
- Huang, G.; Shigesada, K.; Ito, K.; Wee, H.J.; Yokomizo, T.; Ito, Y. Dimerization with PEBP2β protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001, 20, 723–733. [Google Scholar] [CrossRef]
- Wee, H.J.; Voon, D.C.; Bae, S.C.; Ito, Y. PEBP2-β/CBF-β-dependent phosphorylation of RUNX1 and p300 by HIPK2: Implications for leukemogenesis. Blood 2008, 112, 3777–3787. [Google Scholar] [CrossRef]
- Zhang, D.E.; Hetherington, C.J.; Meyers, S.; Rhoades, K.L.; Larson, C.J.; Chen, H.M.; Hiebert, S.W.; Tenen, D.G. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol. Cell. Biol. 1996, 16, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Petrovick, M.S.; Hiebert, S.W.; Friedman, A.D.; Hetherington, C.J.; Tenen, D.G.; Zhang, D.E. Multiple functional domains of AML1: PU.1 and C/EBPalpha synergize with different regions of AML1. Mol. Cell. Biol. 1998, 18, 3915–3925. [Google Scholar] [CrossRef]
- Imai, Y.; Kurokawa, M.; Yamaguchi, Y.; Izutsu, K.; Nitta, E.; Mitani, K.; Satake, M.; Noda, T.; Ito, Y.; Hirai, H. The corepressor mSin3A regulates phosphorylation-induced activation, intranuclear location, and stability of AML1. Mol. Cell. Biol. 2004, 24, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, I.; Yokoyama, A.; Shimizu, K.; Ohki, M. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. EMBO J. 1998, 17, 2994–3004. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yu, M.; Akie, T.E.; Moran, T.B.; Woo, A.J.; Tu, N.; Waldon, Z.; Lin, Y.Y.; Steen, H.; Cantor, A.B. Differentiation-dependent interactions between RUNX-1 and fli-1 during megakaryocyte development. Mol. Cell. Biol. 2009, 29, 4103–4115. [Google Scholar] [CrossRef]
- Michaud, J.; Scott, H.S.; Escher, R. AML1 interconnected pathways of leukemogenesis. Cancer Investig. 2003, 21, 105–136. [Google Scholar] [CrossRef]
- Ito, Y. Oncogenic potential of the RUNX gene family: ‘Overview’. Oncogene 2004, 23, 4198–4208. [Google Scholar] [CrossRef]
- Miyoshi, H.; Kozu, T.; Shimizu, K.; Enomoto, K.; Maseki, N.; Kaneko, Y.; Kamada, N.; Ohki, M. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993, 12, 2715–2721. [Google Scholar] [CrossRef]
- Muller, A.M.; Duque, J.; Shizuru, J.A.; Lubbert, M. Complementing mutations in core binding factor leukemias: From mouse models to clinical applications. Oncogene 2008, 27, 5759–5773. [Google Scholar] [CrossRef]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The importance of diagnostic cytogenetics on outcome in aml: Analysis of 1,612 patients entered into the MRC AML 10 trial. The medical research council adult and children’s leukaemia working parties. Blood 1998, 92, 2322–2333. [Google Scholar] [PubMed]
- Byrd, J.C.; Mrozek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from cancer and leukemia group b (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef]
- Kuchenbauer, F.; Schnittger, S.; Look, T.; Gilliland, G.; Tenen, D.; Haferlach, T.; Hiddemann, W.; Buske, C.; Schoch, C. Identification of additional cytogenetic and molecular genetic abnormalities in acute myeloid leukaemia with t(8;21)/AML1-ETO. Br. J. Haematol. 2006, 134, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Boissel, N.; Reutenauer, S.; Thomas, X.; Delaunay, J.; Cahn, J.Y.; Pigneux, A.; Quesnel, B.; Witz, F.; Thepot, S.; et al. Acute myeloid leukemia with translocation (8;21) or inversion (16) in elderly patients treated with conventional chemotherapy: A collaborative study of the french cbf-aml intergroup. J. Clin. Oncol. 2009, 27, 4747–4753. [Google Scholar] [CrossRef] [PubMed]
- Wolford, J.K.; Prochazka, M. Structure and expression of the human MTG8/ETO gene. Gene 1998, 212, 103–109. [Google Scholar] [CrossRef]
- Kozu, T.; Fukuyama, T.; Yamami, T.; Akagi, K.; Kaneko, Y. Mynd-less splice variants of AML1-MTG8 (RUNX1-CBFA2T1) are expressed in leukemia with t(8;21). Gene. Chromosome Cancer 2005, 43, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.N.; McGhee, L.; Meyers, S. The ETO (MTG8) gene family. Gene 2003, 303, 1–10. [Google Scholar] [CrossRef]
- Feinstein, P.G.; Kornfeld, K.; Hogness, D.S.; Mann, R.S. Identification of homeotic target genes in drosophila melanogaster including nervy, a proto-oncogene homologue. Genetics 1995, 140, 573–586. [Google Scholar]
- Zhang, J.; Hug, B.A.; Huang, E.Y.; Chen, C.W.; Gelmetti, V.; Maccarana, M.; Minucci, S.; Pelicci, P.G.; Lazar, M.A. Oligomerization of eto is obligatory for corepressor interaction. Mol. Cell. Biol. 2001, 21, 156–163. [Google Scholar] [CrossRef]
- Lutterbach, B.; Westendorf, J.J.; Linggi, B.; Patten, A.; Moniwa, M.; Davie, J.R.; Huynh, K.D.; Bardwell, V.J.; Lavinsky, R.M.; Rosenfeld, M.G.; et al. ETO, a target of t(8;21) in acute leukemia, interacts with the n-cor and msin3 corepressors. Mol. Cell. Biol. 1998, 18, 7176–7184. [Google Scholar] [CrossRef]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed]
- Erickson, P.F.; Robinson, M.; Owens, G.; Drabkin, H.A. The ETO portion of acute myeloid leukemia t(8;21) fusion transcript encodes a highly evolutionarily conserved, putative transcription factor. Cancer Res. 1994, 54, 1782–1786. [Google Scholar] [PubMed]
- Zhang, J.; Kalkum, M.; Yamamura, S.; Chait, B.T.; Roeder, R.G. E protein silencing by the leukemogenic AML1-ETO fusion protein. Science 2004, 305, 1286–1289. [Google Scholar] [CrossRef] [PubMed]
- Plevin, M.J.; Zhang, J.; Guo, C.; Roeder, R.G.; Ikura, M. The acute myeloid leukemia fusion protein AML1-ETO targets e proteins via a paired amphipathic helix-like TBP-associated factor homology domain. Proc. Natl. Acad. Sci. USA 2006, 103, 10242–10247. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, I.; Ida, K.; Morohoshi, F.; Yokoyama, A.; Mitsuhashi, N.; Shimizu, K.; Nomura, N.; Hayashi, Y.; Ohki, M. The AML1-MTG8 leukemic fusion protein forms a complex with a novel member of the MTG8(ETO/CDR) family, mtgr1. Mol. Cell. Biol. 1998, 18, 846–858. [Google Scholar] [CrossRef]
- Liu, Y.; Cheney, M.D.; Gaudet, J.J.; Chruszcz, M.; Lukasik, S.M.; Sugiyama, D.; Lary, J.; Cole, J.; Dauter, Z.; Minor, W.; et al. The tetramer structure of the nervy homology two domain, NHR2, is critical for AML1/ETO’S activity. Cancer Cell 2006, 9, 249–260. [Google Scholar] [CrossRef]
- Hug, B.A.; Lazar, M.A. ETO interacting proteins. Oncogene 2004, 23, 4270–4274. [Google Scholar] [CrossRef]
- Odaka, Y.; Mally, A.; Elliott, L.T.; Meyers, S. Nuclear import and subnuclear localization of the proto-oncoprotein ETO (MTG8). Oncogene 2000, 19, 3584–3597. [Google Scholar] [CrossRef]
- Barseguian, K.; Lutterbach, B.; Hiebert, S.W.; Nickerson, J.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Multiple subnuclear targeting signals of the leukemia-related AML1/ETO and ETO repressor proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 15434–15439. [Google Scholar] [CrossRef]
- Tighe, J.E.; Calabi, F. T(8;21) breakpoints are clustered between alternatively spliced exons of MTG8. Clin. Sci. (Lond.) 1995, 89, 215–218. [Google Scholar] [CrossRef]
- Zhang, Y.; Strissel, P.; Strick, R.; Chen, J.; Nucifora, G.; Le Beau, M.M.; Larson, R.A.; Rowley, J.D. Genomic DNA breakpoints in AML1/RUNX1 and ETO cluster with topoisomerase ii DNA cleavage and dnase i hypersensitive sites in t(8;21) leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 3070–3075. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, G.D.; Vargas, M.F.; Medina, M.A.; Leon, P.; Necunir, D.; Elorza, A.A.; Gutierrez, S.E.; Moon, R.T.; Loyola, A.; De Ferrari, G.V. Wnt signaling induces transcription, spatial proximity, and translocation of fusion gene partners in human hematopoietic cells. Blood 2015, 126, 1785–1789. [Google Scholar] [CrossRef]
- Kwok, C.; Zeisig, B.B.; Qiu, J.; Dong, S.; So, C.W. Transforming activity of AML1-ETO is independent of CBFbeta and eto interaction but requires formation of homo-oligomeric complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 2853–2858. [Google Scholar] [CrossRef] [PubMed]
- Hug, B.A.; Lee, S.Y.; Kinsler, E.L.; Zhang, J.; Lazar, M.A. Cooperative function of AML1-ETO corepressor recruitment domains in the expansion of primary bone marrow cells. Cancer Res. 2002, 62, 2906–2912. [Google Scholar] [PubMed]
- DeKelver, R.C.; Yan, M.; Ahn, E.Y.; Shia, W.J.; Speck, N.A.; Zhang, D.E. Attenuation of AML1-ETO cellular dysregulation correlates with increased leukemogenic potential. Blood 2013, 121, 3714–3717. [Google Scholar] [CrossRef]
- Yan, M.; Kanbe, E.; Peterson, L.F.; Boyapati, A.; Miao, Y.; Wang, Y.; Chen, I.M.; Chen, Z.; Rowley, J.D.; Willman, C.L.; et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat. Med. 2006, 12, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.L.; Hou, H.A.; Chen, C.Y.; Liu, C.Y.; Chou, W.C.; Tseng, M.H.; Huang, C.F.; Lee, F.Y.; Liu, M.C.; Yao, M.; et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: Prognostic implication and interaction with other gene alterations. Blood 2009, 114, 5352–5361. [Google Scholar] [CrossRef]
- Ben-Ami, O.; Friedman, D.; Leshkowitz, D.; Goldenberg, D.; Orlovsky, K.; Pencovich, N.; Lotem, J.; Tanay, A.; Groner, Y. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013, 4, 1131–1143. [Google Scholar] [CrossRef]
- Goyama, S.; Schibler, J.; Cunningham, L.; Zhang, Y.; Rao, Y.; Nishimoto, N.; Nakagawa, M.; Olsson, A.; Wunderlich, M.; Link, K.A.; et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Investig. 2013, 123, 3876–3888. [Google Scholar] [CrossRef]
- Staber, P.B.; Zhang, P.; Ye, M.; Welner, R.S.; Levantini, E.; Di Ruscio, A.; Ebralidze, A.K.; Bach, C.; Zhang, H.; Zhang, J.; et al. The RUNX-PU.1 pathway preserves normal and AML/ETO9a leukemic stem cells. Blood 2014, 124, 2391–2399. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Wang, X.; Jin, W.; Tan, Y.; Fang, H.; Chen, S.; Chen, Z.; Wang, K. Genome-wide studies identify a novel interplay between AML1 and AML1/ETO in t(8;21) acute myeloid leukemia. Blood 2016, 127, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2000; Volume 65, pp. 127–133. [Google Scholar]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 g.H.A. Clowes memorial award lecture. Mol. Cancer Res. MCR 2008, 6, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef]
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. P53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Forster, V.J.; Nahari, M.H.; Martinez-Soria, N.; Bradburn, A.K.; Ptasinska, A.; Assi, S.A.; Fordham, S.E.; McNeil, H.; Bonifer, C.; Heidenreich, O.; et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia 2016, 30, 250–253. [Google Scholar] [CrossRef]
- Liddiard, K.; Hills, R.; Burnett, A.K.; Darley, R.L.; Tonks, A. OGG1 is a novel prognostic indicator in acute myeloid leukaemia. Oncogene 2010, 29, 2005–2012. [Google Scholar] [CrossRef]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by parp inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Satoh, Y.; Matsumura, I.; Tanaka, H.; Harada, H.; Harada, Y.; Matsui, K.; Shibata, M.; Mizuki, M.; Kanakura, Y. C-terminal mutation of RUNX1 attenuates the DNA-damage repair response in hematopoietic stem cells. Leukemia 2012, 26, 303–311. [Google Scholar] [CrossRef]
- Zuber, J.; Radtke, I.; Pardee, T.S.; Zhao, Z.; Rappaport, A.R.; Luo, W.; McCurrach, M.E.; Yang, M.M.; Dolan, M.E.; Kogan, S.C.; et al. Mouse models of human aml accurately predict chemotherapy response. Genes Dev. 2009, 23, 877–889. [Google Scholar] [CrossRef]
- Bullinger, L.; Rucker, F.G.; Kurz, S.; Du, J.; Scholl, C.; Sander, S.; Corbacioglu, A.; Lottaz, C.; Krauter, J.; Frohling, S.; et al. Gene-expression profiling identifies distinct subclasses of core binding factor acute myeloid leukemia. Blood 2007, 110, 1291–1300. [Google Scholar] [CrossRef]
- Mrozek, K.; Bloomfield, C.D. Clinical significance of the most common chromosome translocations in adult acute myeloid leukemia. J. Natl. Cancer Inst. Monogr. 2008, 39, 52–57. [Google Scholar] [CrossRef]
- Krauth, M.T.; Eder, C.; Alpermann, T.; Bacher, U.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1t1: Frequency and impact on clinical outcome. Leukemia 2014, 28, 1449–1458. [Google Scholar] [CrossRef]
- Matsuura, S.; Yan, M.; Lo, M.C.; Ahn, E.Y.; Weng, S.; Dangoor, D.; Matin, M.; Higashi, T.; Feng, G.S.; Zhang, D.E. Negative effects of gm-csf signaling in a murine model of t(8;21)-induced leukemia. Blood 2012, 119, 3155–3163. [Google Scholar] [CrossRef]
- Dayyani, F.; Wang, J.; Yeh, J.R.; Ahn, E.Y.; Tobey, E.; Zhang, D.E.; Bernstein, I.D.; Peterson, R.T.; Sweetser, D.A. Loss of TLE1 and TLE4 from the del(9q) commonly deleted region in aml cooperates with AML1-ETO to affect myeloid cell proliferation and survival. Blood 2008, 111, 4338–4347. [Google Scholar] [CrossRef]
- Marcucci, G.; Mrozek, K.; Ruppert, A.S.; Maharry, K.; Kolitz, J.E.; Moore, J.O.; Mayer, R.J.; Pettenati, M.J.; Powell, B.L.; Edwards, C.G.; et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): A cancer and leukemia group b study. J. Clin. Oncol. 2005, 23, 5705–5717. [Google Scholar] [CrossRef]
- Wakita, S.; Yamaguchi, H.; Miyake, K.; Mitamura, Y.; Kosaka, F.; Dan, K.; Inokuchi, K. Importance of c-kit mutation detection method sensitivity in prognostic analyses of t(8;21)(q22;q22) acute myeloid leukemia. Leukemia 2011, 25, 1423–1432. [Google Scholar] [CrossRef]
- Fu, L.; Fu, H.; Tian, L.; Xu, K.; Hu, K.; Wang, J.; Wang, J.; Jing, H.; Shi, J.; Ke, X. High expression of RUNX1 is associated with poorer outcomes in cytogenetically normal acute myeloid leukemia. Oncotarget 2016, 7, 15828–15839. [Google Scholar] [CrossRef]
- Bird, A.P. CPG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Suzuki, T.; Shimizu, Y.; Furuhata, E.; Maeda, S.; Kishima, M.; Nishimura, H.; Enomoto, S.; Hayashizaki, Y.; Suzuki, H. RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv. 2017, 1, 1699–1711. [Google Scholar] [CrossRef]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The ten-eleven translocation-2 (tet2) gene in hematopoiesis and hematopoietic diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef]
- Faber, Z.J.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Paschka, P.; Spath, D.; Habdank, M.; Kohne, C.H.; Germing, U.; von Lilienfeld-Toal, M.; Held, G.; Horst, H.A.; Haase, D.; et al. TET2 mutations in acute myeloid leukemia (AML): Results from a comprehensive genetic and clinical analysis of the aml study group. J. Clin. Oncol. 2012, 30, 1350–1357. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Liu, S.; Shen, T.; Huynh, L.; Klisovic, M.I.; Rush, L.J.; Ford, J.L.; Yu, J.; Becknell, B.; Li, Y.; Liu, C.; et al. Interplay of RUNX1/mtg8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 2005, 65, 1277–1284. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, L.; Lv, N.; Liu, J.; Li, Y.; Chen, X.; Xu, Q.; Chen, G.; Pang, B.; Wang, L.; et al. Methylation-associated silencing of BASP1 contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Exp. Mol. Med. 2018, 50, 44. [Google Scholar] [CrossRef]
- Gao, X.N.; Yan, F.; Lin, J.; Gao, L.; Lu, X.L.; Wei, S.C.; Shen, N.; Pang, J.X.; Ning, Q.Y.; Komeno, Y.; et al. AML1/ETO cooperates with HIF1alpha to promote leukemogenesis through DNMT3a transactivation. Leukemia 2015, 29, 1730–1740. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Meyers, S.; Lenny, N.; Hiebert, S.W. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol. Cell. Biol. 1995, 15, 1974–1982. [Google Scholar] [CrossRef]
- Gelmetti, V.; Zhang, J.; Fanelli, M.; Minucci, S.; Pelicci, P.G.; Lazar, M.A. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner eto. Mol. Cell. Biol. 1998, 18, 7185–7191. [Google Scholar] [CrossRef]
- Amann, J.M.; Nip, J.; Strom, D.K.; Lutterbach, B.; Harada, H.; Lenny, N.; Downing, J.R.; Meyers, S.; Hiebert, S.W. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds msin3a through its oligomerization domain. Mol. Cell. Biol. 2001, 21, 6470–6483. [Google Scholar] [CrossRef]
- Mandoli, A.; Singh, A.A.; Prange, K.H.M.; Tijchon, E.; Oerlemans, M.; Dirks, R.; Ter Huurne, M.; Wierenga, A.T.J.; Janssen-Megens, E.M.; Berentsen, K.; et al. The hematopoietic transcription factors RUNX1 and erg prevent AML1-ETO oncogene overexpression and onset of the apoptosis program in t(8;21) AMLs. Cell Rep. 2016, 17, 2087–2100. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of hdac inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Blobel, G.A. Creb-binding protein and p300: Molecular integrators of hematopoietic transcription. Blood 2000, 95, 745–755. [Google Scholar]
- Kasper, L.H.; Boussouar, F.; Ney, P.A.; Jackson, C.W.; Rehg, J.; van Deursen, J.M.; Brindle, P.K. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature 2002, 419, 738–743. [Google Scholar] [CrossRef]
- Rebel, V.I.; Kung, A.L.; Tanner, E.A.; Yang, H.; Bronson, R.T.; Livingston, D.M. Distinct roles for creb-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2002, 99, 14789–14794. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kurokawa, M.; Imai, Y.; Izutsu, K.; Asai, T.; Ichikawa, M.; Yamamoto, G.; Nitta, E.; Yamagata, T.; Sasaki, K.; et al. AML1 is functionally regulated through p300-mediated acetylation on specific lysine residues. J. Biol. Chem. 2004, 279, 15630–15638. [Google Scholar] [CrossRef]
- Yoshida, H.; Kitabayashi, I. Chromatin regulation by AML1 complex. Int. J. Hematol. 2008, 87, 19–24. [Google Scholar] [CrossRef]
- Wang, L.; Gural, A.; Sun, X.J.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.A.; Vu, L.; Liu, F.; Xu, H.; et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef]
- Saeed, S.; Logie, C.; Francoijs, K.J.; Frige, G.; Romanenghi, M.; Nielsen, F.G.; Raats, L.; Shahhoseini, M.; Huynen, M.; Altucci, L.; et al. Chromatin accessibility, p300, and histone acetylation define PML-RARalpha and AML1-ETO binding sites in acute myeloid leukemia. Blood 2012, 120, 3058–3068. [Google Scholar] [CrossRef]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. Prmt1 is the predominant type i protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar] [CrossRef]
- An, W.; Kim, J.; Roeder, R.G. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 2004, 117, 735–748. [Google Scholar] [CrossRef]
- Zhao, X.; Jankovic, V.; Gural, A.; Huang, G.; Pardanani, A.; Menendez, S.; Zhang, J.; Dunne, R.; Xiao, A.; Erdjument-Bromage, H.; et al. Methylation of RUNX1 by PRMT1 abrogates sin3a binding and potentiates its transcriptional activity. Genes Dev. 2008, 22, 640–653. [Google Scholar] [CrossRef]
- Shia, W.J.; Okumura, A.J.; Yan, M.; Sarkeshik, A.; Lo, M.C.; Matsuura, S.; Komeno, Y.; Zhao, X.; Nimer, S.D.; Yates, J.R., 3rd; et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 2012, 119, 4953–4962. [Google Scholar] [CrossRef]
- Sun, X.J.; Wang, Z.; Wang, L.; Jiang, Y.; Kost, N.; Soong, T.D.; Chen, W.Y.; Tang, Z.; Nakadai, T.; Elemento, O.; et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature 2013, 500, 93–97. [Google Scholar] [CrossRef]
- Barrero, M.J.; Malik, S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol. Cell 2006, 24, 233–243. [Google Scholar] [CrossRef]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Wang, L.; Zeng, H.; Wang, Q.; Zhao, Z.; Boyer, T.G.; Bian, X.; Xu, W. MED12 methylation by carm1 sensitizes human breast cancer cells to chemotherapy drugs. Sci. Adv. 2015, 1, e1500463. [Google Scholar] [CrossRef]
- Schurter, B.T.; Koh, S.S.; Chen, D.; Bunick, G.J.; Harp, J.M.; Hanson, B.L.; Henschen-Edman, A.; Mackay, D.R.; Stallcup, M.R.; Aswad, D.W. Methylation of histone h3 by coactivator-associated arginine methyltransferase 1. Biochemistry 2001, 40, 5747–5756. [Google Scholar] [CrossRef]
- Shishkova, E.; Zeng, H.; Liu, F.; Kwiecien, N.W.; Hebert, A.S.; Coon, J.J.; Xu, W. Global mapping of CARM1 substrates defines enzyme specificity and substrate recognition. Nat. Commun. 2017, 8, 15571. [Google Scholar] [CrossRef]
- Daujat, S.; Bauer, U.M.; Shah, V.; Turner, B.; Berger, S.; Kouzarides, T. Crosstalk between CARM1 methylation and CBP acetylation on histone h3. Curr. Biol. CB 2002, 12, 2090–2097. [Google Scholar] [CrossRef]
- Ptasinska, A.; Assi, S.A.; Martinez-Soria, N.; Imperato, M.R.; Piper, J.; Cauchy, P.; Pickin, A.; James, S.R.; Hoogenkamp, M.; Williamson, D.; et al. Identification of a dynamic core transcriptional network in t(8;21) aml that regulates differentiation block and self-renewal. Cell Rep. 2014, 8, 1974–1988. [Google Scholar] [CrossRef]
- Ptasinska, A.; Assi, S.A.; Mannari, D.; James, S.R.; Williamson, D.; Dunne, J.; Hoogenkamp, M.; Wu, M.; Care, M.; McNeill, H.; et al. Depletion of RUNX1/ETO in t(8;21) aml cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia 2012, 26, 1829–1841. [Google Scholar] [CrossRef]
- Trombly, D.J.; Whitfield, T.W.; Padmanabhan, S.; Gordon, J.A.; Lian, J.B.; van Wijnen, A.J.; Zaidi, S.K.; Stein, J.L.; Stein, G.S. Genome-wide co-occupancy of AML1-ETO and N-CoR defines the t(8;21) AML signature in leukemic cells. BMC Genom. 2015, 16, 309. [Google Scholar] [CrossRef]
- Hoogenkamp, M.; Lichtinger, M.; Krysinska, H.; Lancrin, C.; Clarke, D.; Williamson, A.; Mazzarella, L.; Ingram, R.; Jorgensen, H.; Fisher, A. Early chromatin unfolding by RUNX1: A molecular explanation for differential requirements during specification versus maintenance of the hematopoietic gene expression program. Blood 2009, 114, 299–309. [Google Scholar] [CrossRef]
- Vangala, R.K.; Heiss-Neumann, M.S.; Rangatia, J.S.; Singh, S.M.; Schoch, C.; Tenen, D.G.; Hiddemann, W.; Behre, G. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 2003, 101, 270–277. [Google Scholar] [CrossRef]
- Rosenbauer, F.; Wagner, K.; Kutok, J.L.; Iwasaki, H.; Le Beau, M.M.; Okuno, Y.; Akashi, K.; Fiering, S.; Tenen, D.G. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 2004, 36, 624–630. [Google Scholar] [CrossRef]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell 2018, 34, 626–642. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Kouwe, E.; Staber, P.B. RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia. Int. J. Mol. Sci. 2019, 20, 350. https://doi.org/10.3390/ijms20020350
van der Kouwe E, Staber PB. RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia. International Journal of Molecular Sciences. 2019; 20(2):350. https://doi.org/10.3390/ijms20020350
Chicago/Turabian Stylevan der Kouwe, Emiel, and Philipp Bernhard Staber. 2019. "RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia" International Journal of Molecular Sciences 20, no. 2: 350. https://doi.org/10.3390/ijms20020350
APA Stylevan der Kouwe, E., & Staber, P. B. (2019). RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia. International Journal of Molecular Sciences, 20(2), 350. https://doi.org/10.3390/ijms20020350