Galectin-9 Is a Possible Promoter of Immunopathology in Rheumatoid Arthritis by Activation of Peptidyl Arginine Deiminase 4 (PAD-4) in Granulocytes


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
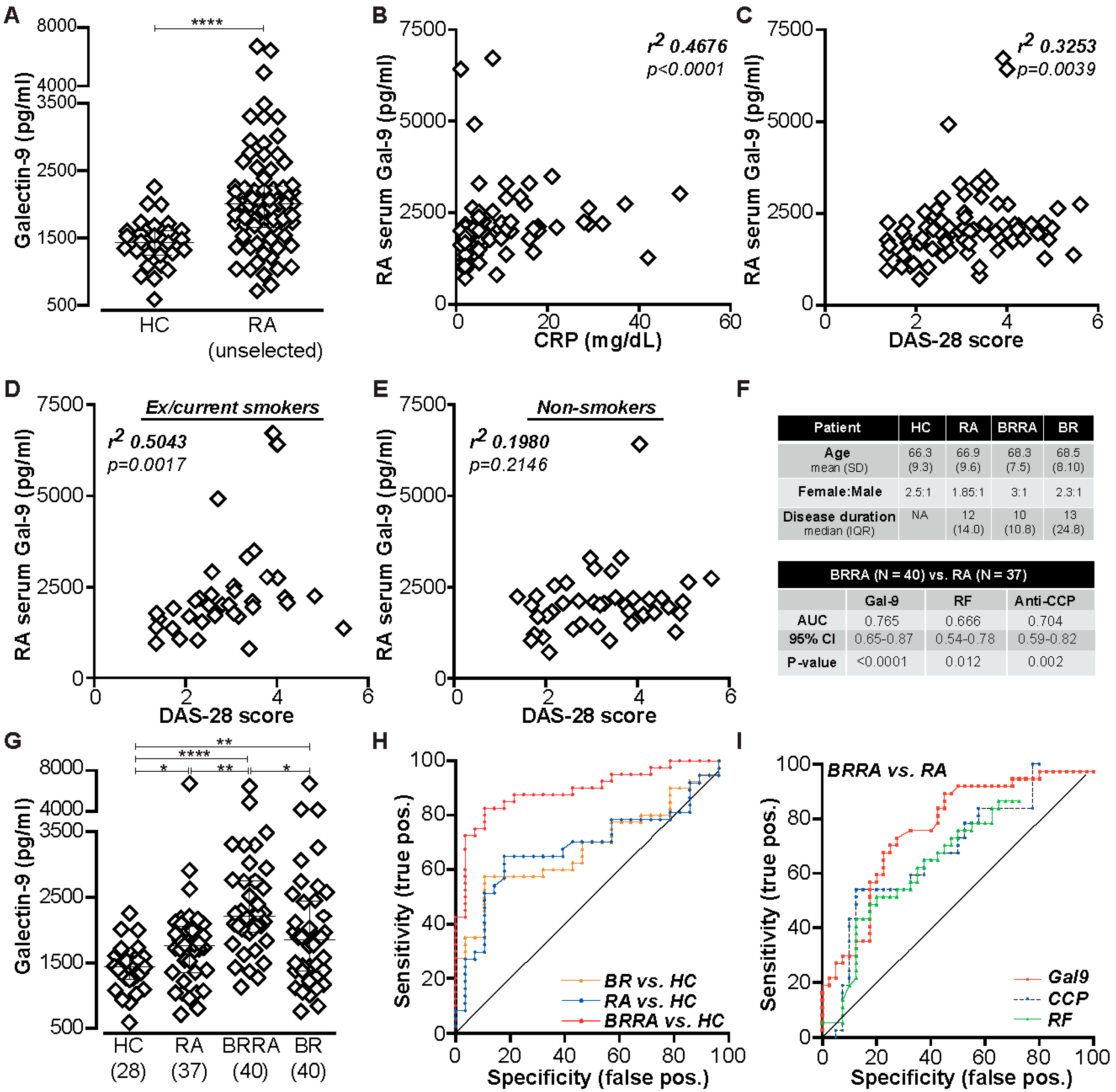
2.1. Gal-9 Is Elevated in Serum of RA Patients and Correlates with Certain Clinical Parameters
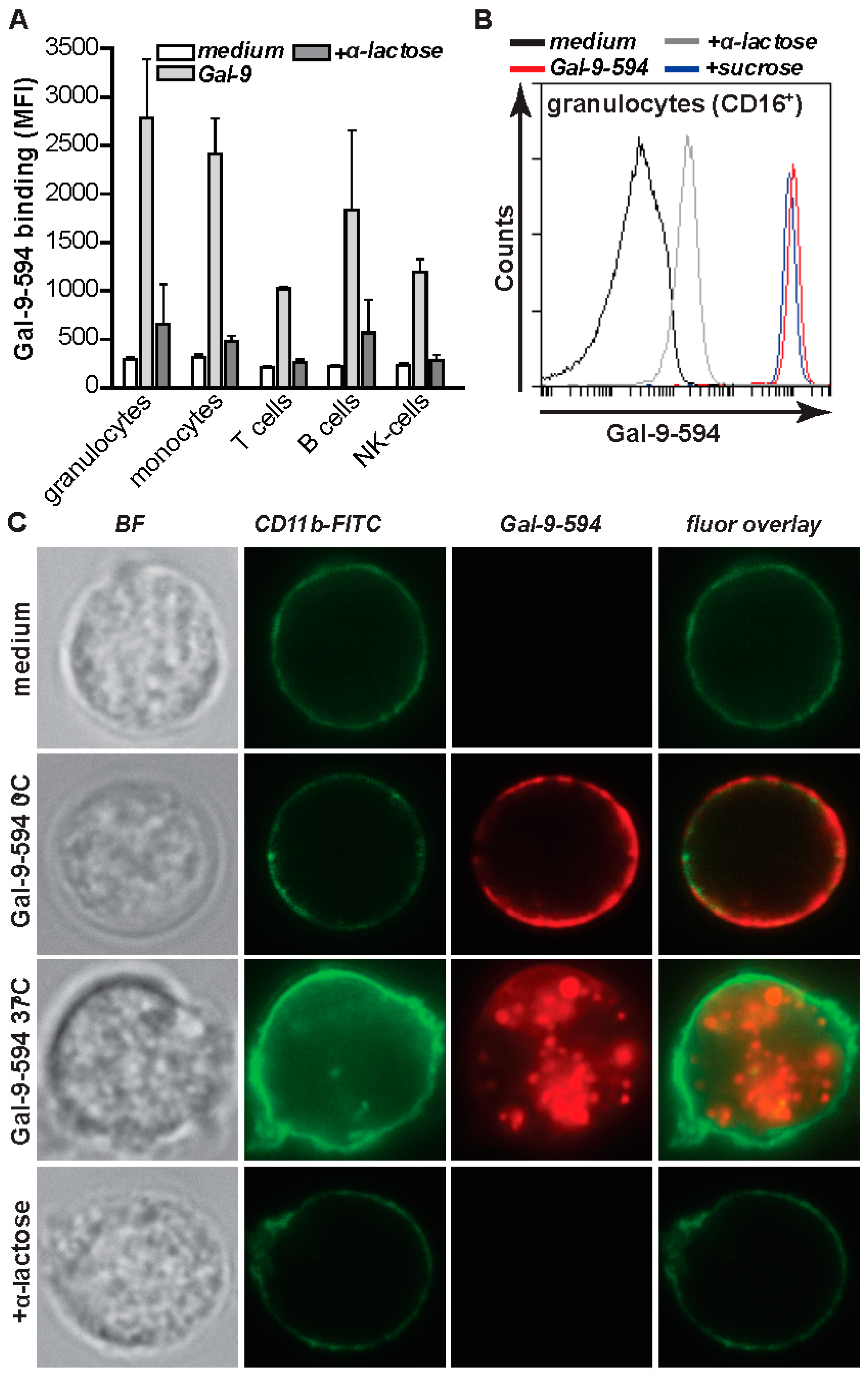
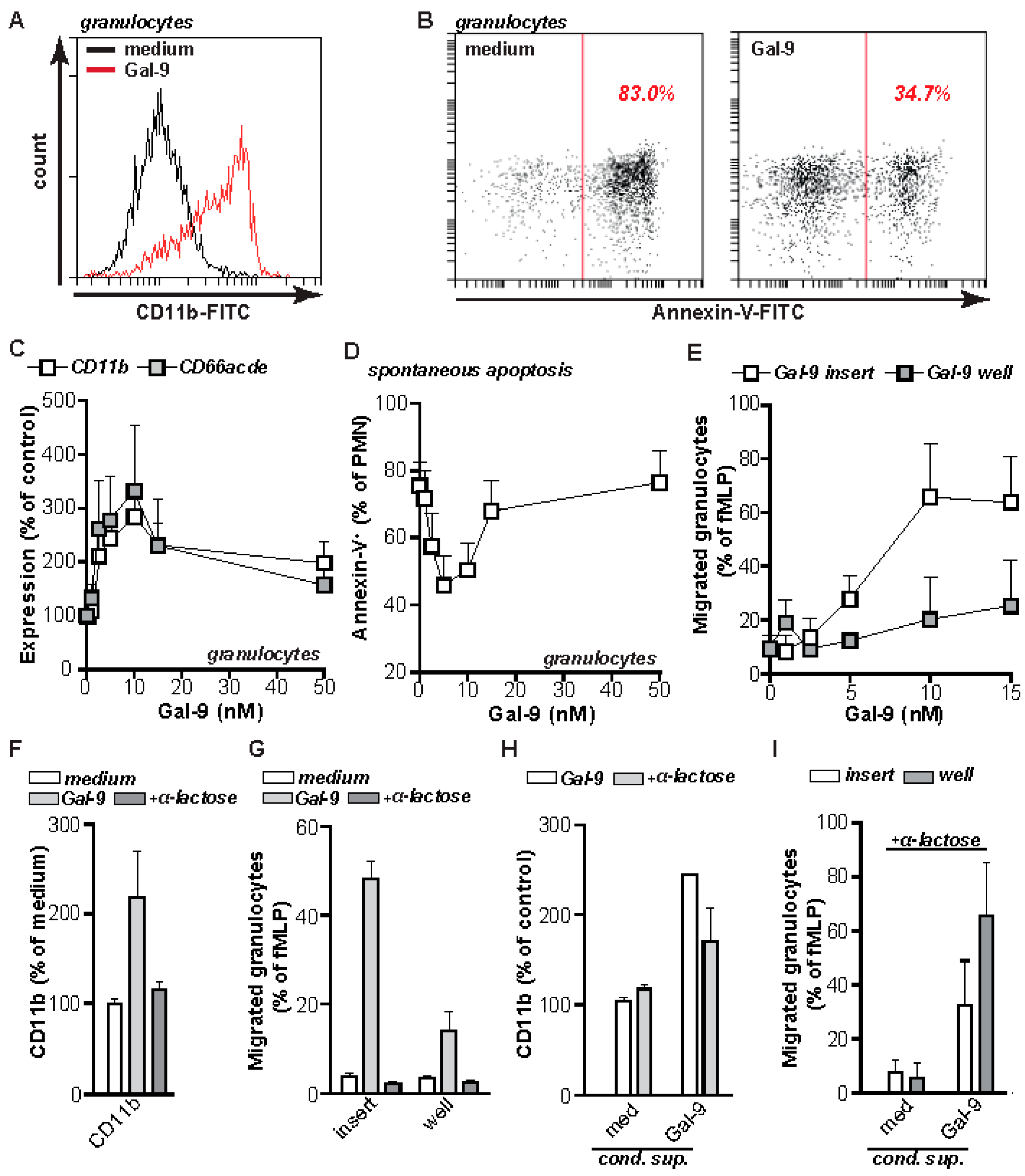
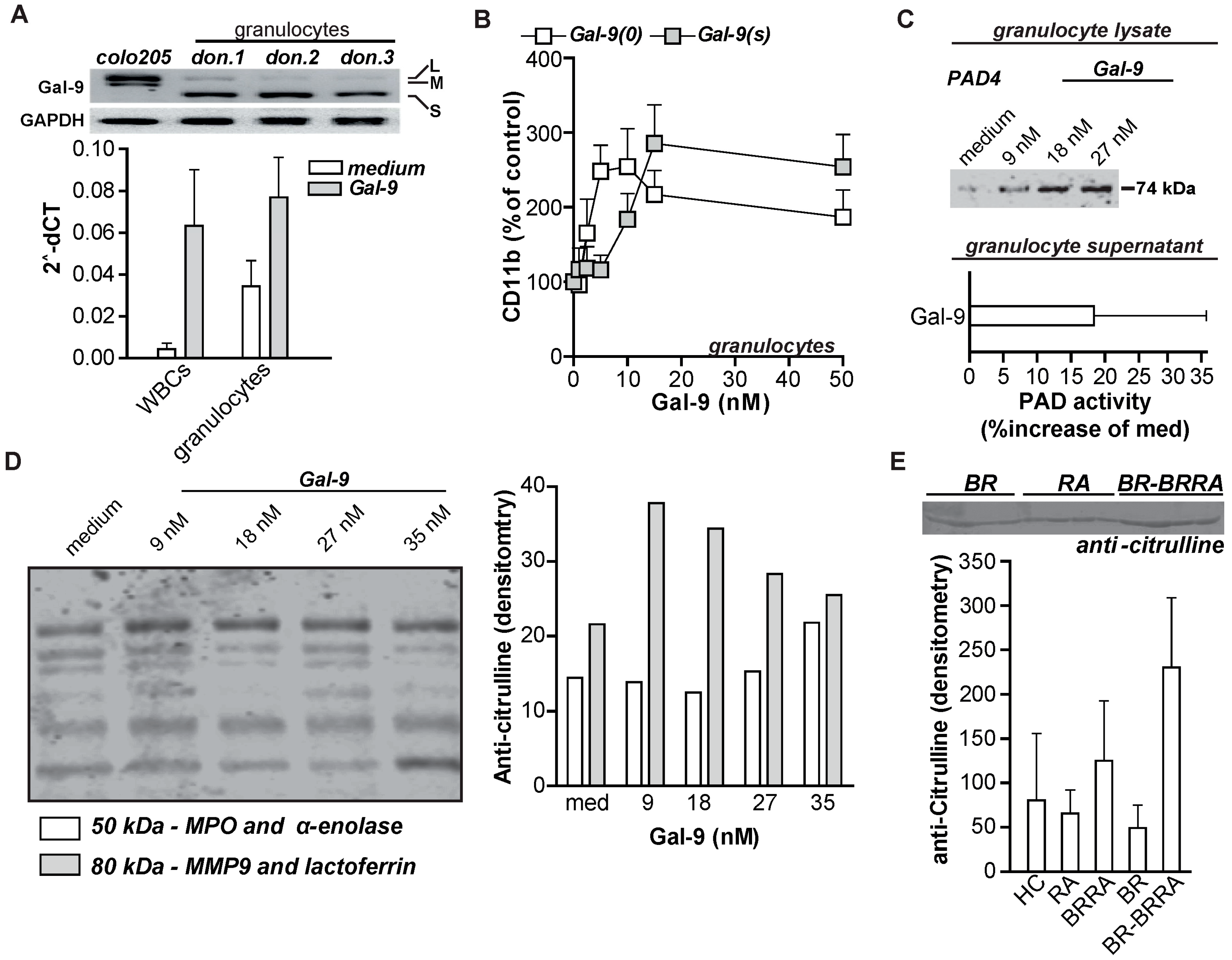
2.2. Gal-9 Induces CRD-Dependent Activation of Granulocytes
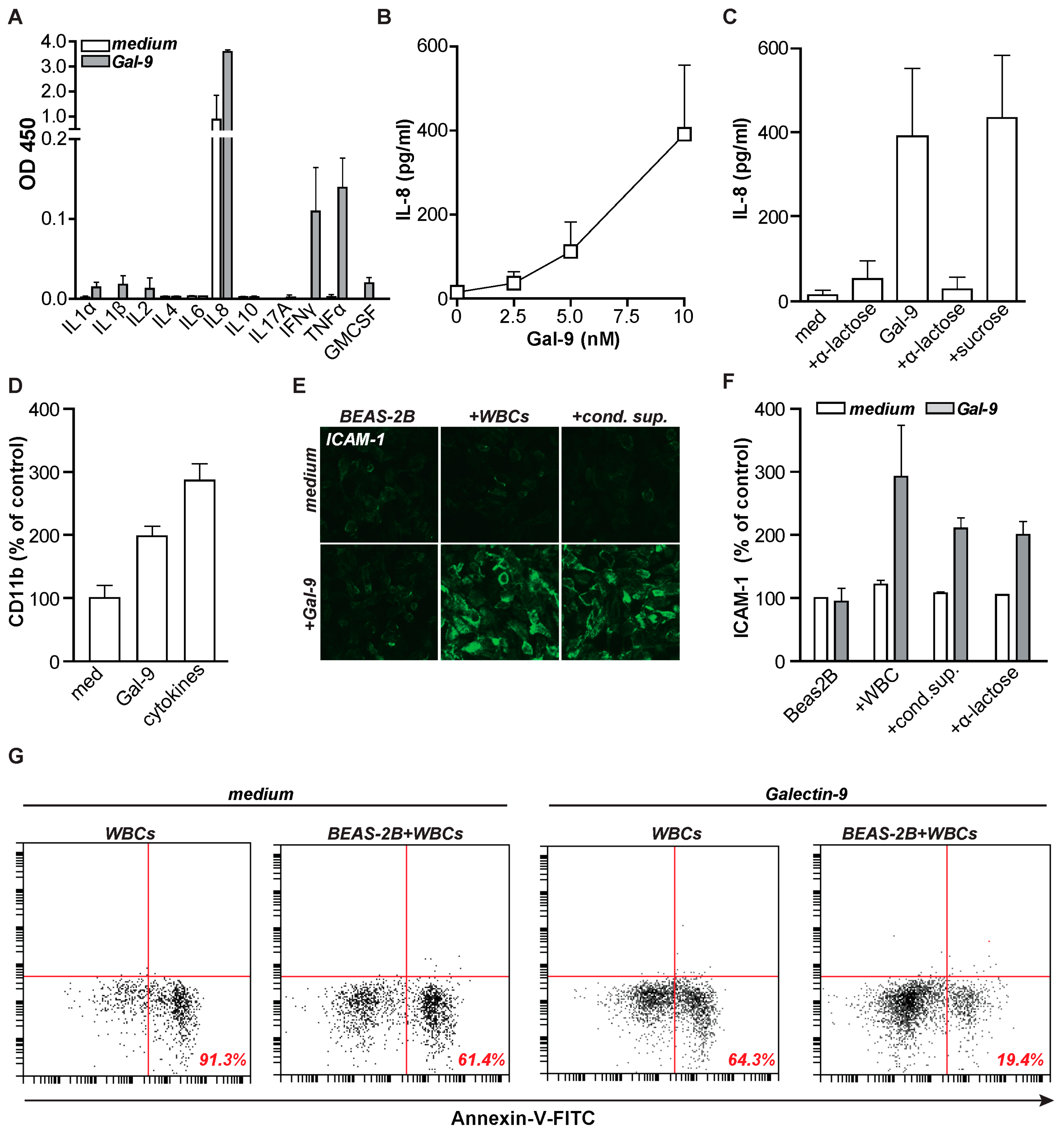
2.3. Gal-9 Activates Pro-Inflammatory Cytokines Leading to Intercellular Adhesion Molecule 1 (ICAM-1) Expression
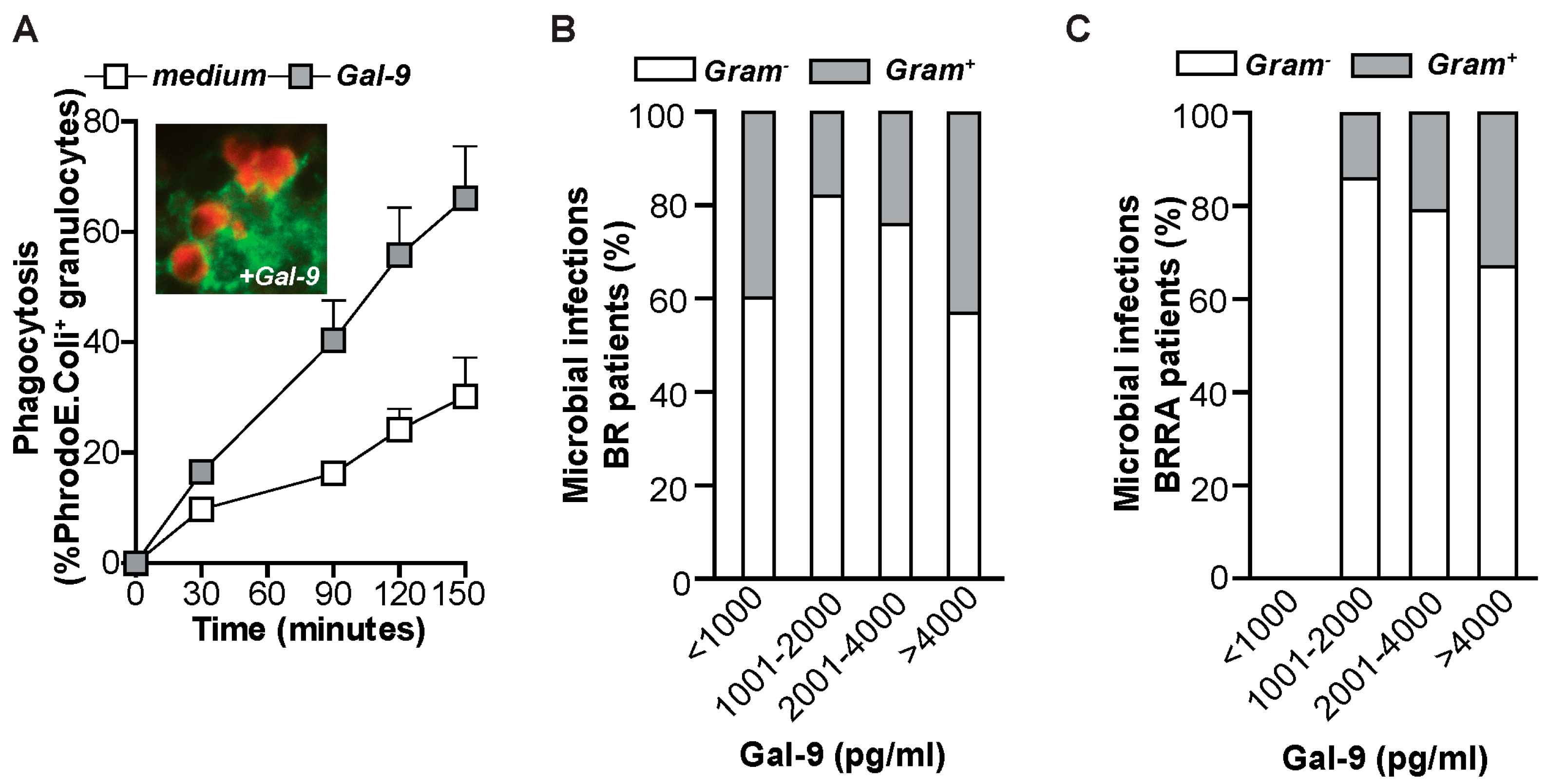
2.4. Gal-9 Induces Anti-Microbial Inflammatory Responses In Vitro
2.5. Gal-9 Induces Autoimmune Inflammatory Responses In Vitro
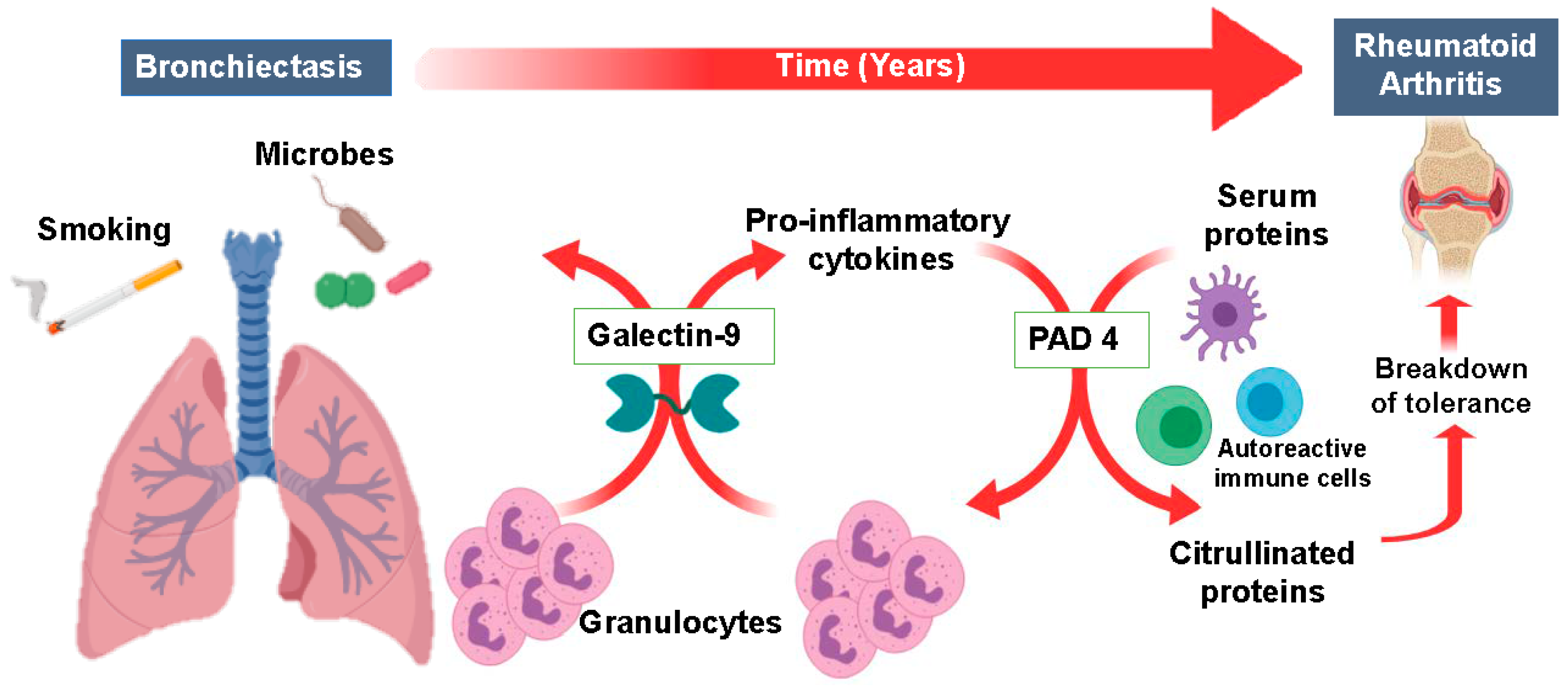
3. Discussion
4. Patients, Materials, and Methods
4.1. Patients and Control Subjects
4.2. Serology
4.3. Isolation of Human Leukocytes
4.4. Galectin-9 (Gal-9)
4.5. Gal-9 Binding Assays
4.6. Granulocyte Activation and Viability Assays
4.7. ICAM-1 Expression Assays
4.8. Cell Migration Assays
4.9. Phagocytosis Assays
4.10. ELISAs on Granulocyte Supernatants
4.11. RTqPCR, PCR, and Gel Electrophoresis
4.12. PAD-4 and Citrulline Western Blots and Mass Spectrometry
4.13. PAD-4 Activity Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rabinovich, G.A.; Toscano, M.A. Turning ‘sweet’ on immunity: Galectin-glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 2009, 9, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.R.; de Bruyn, M.; Helfrich, W.; Bremer, E. Therapeutic potential of galectin-9 in human disease. Med. Res. Rev. 2013, 33, E102–E126. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.R.; de Bruyn, M.; van Ginkel, R.J.; Sigar, E.; Hirashima, M.; Niki, T.; Nishi, N.; Samplonius, D.F.; Helfrich, W.; Bremer, E. The glycan-binding protein galectin-9 has direct apoptotic activity toward melanoma cells. J. Investig. Dermatol. 2012, 132, 2302–2305. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.R.; de Bruyn, M.; Wei, Y.; van Ginkel, R.J.; Hirashima, M.; Niki, T.; Nishi, N.; Zhou, J.; Pouwels, S.D.; Samplonius, D.F.; et al. The epithelial polarity regulator lgals9/galectin-9 induces fatal frustrated autophagy in kras mutant colon carcinoma that depends on elevated basal autophagic flux. Autophagy 2015, 11, 1373–1388. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Oomizu, S.; Arikawa, T.; Niki, T.; Kadowaki, T.; Ueno, M.; Nishi, N.; Yamauchi, A.; Hirashima, M. Galectin-9 suppresses Th17 cell development in an il-2-dependent but tim-3-independent manner. Clin. Immunol. 2012, 143, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Oomizu, S.; Sakata, K.-M.; Sakata, A.; Arikawa, T.; Watanabe, K.; Ito, K.; Takeshita, K.; Niki, T.; Saita, N.; et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory t cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 2008, 127, 78–88. [Google Scholar] [CrossRef]
- Skapenko, A.; Leipe, J.; Lipsky, P.E.; Schulze-Koops, H. The role of the T-cell in autoimmune inflammation. Arthritis Res. Ther. 2005, S4–S14. [Google Scholar] [CrossRef]
- Chou, F.-C.; Shieh, S.-J.; Sytwu, H.-K. Attenuation of th1 response through galectin-9 and T-cell Ig mucin 3 interaction inhibits autoimmune diabetes in nod mice. Eur. J. Immunol. 2009, 39, 2403–2411. [Google Scholar] [CrossRef]
- Kanzaki, M.; Wada, J.; Sugiyama, K.; Nakatsuka, A.; Teshigawara, S.; Murakami, K.; Inoue, K.; Terami, T.; Katayama, A.; Eguchi, J.; et al. Galectin-9 and T-cell immunoglobulin mucin-3 pathway is a therapeutic target for type 1 diabetes. Endocrinology 2012, 153, 612–620. [Google Scholar] [CrossRef]
- Zhang, Q.; Luan, H.; Wang, L.; He, F.; Zhou, H.; Xu, X.; Li, X.; Xu, Q.; Niki, T.; Hirashima, M.; et al. Galectin-9 ameliorates anti-gbm glomerulonephritis by inhibiting Th1 and Th17 immune responses in mice. Am. J. Physiol. Ren. Physiol. 2014, 306, F822–F832. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.M.; Wiersma, V.R.; Samplonius, D.F.; Gerssen, J.; van Ginkel, R.J.; Nijman, H.W.; Hirashima, M.; Niki, T.; Eggleton, P.; Helfrich, W.; et al. Galectin-9 activates and expands human T-helper 1 cells. PLoS ONE 2013, 8, e65616. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Barjon, C.; Niki, T.; Gelin, A.; Praz, F.; Morales, O.; Souquere, S.; Hirashima, M.; Wei, M.; Dellis, O.; et al. Impact of exogenous galectin-9 on human T-cells: Contribution of the t cell receptor complex to antigen-independent activation but not to apoptosis induction. J. Biol. Chem. 2015, 290, 16797–16811. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Mócsai, A. The role of neutrophils in autoimmune diseases. Immunol. Lett. 2012, 143, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Willemze, A.; Trouw, L.A.; Toes, R.E.M.; Huizinga, T.W.J. The influence of ACPA status and characteristics on the course of ra. Nat. Rev. Heumatol. 2012, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Feitsma, A.L.; Toes, R.E.M.; Begovich, A.B.; Chokkalingam, A.P.; de Vries, R.R.P.; Huizinga, T.W.J.; van der Helm-van Mil, A.H.M. Risk of progression from undifferentiated arthritis to rheumatoid arthritis: The effect of the ptpn22 1858t-allele in anti-citrullinated peptide antibody positive patients. Rheumatology 2007, 46, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Kolfenbach, J.R.; Deane, K.D.; Derber, L.A.; O’Donnell, C.I.; Gilliland, W.R.; Edison, J.D.; Rosen, A.; Darrah, E.; Norris, J.M.; Holers, V.M. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, D.; Kagari, T.; Doi, H.; Shimozato, T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology 2006, 119, 195–202. [Google Scholar] [CrossRef]
- Wipke, B.T.; Allen, P.M. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 2001, 167, 1601–1608. [Google Scholar] [CrossRef]
- Eggleton, P.; Wang, L.; Penhallow, J.; Crawford, N.; Brown, K.A. Differences in oxidative response of subpopulations of neutrophils from healthy subjects and patients with rheumatoid arthritis. Ann. Rheum. Dis. 1995, 54, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. Nets are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, ra140–ra178. [Google Scholar] [CrossRef] [PubMed]
- Vega-Carrascal, I.; Bergin, D.A.; McElvaney, O.J.; McCarthy, C.; Banville, N.; Pohl, K.; Hirashima, M.; Kuchroo, V.K.; Reeves, E.P.; McElvaney, N.G. Galectin-9 signaling through tim-3 is involved in neutrophil-mediated gram-negative bacterial killing: An effect abrogated within the cystic fibrosis lung. J. Immunol. 2014, 192, 2418–2431. [Google Scholar] [CrossRef] [PubMed]
- Steichen, A.L.; Simonson, T.J.; Salmon, S.L.; Metzger, D.W.; Mishra, B.B.; Sharma, J. Alarmin function of galectin-9 in murine respiratory tularemia. PLoS ONE 2015, 10, e0123573. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, S.; Huang, S.; Pei, F.; Lu, F. Upregulated tim-3/galectin-9 expressions in acute lung injury in a murine malarial model. Parasitol. Res. 2016, 115, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Ytterberg, A.J.; Reynisdottir, G.; Malmström, V.; Klareskog, L. Lungs, joints and immunity against citrullinated proteins in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Iqbal, K.; Iman-Gutierrez, L.; Evans, P.; Manchegowda, K. Lung involvement in inflammatory rheumatic diseases. Best Pract. Res. Clin. Rheumatol. 2016, 30, 870–888. [Google Scholar] [CrossRef]
- Perry, E.; Kelly, C.; Eggleton, P.; De Soyza, A.; Hutchinson, D. The lung in ACPA-positive rheumatoid arthritis: An initiating site of injury? Rheumatology 2014, 53, 1940–1950. [Google Scholar] [CrossRef]
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engström, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural changes and antibody enrichment in the lungs are early features of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 31–39. [Google Scholar] [CrossRef]
- Hutchinson, D.; Shepstone, L.; Moots, R.; Lear, J.T.; Lynch, M.P. Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann. Rheum. Dis. 2001, 60, 223–227. [Google Scholar] [CrossRef]
- Lugli, E.B.; Correia, R.E.S.M.; Fischer, R.; Lundberg, K.; Bracke, K.R.; Montgomery, A.B.; Kessler, B.M.; Brusselle, G.G.; Venables, P.J. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: Implications for autoimmunity in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Makrygiannakis, D.; Hermansson, M.; Ulfgren, A.K.; Nicholas, A.P.; Zendman, A.J.W.; Eklund, A.; Grunewald, J.; Skold, C.M.; Klareskog, L.; Catrina, A.I. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in bal cells. Ann. Rheum. Dis. 2008, 67, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Perricone, C.; Versini, M.; Ben-Ami, D.; Gertel, S.; Watad, A.; Segel, M.J.; Ceccarelli, F.; Conti, F.; Cantarini, L.; Bogdanos, D.P.; et al. Smoke and autoimmunity: The fire behind the disease. Autoimmun. Rev. 2016, 15, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Gardiner, P. The relationship between rheumatoid arthritis and bronchiectasis. Ann. Rheum. Dis. 1994, 53, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Eggleton, P.; De Soyza, A.; Hutchinson, D.; Kelly, C. Increased disease activity, severity and autoantibody positivity in rheumatoid arthritis patients with co-existent bronchiectasis. Int. J. Rheum. Dis. 2017, 20, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Stenton, C.; Kelly, C.; Eggleton, P.; Hutchinson, D.; De Soyza, A. RA autoantibodies as predictors of rheumatoid arthritis in non-cystic fibrosis bronchiectasis patients. Eur. Respir. J. 2014, 44, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Quirke, A.-M.; Perry, E.; Cartwright, A.; Kelly, C.; De Soyza, A.; Eggleton, P.; Hutchinson, D.; Venables, P.J. Bronchiectasis is a model for chronic bacterial infection inducing autoimmunity in rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, Y.; Koehn, C.D.; Zhang, Z.; Su, K. Galectins in the pathogenesis of rheumatoid arthritis. J. Clin. Cell. Immunol. 2013, 4, 1000164. [Google Scholar]
- Vega-Carrascal, I.; Reeves, E.P.; Niki, T.; Arikawa, T.; McNally, P.; O’Neill, S.J.; Hirashima, M.; McElvaney, N.G. Dysregulation of tim-3-galectin-9 pathway in the cystic fibrosis airways. J. Immunol. 2011, 186, 2897–2909. [Google Scholar] [CrossRef]
- Niki, T.; Fujita, K.; Rosen, H.; Hirashima, M.; Masaki, T.; Hattori, T.; Hoshino, K. Plasma galectin-9 concentrations in normal and diseased condition. Cell. Physiol. Biochem. 2018, 50, 1856–1868. [Google Scholar] [CrossRef]
- Nishi, N.; Itoh, A.; Fujiyama, A.; Yoshida, N.; Araya, S.-i.; Hirashima, M.; Shoji, H.; Nakamura, T. Development of highly stable galectins: Truncation of the linker peptide confers protease-resistance on tandem-repeat type galectins. FEBS Lett. 2005, 579, 2058–2064. [Google Scholar] [CrossRef]
- Su, E.W.; Bi, S.; Kane, L.P. Galectin-9 regulates T helper cell function independently of tim-3. Glycobiology 2011, 21, 1258–1265. [Google Scholar] [CrossRef]
- Burke-Gaffney, A.; Hellewell, P.G. Tumour necrosis factor-alpha-induced icam-1 expression in human vascular endothelial and lung epithelial cells: Modulation by tyrosine kinase inhibitors. Br. J. Pharmacol. 1996, 119, 1149–1158. [Google Scholar] [CrossRef]
- Krunkosky, T.M.; Fischer, B.M.; Martin, L.D.; Jones, N.; Akley, N.J.; Adler, K.B. Effects of tnf-alpha on expression of icam-1 in human airway epithelial cells in vitro. Signaling pathways controlling surface and gene expression. Am. J. Respir. Cell Mol. Biol. 2000, 22, 685–692. [Google Scholar] [CrossRef]
- Seki, M.; Sakata, K.-m.; Oomizu, S.; Arikawa, T.; Sakata, A.; Ueno, M.; Nobumoto, A.; Niki, T.; Saita, N.; Ito, K.; et al. Beneficial effect of galectin 9 on rheumatoid arthritis by induction of apoptosis of synovial fibroblasts. Arthritis Rheum. 2007, 56, 3968–3976. [Google Scholar] [CrossRef]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Hutchinson, D.; Clarke, A.; Heesom, K.; Murphy, D.; Eggleton, P. Carbamylation/citrullination of IgG Fc in bronchiectasis, established RA with bronchiectasis and RA smokers: A potential risk factor for disease. ERJ Open Res. 2017, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Baka, Z.; Barta, P.; Losonczy, G.; Krenacs, T.; Papay, J.; Szarka, E.; Sarmay, G.; Babos, F.; Magyar, A.; Geher, P.; et al. Specific expression of pad-4 and citrullinated proteins in lung cancer is not associated with anti-ccp antibody production. Int. Immunol. 2011, 23, 405–414. [Google Scholar] [CrossRef]
- Kashio, Y.; Nakamura, K.; Abedin, M.J.; Seki, M.; Nishi, N.; Yoshida, N.; Nakamura, T.; Hirashima, M. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunol. 2003, 170, 3631–3636. [Google Scholar] [CrossRef]
- Zhou, Y.; Di Pucchio, T.; Sims, G.P.; Mittereder, N.; Mustelin, T. Characterization of the hypercitrullination reaction in human neutrophils and other leukocytes. Mediat. Inflamm. 2015, 2015, 236451. [Google Scholar] [CrossRef]
- Katoh, S.; Nobumoto, A.; Matsumoto, N.; Matsumoto, K.; Ehara, N.; Niki, T.; Inada, H.; Nishi, N.; Yamauchi, A.; Fukushima, K.; et al. Involvement of galectin-9 in lung eosinophilia in patients with eosinophilic pneumonia. Int. Arch. Allergy Immunol. 2010, 153, 294–302. [Google Scholar] [CrossRef]
- Matsumoto, N.; Katoh, S.; Yanagi, S.; Arimura, Y.; Tokojima, M.; Ueno, M.; Hirashima, M.; Nakazato, M. A possible role of galectin-9 in the pulmonary fibrosis of patients with interstitial pneumonia. Lung 2013, 191, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Chagan-Yasutan, H.; Ndhlovu, L.C.; Lacuesta, T.L.; Kubo, T.; Leano, P.S.A.; Niki, T.; Oguma, S.; Morita, K.; Chew, G.M.; Barbour, J.D.; et al. Galectin-9 plasma levels reflect adverse hematological and immunological features in acute dengue virus infection. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2013, 58, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Shiratori, B.; Chagan-Yasutan, H.; Matsumoto, M.; Niki, T.; Tanaka, M.; Takahashi, Y.; Usami, O.; Ashino, Y.; Hattori, T. Secretion of ifn-γ associated with galectin-9 production by pleural fluid cells from a patient with extrapulmonary tuberculosis. Int. J. Mol. Sci. 2017, 18, 1382. [Google Scholar] [CrossRef] [PubMed]
- Chabot, S.; Kashio, Y.; Seki, M.; Shirato, Y.; Nakamura, K.; Nishi, N.; Nakamura, T.; Matsumoto, R.; Hirashima, M. Regulation of galectin-9 expression and release in jurkat T cell line cells. Glycobiology 2002, 12, 111–118. [Google Scholar] [CrossRef]
- Oomizu, S.; Arikawa, T.; Niki, T.; Kadowaki, T.; Ueno, M.; Nishi, N.; Yamauchi, A.; Hattori, T.; Masaki, T.; Hirashima, M. Cell surface galectin-9 expressing th cells regulate Th17 and FOXP3+ treg development by galectin-9 secretion. PLoS ONE 2012, 7, e48574. [Google Scholar] [CrossRef]
- Imaizumi, T.; Kumagai, M.; Sasaki, N.; Kurotaki, H.; Mori, F.; Seki, M.; Nishi, N.; Fujimoto, K.; Tanji, K.; Shibata, T.; et al. Interferon-gamma stimulates the expression of galectin-9 in cultured human endothelial cells. J. Leukoc. Biol. 2002, 72, 486–491. [Google Scholar] [PubMed]
- Gieseke, F.; Kruchen, A.; Tzaribachev, N.; Bentzien, F.; Dominici, M.; Müller, I. Proinflammatory stimuli induce galectin-9 in human mesenchymal stromal cells to suppress T-cell proliferation. Eur. J. Immunol. 2013, 43, 2741–2749. [Google Scholar] [CrossRef]
- De Kivit, S.; Kostadinova, A.I.; Kerperien, J.; Ayechu Muruzabal, V.; Morgan, M.E.; Knippels, L.M.J.; Kraneveld, A.D.; Garssen, J.; Willemsen, L.E.M. Galectin-9 produced by intestinal epithelial cells enhances aldehyde dehydrogenase activity in dendritic cells in a pi3k- and p38-dependent manner. J. Innate Immun. 2017, 9, 609–620. [Google Scholar] [CrossRef] [PubMed]
- De Kivit, S.; Kraneveld, A.D.; Knippels, L.M.J.; van Kooyk, Y.; Garssen, J.; Willemsen, L.E.M. Intestinal epithelium-derived galectin-9 is involved in the immunomodulating effects of nondigestible oligosaccharides. J. Innate Immun. 2013, 5, 625–638. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; de Fougerolles, A.R.; Stacker, S.A.; Garcia-Aguilar, J.; Hibbs, M.L.; Springer, T.A. Icam-1 (cd54): A counter-receptor for mac-1 (CD11b/CD18). J. Cell Biol. 1990, 111, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- Dapat, I.C.; Pascapurnama, D.N.; Iwasaki, H.; Labayo, H.K.; Chagan-Yasutan, H.; Egawa, S.; Hattori, T. Secretion of galectin-9 as a damp during dengue virus infection in thp-1 cells. Int. J. Mol. Sci. 2017, 18, 1644. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, C.; Sebbag, M.; Clavel, C.; Chapuy-Regaud, S.; Al Badine, R.; Méchin, M.-C.; Vincent, C.; Nachat, R.; Yamada, M.; Takahara, H.; et al. Peptidyl arginine deiminase type 2 (pad-2) and pad-4 but not pad-1, pad-3, and pad-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 2007, 56, 3541–3553. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An american college of rheumatology/european league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef]
- Chalmers, J.D.; Goeminne, P.; Aliberti, S.; McDonnell, M.J.; Lonni, S.; Davidson, J.; Poppelwell, L.; Salih, W.; Pesci, A.; Dupont, L.J.; et al. The bronchiectasis severity index. An international derivation and validation study. Am. J. Respir. Crit. Care Med. 2014, 189, 576–585. [Google Scholar] [CrossRef]
- Donnelly, S.; Roake, W.; Brown, S.; Young, P.; Naik, H.; Wordsworth, P.; Isenberg, D.A.; Reid, K.B.M.; Eggleton, P. Impaired recognition of apoptotic neutrophils by the c1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Eggleton, P.; Gargan, R.; Fisher, D. Rapid method for the isolation of neutrophils in high yield without the use of dextran or density gradient polymers. J. Immunol. Methods 1989, 121, 105–113. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiersma, V.R.; Clarke, A.; Pouwels, S.D.; Perry, E.; Abdullah, T.M.; Kelly, C.; Soyza, A.D.; Hutchinson, D.; Eggleton, P.; Bremer, E. Galectin-9 Is a Possible Promoter of Immunopathology in Rheumatoid Arthritis by Activation of Peptidyl Arginine Deiminase 4 (PAD-4) in Granulocytes. Int. J. Mol. Sci. 2019, 20, 4046. https://doi.org/10.3390/ijms20164046
Wiersma VR, Clarke A, Pouwels SD, Perry E, Abdullah TM, Kelly C, Soyza AD, Hutchinson D, Eggleton P, Bremer E. Galectin-9 Is a Possible Promoter of Immunopathology in Rheumatoid Arthritis by Activation of Peptidyl Arginine Deiminase 4 (PAD-4) in Granulocytes. International Journal of Molecular Sciences. 2019; 20(16):4046. https://doi.org/10.3390/ijms20164046
Chicago/Turabian StyleWiersma, Valerie R., Alex Clarke, Simon D. Pouwels, Elizabeth Perry, Trefa M. Abdullah, Clive Kelly, Anthony De Soyza, David Hutchinson, Paul Eggleton, and Edwin Bremer. 2019. "Galectin-9 Is a Possible Promoter of Immunopathology in Rheumatoid Arthritis by Activation of Peptidyl Arginine Deiminase 4 (PAD-4) in Granulocytes" International Journal of Molecular Sciences 20, no. 16: 4046. https://doi.org/10.3390/ijms20164046
APA StyleWiersma, V. R., Clarke, A., Pouwels, S. D., Perry, E., Abdullah, T. M., Kelly, C., Soyza, A. D., Hutchinson, D., Eggleton, P., & Bremer, E. (2019). Galectin-9 Is a Possible Promoter of Immunopathology in Rheumatoid Arthritis by Activation of Peptidyl Arginine Deiminase 4 (PAD-4) in Granulocytes. International Journal of Molecular Sciences, 20(16), 4046. https://doi.org/10.3390/ijms20164046