Blockade of EGFR Activation Promotes TNF-Induced Lung Epithelial Cell Apoptosis and Pulmonary Injury

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
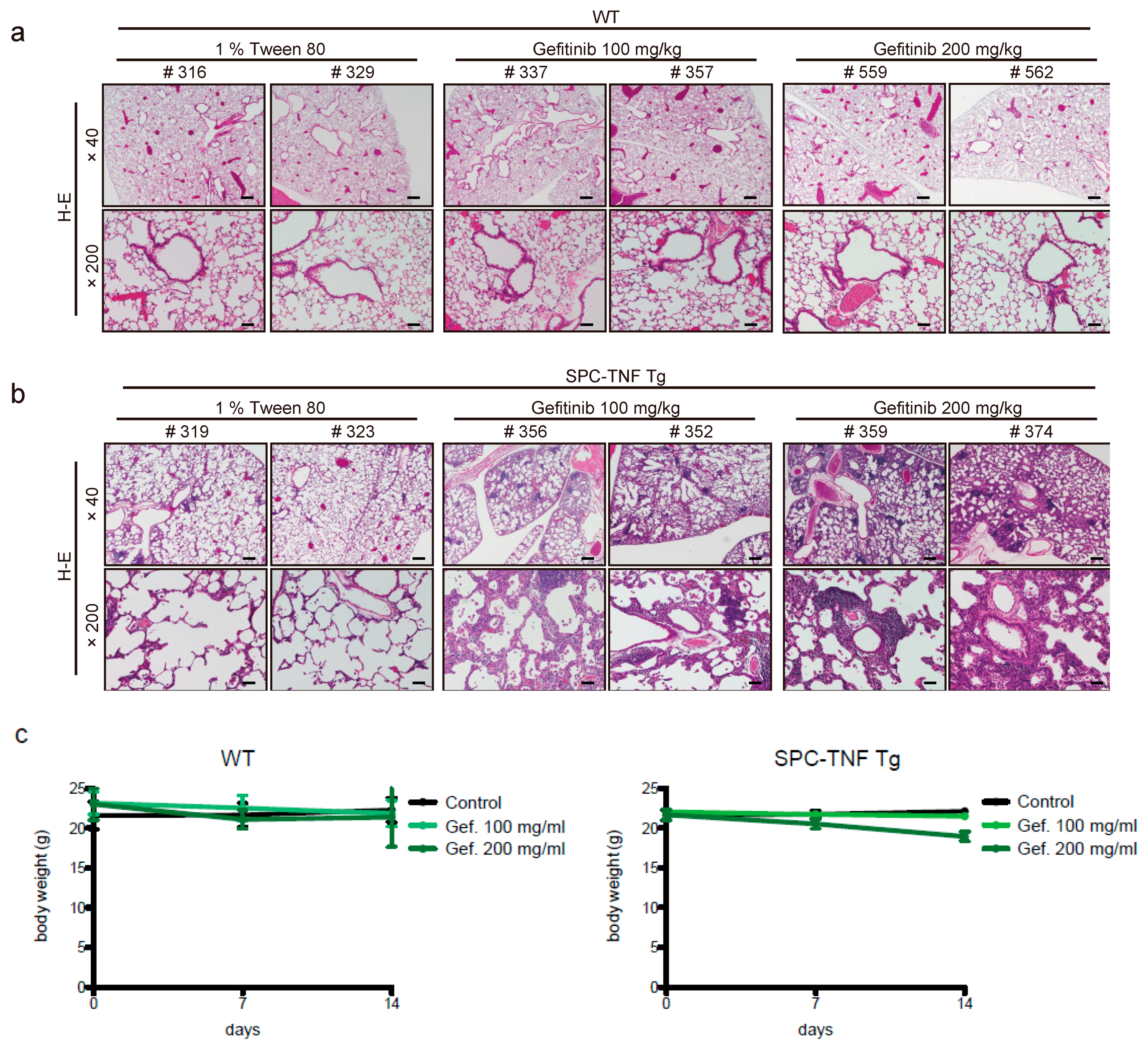
2.1. Gefitinib Treatment Enhanced Lymphocytic Infiltration and Lung Inflammation in SPC-TNF Transgenic Mice
2.2. Indocyanine Green (ICG)-Loaded Liposomes Accumulated in Whole Lungs of Gefitinib-Treated SPC-TNF tg Mice
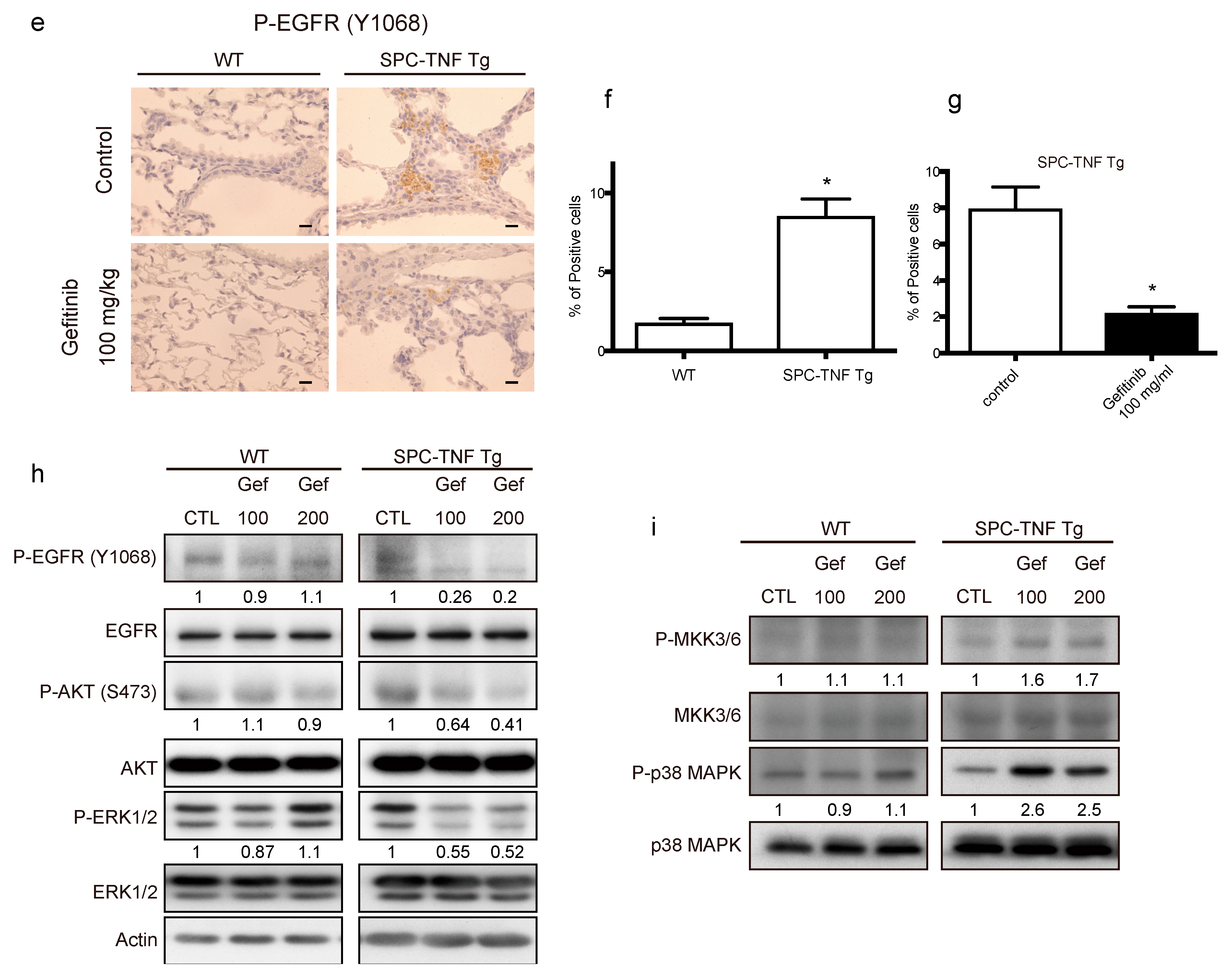
2.3. EGFR Tyrosine Kinase Activity Protected Lung Epithelial Cells from TNF-Induced Apoptosis in the SPC-TNF tg Mice Model
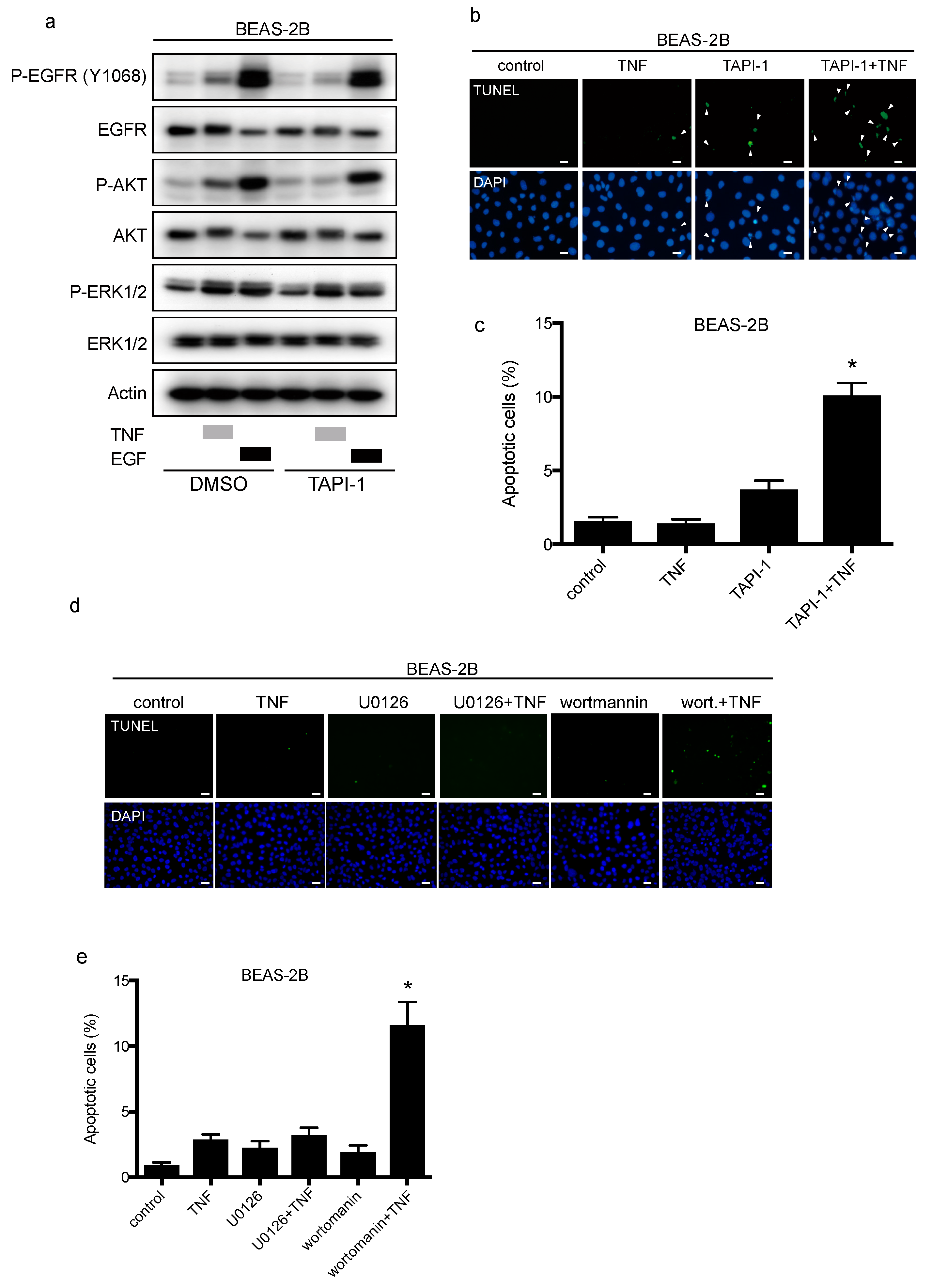
2.4. TNF Stimulated EGFR Phosphorylation and Prevented Apoptosis in Lung Epithelial Cells
2.5. TNF Transactivates EGFR through TACE and Regulates Cell-Survival via the PI3-Kinase/AKT Pathway
2.6. EGFR Ligands, HB-EGF, and TGF-α Dominantly Regulate TNF-Induced EGFR Transactivation via TACE in BEAS-2B Cells
3. Discussion
4. Materials and Methods
4.1. Growth Factors, Antibodies and Inhibitors
4.2. Cell Culture
4.3. Transient Transfection of siRNA
4.4. Cell Lysate Preparation and Western Blot Analysis
4.5. Real-Time RT-PCR
4.6. Mice, EGFR-TKIs Administration, and Tissue Preparations
4.7. Apoptosis Assay
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated egfr. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Yatabe, Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007, 98, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.N.; Lord, S.J.; Gebski, V.; Links, M.; Bray, V.; Gralla, R.J.; Yang, J.C.; Lee, C.K. Risk of treatment-related toxicities from egfr tyrosine kinase inhibitors: A meta-analysis of clinical trials of gefitinib, erlotinib, and afatinib in advanced egfr-mutated non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Okamoto, I.; Yamamoto, N.; Takeda, K.; Tamura, K.; Seto, T.; Ariyoshi, Y.; Fukuoka, M. Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J. Clin. Oncol. 2006, 24, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. Egfr signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Bierman, A.; Yerrapureddy, A.; Reddy, N.M.; Hassoun, P.M.; Reddy, S.P. Epidermal growth factor receptor (egfr) regulates mechanical ventilation-induced lung injury in mice. Transl. Res. 2008, 152, 265–272. [Google Scholar] [CrossRef]
- Carpenter, G. Erbb-4: Mechanism of action and biology. Exp. Cell Res. 2003, 284, 66–77. [Google Scholar] [CrossRef]
- Zwick, E.; Hackel, P.O.; Prenzel, N.; Ullrich, A. The egf receptor as central transducer of heterologous signalling systems. Trends Pharmacol. Sci. 1999, 20, 408–412. [Google Scholar] [CrossRef]
- Shim, J.J.; Dabbagh, K.; Ueki, I.F.; Dao-Pick, T.; Burgel, P.R.; Takeyama, K.; Tam, D.C.; Nadel, J.A. Il-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L134–L140. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Kuo, C.T.; Lin, C.C.; Hsieh, H.L.; Yang, C.M. Il-1beta induces expression of matrix metalloproteinase-9 and cell migration via a c-src-dependent, growth factor receptor transactivation in a549 cells. Br. J. Pharmacol. 2010, 160, 1595–1610. [Google Scholar] [CrossRef]
- Kuwahara, I.; Lillehoj, E.P.; Lu, W.; Singh, I.S.; Isohama, Y.; Miyata, T.; Kim, K.C. Neutrophil elastase induces il-8 gene transcription and protein release through p38/nf-{kappa}b activation via egfr transactivation in a lung epithelial cell line. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L407–L416. [Google Scholar] [CrossRef]
- Lee, C.W.; Lin, C.C.; Lin, W.N.; Liang, K.C.; Luo, S.F.; Wu, C.B.; Wang, S.W.; Yang, C.M. Tnf-alpha induces mmp-9 expression via activation of src/egfr, pdgfr/pi3k/akt cascade and promotion of nf-kappab/p300 binding in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L799–L812. [Google Scholar] [CrossRef]
- Edelblum, K.L.; Yan, F.; Yamaoka, T.; Polk, D.B. Regulation of apoptosis during homeostasis and disease in the intestinal epithelium. Inflamm. Bowel Dis. 2006, 12, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of erk and jnk-p38 map kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Yan, F.; John, S.K.; Wilson, G.; Jones, D.S.; Washington, M.K.; Polk, D.B. Kinase suppressor of ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J. Clin. Investig. 2004, 114, 1272–1280. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the jnk group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Yamaoka, T.; Yan, F.; Cao, H.; Hobbs, S.S.; Dise, R.S.; Tong, W.; Polk, D.B. Transactivation of egf receptor and erbb2 protects intestinal epithelial cells from tnf-induced apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11772–11777. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Araki, K.; Vesin, C.; Garcia, I.; Kapanci, Y.; Whitsett, J.A.; Piguet, P.F.; Vassalli, P. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J. Clin. Invest. 1995, 96, 250–259. [Google Scholar] [CrossRef]
- Hirai, M.; Minematsu, H.; Kondo, N.; Oie, K.; Igarashi, K.; Yamazaki, N. Accumulation of liposome with sialyl lewis x to inflammation and tumor region: Application to in vivo bio-imaging. Biochem. Biophys. Res. Commun. 2007, 353, 553–558. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ask1, a mammalian mapkkk that activates sapk/jnk and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Jarpe, M.B.; Widmann, C.; Knall, C.; Schlesinger, T.K.; Gibson, S.; Yujiri, T.; Fanger, G.R.; Gelfand, E.W.; Johnson, G.L. Anti-apoptotic versus pro-apoptotic signal transduction: Checkpoints and stop signs along the road to death. Oncogene 1998, 17, 1475–1482. [Google Scholar] [CrossRef]
- Grethe, S.; Ares, M.P.; Andersson, T.; Porn-Ares, M.I. P38 mapk mediates tnf-induced apoptosis in endothelial cells via phosphorylation and downregulation of bcl-x(l). Exp. Cell Res. 2004, 298, 632–642. [Google Scholar] [CrossRef]
- Baselga, J. The egfr as a target for anticancer therapy--focus on cetuximab. Eur. J. Cancer 2001, 37, 16–22. [Google Scholar] [CrossRef]
- Shao, M.X.; Ueki, I.F.; Nadel, J.A. Tumor necrosis factor alpha-converting enzyme mediates muc5ac mucin expression in cultured human airway epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11618–11623. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Blanchet, S.; Ramgolam, K.; Baulig, A.; Marano, F.; Baeza-Squiban, A. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 421–427. [Google Scholar] [CrossRef]
- Rumelhard, M.; Ramgolam, K.; Auger, F.; Dazy, A.C.; Blanchet, S.; Marano, F.; Baeza-Squiban, A. Effects of pm2.5 components in the release of amphiregulin by human airway epithelial cells. Toxicol. Lett. 2007, 168, 155–164. [Google Scholar] [CrossRef]
- Kheradmand, F.; Folkesson, H.G.; Shum, L.; Derynk, R.; Pytela, R.; Matthay, M.A. Transforming growth factor-alpha enhances alveolar epithelial cell repair in a new in vitro model. Am. J. Physiol. 1994, 267, L728–L738. [Google Scholar] [CrossRef]
- Ryan, R.M.; Mineo-Kuhn, M.M.; Kramer, C.M.; Finkelstein, J.N. Growth factors alter neonatal type ii alveolar epithelial cell proliferation. Am. J. Physiol. 1994, 266, L17–L22. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef]
- Madtes, D.K.; Rubenfeld, G.; Klima, L.D.; Milberg, J.A.; Steinberg, K.P.; Martin, T.R.; Raghu, G.; Hudson, L.D.; Clark, J.G. Elevated transforming growth factor-alpha levels in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1998, 158, 424–430. [Google Scholar] [CrossRef][Green Version]
- Chesnutt, A.N.; Kheradmand, F.; Folkesson, H.G.; Alberts, M.; Matthay, M.A. Soluble transforming growth factor-alpha is present in the pulmonary edema fluid of patients with acute lung injury. Chest 1997, 111, 652–656. [Google Scholar] [CrossRef]
- Venkataraman, T.; Coleman, C.M.; Frieman, M.B. Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J. Virol. 2017, 91, e00182-17. [Google Scholar] [CrossRef]
- Frey, M.R.; Edelblum, K.L.; Mullane, M.T.; Liang, D.; Polk, D.B. The erbb4 growth factor receptor is required for colon epithelial cell survival in the presence of tnf. Gastroenterology 2009, 136, 217–226. [Google Scholar] [CrossRef]
- Stahn, R.; Grittner, C.; Zeisig, R.; Karsten, U.; Felix, S.B.; Wenzel, K. Sialyl lewis(x)-liposomes as vehicles for site-directed, e-selectin-mediated drug transfer into activated endothelial cells. Cell. Mol. Life Sci. 2001, 58, 141–147. [Google Scholar] [CrossRef]
- Maehara, A.; Nishida, K.; Furutani, M.; Matsumoto, E.; Ohtsuka, A.; Ninomiya, Y.; Oohashi, T. Light and electron microscopic detection of inflammation-targeting liposomes encapsulating high-density colloidal gold in arthritic mice. Inflamm. Res. 2014, 63, 139–147. [Google Scholar] [CrossRef]
- Lee, J.; Ho, W.H.; Maruoka, M.; Corpuz, R.T.; Baldwin, D.T.; Foster, J.S.; Goddard, A.D.; Yansura, D.G.; Vandlen, R.L.; Wood, W.I.; et al. Il-17e, a novel proinflammatory ligand for the il-17 receptor homolog il-17rh1. J. Biol. Chem. 2001, 276, 1660–1664. [Google Scholar] [CrossRef]
- Linden, A.; Hoshino, H.; Laan, M. Airway neutrophils and interleukin-17. Eur. Respir. J. 2000, 15, 973–977. [Google Scholar] [CrossRef]
- Chakir, J.; Shannon, J.; Molet, S.; Fukakusa, M.; Elias, J.; Laviolette, M.; Boulet, L.P.; Hamid, Q. Airway remodeling-associated mediators in moderate to severe asthma: Effect of steroids on tgf-beta, il-11, il-17, and type i and type iii collagen expression. J. Allergy Clin. Immunol. 2003, 111, 1293–1298. [Google Scholar] [CrossRef]
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mrna and protein levels of il-17a in sputum of stable cystic fibrosis patients. Respir. Res. 2010, 11, 177. [Google Scholar] [CrossRef]
- Iyoda, M.; Shibata, T.; Kawaguchi, M.; Hizawa, N.; Yamaoka, T.; Kokubu, F.; Akizawa, T. Il-17a and il-17f stimulate chemokines via mapk pathways (erk1/2 and p38 but not jnk) in mouse cultured mesangial cells: Synergy with tnf-alpha and il-1beta. Am. J. Physiol. Renal. Physiol. 2010, 298, F779–F787. [Google Scholar] [CrossRef]
- Kudoh, S.; Kato, H.; Nishiwaki, Y.; Fukuoka, M.; Nakata, K.; Ichinose, Y.; Tsuboi, M.; Yokota, S.; Nakagawa, K.; Suga, M.; et al. Interstitial lung disease in japanese patients with lung cancer: A cohort and nested case-control study. Am. J. Respir. Crit. Care Med. 2008, 177, 1348–1357. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Andersen-Ranberg, K.; Jeune, B.; Pedersen, A.N.; Skinhoj, P.; Pedersen, B.K. A high plasma concentration of tnf-alpha is associated with dementia in centenarians. J. Gerontol. A Biol. Sci. Med. Sci. 1999, 54, M357–M364. [Google Scholar] [CrossRef]
- Tanni, S.E.; Pelegrino, N.R.; Angeleli, A.Y.; Correa, C.; Godoy, I. Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in copd patients. J. Inflamm. (Lond) 2010, 7, 29. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type | SPC-TNF tg | p | |
|---|---|---|---|
| Ifng | 1 | 0.9 ± 0.1 | 0.53 |
| Il1b | 1 | 2.1 ± 0.4 | 0.011 |
| Il6 | 1 | 0.5 ± 0.0 | <0.0001 |
| Il10 | 1 | 2.6 ± 0.2 | 0.0004 |
| Il17a | 1 | 2.6 ± 0.4 | 0.0088 |
| Il21 | 1 | 0.6 ± 0.1 | 0.0015 |
| Il23a | 1 | 1.9 ± 0.3 | 0.01 |
| Tnf | 1 | 825.3 ± 110 | 0.0002 |
| Tgfb1 | 1 | 1.6 ± 0.4 | <0.0001 |
| Foxp3 | 1 | 5.1 ± 0.6 | 0.0003 |
| Gata3 | 1 | 0.6 ± 0.0 | <0.0001 |
| Tbet | 1 | 0.8 ± 0.1 | 0.37 |
| Gm-csf | 1 | 0.7 ± 0.1 | 0.11 |
| Egfr | 1 | 0.8 ± 0.2 | 0.06 |
| Control | Gef 100 mg/kg | p | Gef 200 mg/kg | p | |
|---|---|---|---|---|---|
| Ifng | 1 | 0.89 ± 0.1 | 0.37 | 0.69 ± 0.2 | 0.086 |
| Il1b | 1 | 1.9 ± 0.1 | <0.0001 | 13.7 ± 2.3 | 0.0008 |
| Il6 | 1 | 1.1 ± 0.1 | 0.31 | 1.7 ± 0.1 | <0.0001 |
| Il10 | 1 | 1.3 ± 0.1 | 0.0093 | 1.7 ± 0.1 | 0.0005 |
| Il17a | 1 | 1.0 ± 0.1 | 0.93 | 2.6 ± 0.5 | 0.024 |
| Il21 | 1 | 1.1 ± 0.1 | 0.27 | 1.1 ± 0.1 | 0.15 |
| Il23a | 1 | 1.1 ± 0.0 | 0.086 | 1.0 ± 0.0 | 0.86 |
| Tnf | 1 | 1.4 ± 0.0 | 0.17 | 1.4 ± 0.2 | 0.093 |
| Tgfb1 | 1 | 0.93 ± 0.0 | 0.076 | 0.97 ± 0.0 | 0.34 |
| Foxp3 | 1 | 1.6 ± 0.1 | <0.0001 | 1.6 ± 0.1 | 0.0051 |
| Gata3 | 1 | 0.99 ± 0.1 | 0.97 | 0.6 ± 0.1 | 0.0015 |
| Tbet | 1 | 0.82 ± 0.1 | 0.017 | 0.5 ± 0.1 | 0.0012 |
| Gm-csf | 1 | 0.80 ± 0.1 | 0.057 | 0.8 ± 0.1 | 0.16 |
| Egfr | 1 | 1.0 ± 0.1 | 0.94 | 1.0 ± 0.2 | 0.87 |
| Control | Gef 100 mg/kg | p | Gef 200 mg/kg | p | |
|---|---|---|---|---|---|
| Ifng | 1 | 0.8 ± 0.0 | 0.0004 | 0.8 ± 0.1 | 0.034 |
| Il1b | 1 | 0.8 ± 0.1 | 0.095 | 1.2 ± 0.1 | 0.0011 |
| Il6 | 1 | 1.3 ± 0.0 | <0.0001 | 1.9 ± 0.2 | 0.001 |
| Il10 | 1 | 1.0 ± 0.1 | 0.96 | 1.6 ± 0.1 | 0.0003 |
| Il17a | 1 | 1.5 ± 0.1 | 0.0018 | 5.8 ± 0.4 | 0.014 |
| Il21 | 1 | 1.3 ± 0.1 | 0.027 | 1.5 ± 0.1 | 0.0046 |
| Il23a | 1 | 0.9 ± 0.1 | 0.16 | 1.1 ± 0.1 | 0.11 |
| Tnf | 1 | 0.9 ± 0.0 | 0.0057 | 1.2 ± 0.1 | 0.0001 |
| Tgfb1 | 1 | 1.2 ± 0.1 | 0.0042 | 1.9 ± 0.2 | 0.003 |
| Foxp3 | 1 | 0.9 ± 0.0 | 0.24 | 1.1 ± 0.2 | 0.042 |
| Gata3 | 1 | 1.0 ± 0.1 | 0.69 | 0.8 ± 0.0 | <0.0001 |
| Tbet | 1 | 1.1 ± 0.0 | 0.32 | 1.2 ± 0.2 | 0.49 |
| Gm-csf | 1 | 0.7 ± 0.0 | <0.0001 | 1.1 ± 0.1 | 0.22 |
| Egfr | 1 | 1.1 ± 0.1 | 0.53 | 1.1 ± 0.0 | 0.14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaoka, T.; Arata, S.; Homma, M.; Homma, T.; Kusumoto, S.; Ando, K.; Manabe, R.; Kishino, Y.; Ohba, M.; Tsurutani, J.; et al. Blockade of EGFR Activation Promotes TNF-Induced Lung Epithelial Cell Apoptosis and Pulmonary Injury. Int. J. Mol. Sci. 2019, 20, 4021. https://doi.org/10.3390/ijms20164021
Yamaoka T, Arata S, Homma M, Homma T, Kusumoto S, Ando K, Manabe R, Kishino Y, Ohba M, Tsurutani J, et al. Blockade of EGFR Activation Promotes TNF-Induced Lung Epithelial Cell Apoptosis and Pulmonary Injury. International Journal of Molecular Sciences. 2019; 20(16):4021. https://doi.org/10.3390/ijms20164021
Chicago/Turabian StyleYamaoka, Toshimitsu, Satoru Arata, Mayumi Homma, Tetsuya Homma, Sojiro Kusumoto, Koichi Ando, Ryou Manabe, Yasunari Kishino, Motoi Ohba, Junji Tsurutani, and et al. 2019. "Blockade of EGFR Activation Promotes TNF-Induced Lung Epithelial Cell Apoptosis and Pulmonary Injury" International Journal of Molecular Sciences 20, no. 16: 4021. https://doi.org/10.3390/ijms20164021
APA StyleYamaoka, T., Arata, S., Homma, M., Homma, T., Kusumoto, S., Ando, K., Manabe, R., Kishino, Y., Ohba, M., Tsurutani, J., Takimoto, M., Ohmori, T., & Sagara, H. (2019). Blockade of EGFR Activation Promotes TNF-Induced Lung Epithelial Cell Apoptosis and Pulmonary Injury. International Journal of Molecular Sciences, 20(16), 4021. https://doi.org/10.3390/ijms20164021