Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC)


Abstract
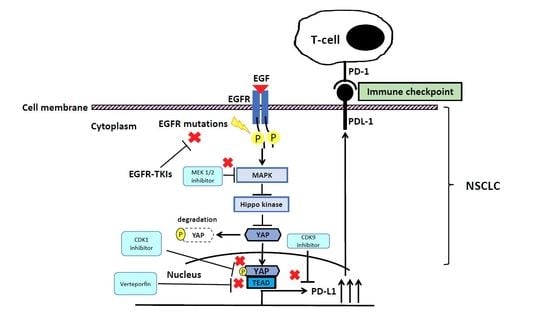
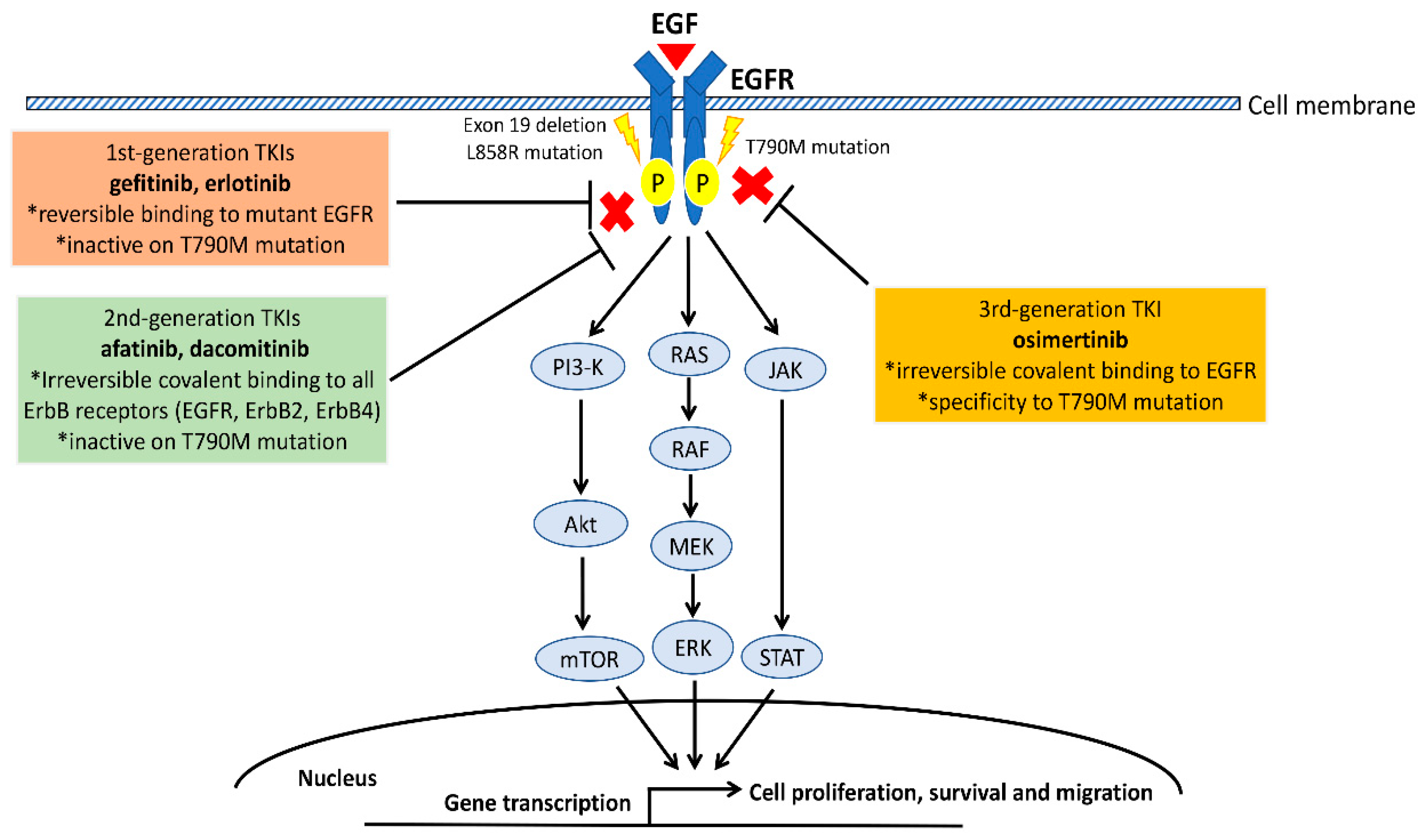
1. The Epidermal Growth Factor Receptor (EGFR) Pathway in Non-Small Cell Lung Cancer (NSCLC)
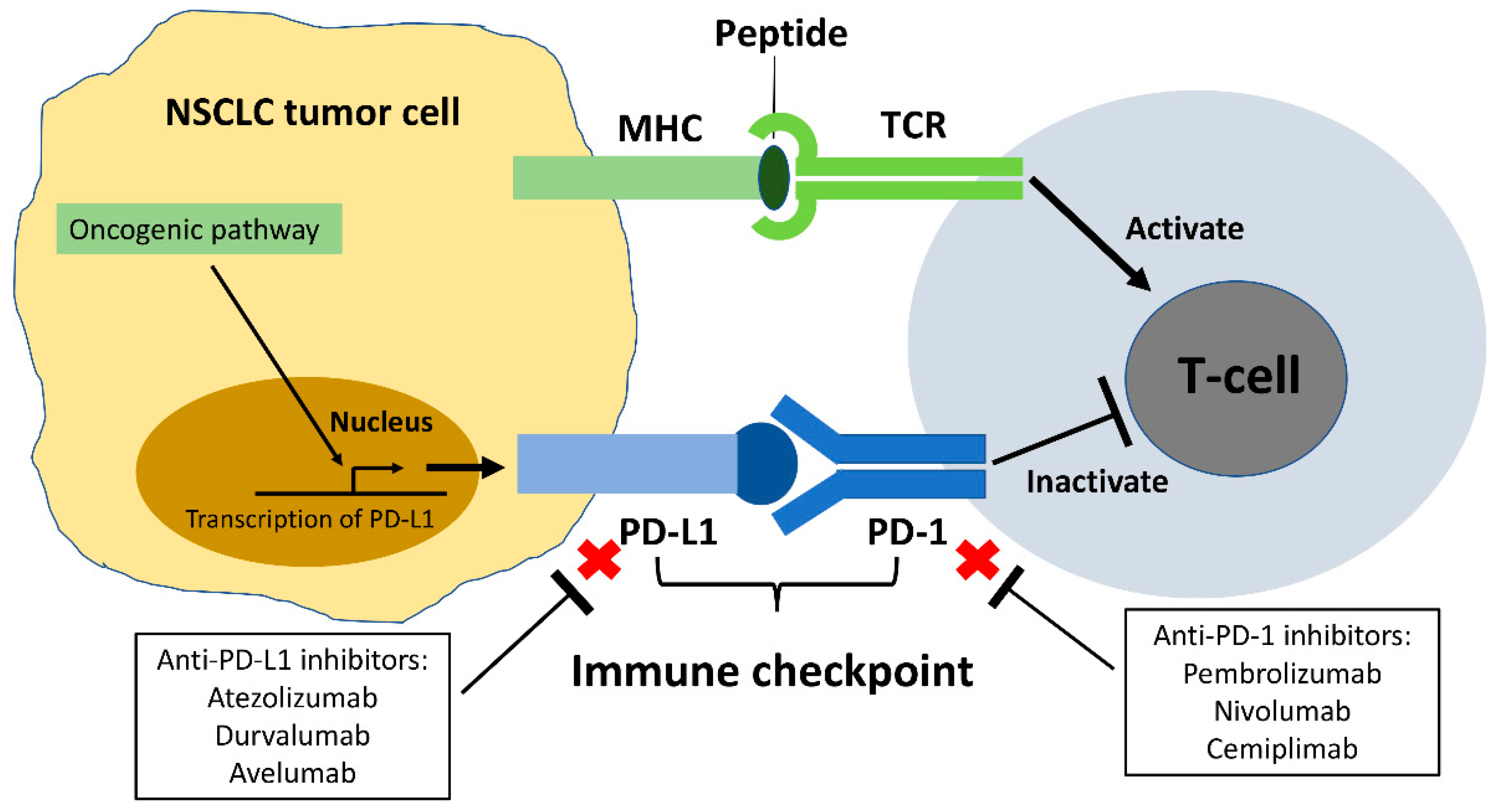
2. Programmed Cell Death Protein-1 (PD-1)/Programmed Death-Ligand 1 (PD-L1) Immune Checkpoint in NSCLC
3. Yes-Associated Protein (YAP) in Human NSCLC
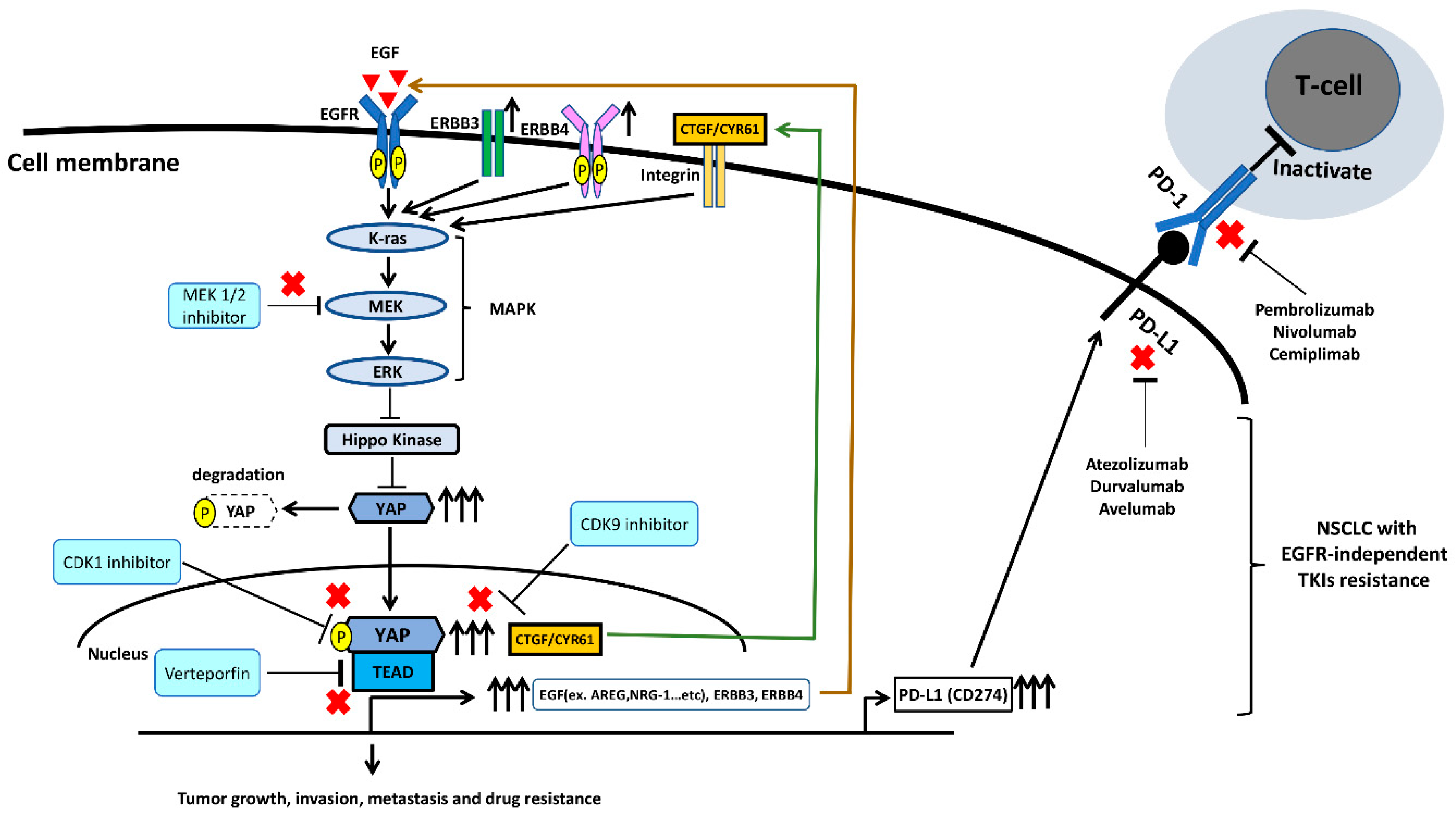
4. The Interaction between EGFR and YAP Signaling Pathways in NSCLC
5. The EGFR and YAP Signaling Pathways Regulate PD-L1 Expression in NSCLC
6. Future Perspectives
6.1. The Role of Anti-PD-1/PD-L1 ICIs in EGFR Mutant NSCLC
6.2. Targeting YAP Thrapy in Combination with ICIs in EGFR-TKI Resistant NSCLC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, S9–S15. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; Di Maria, M.V.; Veve, R.; Bremmes, R.M.; Barón, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Ha, S.Y.; Choi, S.J.; Cho, J.H.; Choi, H.J.; Lee, J.; Jung, K.; Irwin, D.; Liu, X.; Lira, M.E.; Mao, M.; et al. Lung cancer in never-smoker Asian females is driven by oncogenic mutations, most often involving EGFR. Oncotarget 2015, 6, 5465–5474. [Google Scholar] [CrossRef]
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: Perplexity and solution (Review). Oncol. Rep. 2017, 37, 1347–1358. [Google Scholar] [CrossRef]
- Saber, A.; van der Wekken, A.; Hiltermann, T.J.; Kok, K.; Van den Berg, A.; Groen, H. Genomic aberrations guiding treatment of non-small cell lung cancer patients. Cancer Treat. Commun. 2015, 4, 23–33. [Google Scholar] [CrossRef]
- Chapman, A.M.; Sun, K.Y.; Ruestow, P.; Cowan, D.M.; Mad, A.K. Lung cancer mutation profile of EGFR, ALK, and KRAS: Meta-analysis and comparison of never and ever smokers. Lung Cancer 2016, 102, 122–134. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Soulières, D.; Moecks, J.; Bara, I.; Mok, T.; Klughammer, B. Pooled analysis of clinical outcome for EGFR TKI-treated patients with EGFR mutation-positive NSCLC. J. Cell. Mol. Med. 2014, 18, 1519–1539. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tamiya, A.; Nakahama, K.; Naoki, Y.; Kanazu, M.; Omachi, N.; Okishio, K.; Kasai, T.; Atagi, S. Impact of metastatic status on the prognosis of EGFR mutation-positive non-small cell lung cancer patients treated with first-generation EGFR-tyrosine kinase inhibitors. Oncol. Lett. 2017, 14, 7589–7596. [Google Scholar] [CrossRef]
- Hsu, P.C.; Liu, C.Y.; Li, S.H.; Huang, S.H.; Wang, C.L.; Kou, C.H.; Chung, F.T.; Chen, C.H.; Yu, C.T.; Yang, C.T. Efficacy of platinum-based combination chemotherapy in advanced lung adenocarcinoma harboring sensitive epidermal growth factor receptor (EGFR) mutations with acquired resistance to first-line EGFR tyrosine kinase inhibitor (TKI). Cancer Treat. Commun. 2016, 9, 48–55. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in this Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Togashi, Y.; Yatabe, Y.; Mizuuchi, H.; Jangchul, P.; Kondo, C.; Shimoji, M.; Sato, K.; Suda, K.; Tomizawa, K.; et al. EGFR Exon 18 Mutations in Lung Cancer: Molecular Predictors of Augmented Sensitivity to Afatinib or Neratinib as Compared with First- or Third-Generation TKIs. Clin. Cancer Res. 2015, 21, 5305–5313. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomized phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; Obyrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients With Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Yoshida, T.; Zhang, G.; Haura, E.B. Targeting epidermal growth factor receptor: Central signaling kinase in lung cancer. Biochem. Pharm. 2010, 80, 613–623. [Google Scholar] [CrossRef]
- Hashida, S.; Soh, J.; Toyooka, S.; Tanaka, T.; Furukawa, M.; Shien, K.; Yamamoto, H.; Asano, H.; Tsukuda, K.; Hagiwara, K.; et al. Presence of the minor EGFR T790M mutation is associated with drug-sensitive EGFR mutations in lung adenocarcinoma patients. Oncol. Rep. 2014, 32, 145–152. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; Obyrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.; Xiong, Z.C.; Sun, X.; Zhang, S.L.; Ma, J.T.; Han, C.B. Meta-analysis of the impact of de novo and acquired EGFR T790M mutations on the prognosis of patients with non-small cell lung cancer receiving EGFR-TKIs. Onco. Targets Ther. 2017, 10, 2267–2279. [Google Scholar] [CrossRef]
- Jänne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T Cell Co-receptors for Cancer Therapy. Immun. 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Perrotta, F.; Rocco, D.; Vitiello, F.; De Palma, R.; Guerra, G.; De Luca, A.; Navani, N.; Bianco, A. Immune Checkpoint Blockade for Advanced NSCLC: A New Landscape for Elderly Patients. Int. J. Mol. Sci. 2019, 20, 2258. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Zielinski, C.; Knapp, S.; Mascaux, C.; Hirsch, F. Rationale for targeting the immune system through checkpoint molecule blockade in the treatment of non-small-cell lung cancer. Ann. Oncol. 2013, 24, 1170–1179. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Tunger, A.; Kießler, M.; Wehner, R.; Temme, A.; Meier, F.; Bachmann, M.; Schmitz, M. Immune Monitoring of Cancer Patients Prior to and During CTLA-4 or PD-1/PD-L1 Inhibitor Treatment. Biomedicines 2018, 6, 26. [Google Scholar] [CrossRef]
- Cooper, W.A.; Tran, T.; Vilain, R.E.; Madore, J.; Selinger, C.I.; Kohonen-Corish, M.; Yip, P.; Yu, B.; O’Toole, S.A.; McCaughan, B.C.; et al. PD-L1 expression is a favorable prognostic factor in early stage non-small cell carcinoma. Lung Cancer 2015, 89, 181–188. [Google Scholar] [CrossRef]
- Ogunleyea, F.; Blankenshipa, L.; Millisorb, V.; Anderson, J.; Jaiyesimi, I. Programmed cell death-1/Programmed cell death ligand-1(PD-1/PD-L1) inhibitors, heralding a new era of immunotherapy in the management of advanced Non-Small Cell Lung Cancer (NSCLC). Cancer Treat. Res. Commun. 2017, 12, 6–13. [Google Scholar] [CrossRef]
- Tang, D.; Zhao, D.; Wu, Y.; Yao, R.; Zhou, L.; Lu, L.; Gao, W.; Sun, Y. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef]
- Passiglia, F.; Bronte, G.; Bazan, V.; Natoli, C.; Rizzo, S.; Galvano, A.; Listì, A.; Cicero, G.; Rolfo, C.; Santini, D.; et al. PD-L1 expression as predictive biomarker in patients with NSCLC: A pooled analysis. Oncotarget 2016, 7, 19738–19747. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Perez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Corrales, L.; Scilla, K.; Caglevic, C.; Miller, K.; Oliveira, J.; Rolfo, C. Immunotherapy in Lung Cancer: A New Age in Cancer Treatment. Adv. Exp. Med. Biol. 2018, 995, 65–95. [Google Scholar]
- Assi, H.I.; Kamphorst, A.O.; Moukalled, N.M.; Ramalingam, S.S. Immune checkpoint inhibitors in advanced non-small cell lung cancer. Cancer. 2018, 124, 248–261. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, Q.; Liu, A.M.; Tang, C.; Gong, Y.; Bian, J.; Luk, J.M.; Xu, Z.; Chen, J. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol Rep. 2013, 29, 840–846. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef]
- Guo, L.; Teng, L. YAP/TAZ for cancer therapy: Opportunities and challenges (review). Int. J. Oncol. 2015, 46, 1444–1452. [Google Scholar] [CrossRef]
- Chen, L.; Feng, P.; Peng, A.; Qiu, X.; Zhu, X.; He, S.; Zhou, D. cAMP response element-binding protein and Yes-associated protein form a feedback loop that promotes neurite outgrowth. J. Cell. Mol. Med. 2018, 22, 374–381. [Google Scholar] [CrossRef]
- Dai, Y.; Jablons, D.; You, L. Hippo pathway in lung development. J. Thorac. Dis. 2017, 9, 2246–2250. [Google Scholar] [CrossRef]
- Fu, S.L.; Zhao, W.Y.; Zhang, W.J.; Song, H.; Ji, H.B.; Tang, N. Hippo signaling pathway in lung development, regeneration, and diseases. Yi. Chuan. 2017, 39, 597–606. [Google Scholar]
- Wang, Y.; Dong, Q.; Zhang, Q.; Li, Z.; Wang, E.; Qiu, X. Overexpression of Yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010, 101, 1279–1285. [Google Scholar] [CrossRef]
- Guo, J.; Wu, Y.; Yang, L.; Du, J.; Gong, K.; Chen, W.; Dai, J.; Li, X.; Xi, S. Repression of YAP by NCTD disrupts NSCLC progression. Oncotarget 2017, 8, 2307–2319. [Google Scholar] [CrossRef]
- Ye, X.Y.; Luo, Q.Q.; Xu, Y.H.; Tang, N.W.; Niu, X.M.; Li, Z.M.; Shen, S.P.; Lu, S.; Chen, Z.W. 17-AAG suppresses growth and invasion of lung adenocarcinoma cells via regulation of the LATS1/YAP pathway. J. Cell. Mol. Med. 2015, 19, 651–663. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef]
- Jin, D.; Wu, Y.; Shao, C.; Gao, Y.; Wang, D.; Guo, J. Norcantharidin reverses cisplatin resistance and inhibits the epithelial mesenchymal transition of human non-small lung cancer cells by regulating the YAP pathway. Oncol. Rep. 2018, 40, 609–620. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Hsu, P.C.; Tian, B.; Yang, Y.L.; Wang, Y.C.; Liu, S.; Urisman, A.; Yang, C.T.; Xu, Z.; Jablons, D.M.; You, L. Cucurbitacin E inhibits the Yes-associated protein signaling pathway and suppresses brain metastasis of human non-small cell lung cancer in a murine model. Oncol. Rep. 2019. [Google Scholar] [CrossRef]
- Dubois, F.; Keller, M.; Calvayrac, O.; Soncin, F.; Hoa, L.; Hergovich, A.; Parrini, M.C.; Mazières, J.; Vaisse-Lesteven, M.; Camonis, J.; et al. RASSF1A Suppresses the Invasion and Metastatic Potential of Human Non-Small Cell Lung Cancer Cells by Inhibiting YAP Activation through the GEF-H1/RhoB Pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massagué, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009, 138, 51–62. [Google Scholar] [CrossRef]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014, 156, 1002–1016. [Google Scholar] [CrossRef]
- Hsu, P.C.; Miao, J.; Huang, Z.; Yang, Y.L.; Xu, Z.; You, J.; Dai, Y.; Yeh, C.C.; Chan, G.; Liu, S.; et al. Inhibition of yes-associated protein suppresses brain metastasis of human lung adenocarcinoma in a murine model. J. Cell. Mol. Med. 2018, 22, 3073–3085. [Google Scholar] [CrossRef]
- Kitano, H.; Chung, J.Y.; Ylaya, K.; Conway, C.; Takikita, M.; Fukuoka, J.; Doki, Y.; Hanaoka, J.; Hewitt, S.M. Profiling of phospho-AKT, phospho-mTOR, phospho-MAPK and EGFR in non-small cell lung cancer. J Histochem Cytochem. 2014, 62, 335–346. [Google Scholar] [CrossRef]
- Sun, K.K.; Zhong, N.; Yang, Y.; Zhao, L.; Jiao, Y. Enhanced radiosensitivity of NSCLC cells by transducer of erbB2.1 (TOB1) through modulation of the MAPK/ERK pathway. Oncol Rep. 2013, 29, 2385–2391. [Google Scholar] [CrossRef]
- Lu, M.; Liu, B.; Xiong, H.; Wu, F.; Hu, C.; Liu, P. Trans-3,5,4′-trimethoxystilbene reduced gefitinib resistance in NSCLCs via suppressing MAPK/Akt/Bcl-2 pathway by upregulation of miR-345 and miR-498. J Cell Mol Med. 2019, 23, 2431–2441. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Wargo, J.A.; Bivona, T.G. YAP in MAPK pathway targeted therapy resistance. Cell Cycle 2015, 14, 1765–1766. [Google Scholar] [CrossRef]
- You, B.; Yang, Y.L.; Xu, Z.; Dai, Y.; Liu, S.; Mao, J.H.; Tetsu, O.; Li, H.; Jablons, D.M.; You, L. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015, 6, 4357–4368. [Google Scholar] [CrossRef]
- McGowan, M.; Kleinberg, L.; Halvorsen, A.R.; Helland, A.; Brustugun, O.T. NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer 2017, 8, 497–504. [Google Scholar]
- Huang, J.M.; Nagatomo, I.; Suzuki, E.; Mizuno, T.; Kumagai, T.; Berezov, A.; Zhang, H.; Karlan, B.; Greene, M.I.; Wang, Q. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene. 2013, 32, 2220–2229. [Google Scholar] [CrossRef]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef]
- Haskins, J.W.; Nguyen, D.X.; Stern, D.F. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci. Signal. 2014, 7, ra116. [Google Scholar] [CrossRef]
- Sudol, M. Neuregulin 1-activated ERBB4 as a "dedicated" receptor for the Hippo-YAP pathway. Sci. Signal. 2014, 7, pe29. [Google Scholar] [CrossRef]
- He, C.; Lv, X.; Hua, G.; Lele, S.M.; Remmenga, S.; Dong, J.; Davis, J.S.; Wang, C. YAP forms autocrine loops with the ERBB pathway to regulate ovarian cancer initiation and progression. Oncogene. 2015, 34, 6040–6054. [Google Scholar] [CrossRef]
- Reddy, B.V.; Irvine, K.D. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell 2013, 24, 459–471. [Google Scholar] [CrossRef]
- Yang, S.; Ji, M.; Zhang, L.; Chen, Y.; Wennmann, D.O.; Kremerskothen, J.; Dong, J. Phosphorylation of KIBRA by the extracellular signal-regulated kinase (ERK)-ribosomal S6 kinase (RSK) cascade modulates cell proliferation and migration. Cell Signal. 2014, 26, 343–351. [Google Scholar] [CrossRef]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef]
- Tan, W.L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.T.; Tan, E.H.; Zhai, W.; Hillmer, A.M.; Tam, W.L.; et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Greten, F.R. YAP1 takes over when oncogenic K-Ras slumbers. Cell 2014, 158, 11–12. [Google Scholar] [CrossRef]
- Thakur, A.; Schalk, D.; Tomaszewski, E.; Kondadasula, S.V.; Yano, H.; Sarkar, F.H.; Lum, L.G. Microenvironment generated during EGFR targeted killing of pancreatic tumor cells by ATC inhibits myeloid-derived suppressor cells through COX2 and PGE2 dependent pathway. J. Transl. Med. 2013, 11, 35. [Google Scholar] [CrossRef]
- Feng, P.H.; Yu, C.T.; Chen, K.Y.; Luo, C.S.; Wu, S.M.; Liu, C.Y.; Kuo, L.W.; Chan, Y.F.; Chen, T.T.; Chang, C.C.; et al. S100A9+ MDSC and TAM-mediated EGFR-TKI resistance in lung adenocarcinoma: The role of RELB. Oncotarget 2018, 9, 7631. [Google Scholar]
- Gong, X.; Li, X.; Jiang, T.; Xie, H.; Zhu, Z.; Zhou, F.; Zhou, C. Combined Radiotherapy and Anti-PD-L1 Antibody Synergistically Enhances Antitumor Effect in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1085–1097. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.A. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef]
- Zhang, H.; Dutta, P.; Liu, J.; Sabri, N.; Song, Y.; Li, W.X.; Li, J. Tumour cell-intrinsic CTLA4 regulates PD-L1 expression in non-small cell lung cancer. J. Cell Mol. Med. 2019, 23, 535–542. [Google Scholar] [CrossRef]
- Gebhardt, T.; Harvey, K.F. Hippo Wades into Cancer Immunology. Dev. Cell 2016, 39, 635–637. [Google Scholar] [CrossRef]
- Wang, S.; Xie, F.; Chu, F.; Zhang, Z.; Yang, B.; Dai, T.; Gao, L.; Wang, L.; Ling, L.; Jia, J.; et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKε-mediated phosphorylation. Nat. Immunol. 2017, 18, 733–743. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef]
- Hsu, P.C.; Miao, J.; Wang, Y.C.; Zhang, W.Q.; Yang, Y.L.; Wang, C.W.; Yang, C.T.; Huang, Z.; You, J.; Xu, Z.; et al. Inhibition of yes-associated protein down-regulates PD-L1 (CD274) expression in human malignant pleural mesothelioma. J. Cell. Mol. Med. 2018, 22, 3139–3148. [Google Scholar] [CrossRef]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef]
- Hsu, P.C.; Yang, C.T.; Jablons, D.M.; You, L. The Role of Yes-Associated Protein (YAP) in Regulating Programmed Death-Ligand 1 (PD-L1) in Thoracic Cancer. Biomedicines 2018, 6, 114. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Taha, Z.; Janse van Rensburg, H.J.; Yang, X. The Hippo Pathway: Immunity and Cancer. Cancers (Basel) 2018, 10, 94. [Google Scholar] [CrossRef]
- Danovi, S.A.; Rossi, M.; Gudmundsdottir, K.; Yuan, M.; Melino, G.; Basu, S. Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death Differ. 2008, 15, 217–219. [Google Scholar] [CrossRef]
- Qiao, Y.; Qian, Y.; Wang, J.; Tang, X. Melanoma cell adhesion molecule stimulates yes-associated protein transcription by enhancing CREB activity via c-Jun/c-Fos in hepatocellular carcinoma cells. Oncol. Lett. 2016, 11, 3702–3708. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. 2018, 37, e98642. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef]
- Yang, J.C.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.L.; Papadimitrakopoulou, V.A.; Su, W.C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in Combination With Erlotinib or Gefitinib as First-Line Therapy for Advanced NSCLC With Sensitizing EGFR Mutation. J Thorac Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef]
- Soo, R.A.; Lim, S.M.; Syn, N.L.; Teng, R.; Soong, R.; Mok, T.S.K.; Cho, B.C. Immune checkpoint inhibitors in epidermal growth factor receptor mutant non-small cell lung cancer: Current controversies and future directions. Lung Cancer 2018, 115, 12–20. [Google Scholar] [CrossRef]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef]
- Yang, L.; Yang, S.; Liu, Y.; Li, J.; Hu, X.; Wang, Y.; Zhang, Y.; Wang, Y. Combination TS-1 plus EGFR-tyrosine kinase inhibitors (TKIs) for the treatment of non-small cell lung cancer after progression on first-line or further EGFR-TKIs: A phase II, single-arm trial. Thorac. Cancer 2018, 9, 693–698. [Google Scholar] [CrossRef]
- Dong, L.; Han, Z.F.; Feng, Z.H.; Jia, Z.Y. Comparison of pemetrexed and docetaxel as salvage chemotherapy for the treatment for nonsmall-cell lung cancer after the failure of epidermal growth factor receptor-tyrosine kinase inhibitors. J. Int. Med. Res. 2014, 42, 191–197. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, Y.; Yang, Y.; Yue, Z.; Pan, Z. Efficacy of pemetrexed and carboplatin with or without bevacizumab in lung adenocarcinoma patients with EGFR non-T790M mutations after progression on first-line EGFR-tyrosine kinase inhibitors. Thorac. Cancer 2018, 9, 1151–1155. [Google Scholar] [CrossRef]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef]
- Garassino, M.C.; Cho, B.C.; Kim, J.H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Corral Jaime, J.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521–536. [Google Scholar] [CrossRef]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann Oncol. 2017, 28, 1532–1539. [Google Scholar] [CrossRef]
- Kuo, C.S.; Wang, C.C.; Huang, Y.C.; Pavlidis, S.; Liu, C.Y.; Ko, H.W.; Chung, F.T.; Lin, T.Y.; Wang, C.L.; Guo, Y.K.; et al. Comparison of a combination of chemotherapy and immune checkpoint inhibitors and immune checkpoint inhibitors alone for the treatment of advanced and metastatic non-small cell lung cancer. Thorac. Cancer 2019, 10, 1158–1166. [Google Scholar] [CrossRef]
- Lee, T.F.; Tseng, Y.C.; Nguyen, P.A.; Li, Y.C.; Ho, C.C.; Wu, C.W. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef]
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Tang, Z.; Ma, Q.; Wang, L.; Liu, C.; Gao, H.; Yang, Z.; Liu, Z.; Zhang, H.; Ji, L.; Jiang, G. A brief review: Some compounds targeting YAP against malignancies. Future Oncol. 2019, 15, 1535–1543. [Google Scholar] [CrossRef]
- Lee, J.W.; Zhang, Y.; Eoh, K.J.; Sharma, R.; Sanmamed, M.F.; Wu, J.; Choi, J.; Park, H.S.; Iwasaki, A.; Kaftan, E.; et al. The Combination of MEK Inhibitor With Immunomodulatory Antibodies Targeting Programmed Death 1 and Programmed Death Ligand 1 Results in Prolonged Survival in Kras/p53-Driven Lung Cancer. J. Thorac. Oncol. 2019, 14, 1046–1060. [Google Scholar] [CrossRef]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo Pathway and YAP/TAZ-TEAD Protein-Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef]
- Woodard, G.A.; Yang, Y.L.; You, L.; Jablons, D.M. Drug development against the hippo pathway in mesothelioma. Transl. Lung Cancer Res. 2017, 6, 335–342. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L.; Liu, M.; Chong, R.; Ding, S.J.; Chen, Y.; Dong, J. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013, 73, 6722–6733. [Google Scholar] [CrossRef]
- Brinda, B.; Khan, I.; Parkin, B.; Konig, H. The rocky road to personalized medicine in acute myeloid leukaemia. J. Cell. Mol. Med. 2018, 22, 1411–1427. [Google Scholar] [CrossRef]
- Zhang, W.Q.; Dai, Y.Y.; Hsu, P.C.; Wang, H.; Cheng, L.; Yang, Y.L.; Wang, Y.C.; Xu, Z.; Liu, S.; Chan, G.; et al. Targeting YAP in malignant pleural mesothelioma. J. Cell. Mol. Med. 2017, 21, 2663–2676. [Google Scholar] [CrossRef]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 2018, 175, 1244–1258. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial name | Treatment Arms | N | Mutation | Response Rate (%) | Median PFS, Months (HR [95%CI]) | Median OS, Months (HR [95%CI]) |
|---|---|---|---|---|---|---|
| IPASS [18] | Gefitinib vs. Cp/Pac | 132 vs. 129 | Exon 18–21 | 71.2 vs. 7.3 (p < 0.001) | 9.5 vs. 6.3 (0.48 [0.36–0.64]; p < 0.001) | 21.6 vs. 21.9 (1.00 [0.76–1.33]; p = 0.990) |
| WJTOG3405 [19] | Gefitinib vs. Cis/Doc | 86 vs. 86 | Exon 19 and 21 | 62.1 vs. 32.2 (p < 0.0001) | 9.6 vs. 6.6 (0.52 [0.38–0.72]; p < 0.001) | 35.5 vs. 38.8 (1.19 [0.77–1.83]; p = 0.443) |
| NEJ002 [20] | Gefitinib vs. Cp/Pac | 114 vs. 114 | Exon 18–21 | 73.7 vs. 30.7 (p < 0.001) | 10.8 vs. 5.4 (0.32 [0.24–0.44]; p < 0.001) | 27.7 vs. 26.6 (0.89 [0.63–1.24]; p = 0.483) |
| OPTIMAL [21] | Erlotinib vs. Cp/Gem | 82 vs. 72 | Exon 19 and 21 | 83 vs. 36 (p < 0.0001) | 13.7 vs. 4.6 (0.16 [0.10–0.26]; p < 0.0001) | 22.7 vs. 28.9 (1.04 [0.69–1.58]; p = 0.692) |
| EURTAC [22] | Erlotinib vs. Cis/Gem or Cp/Doc | 86 vs. 87 | Exon 19 and 21 | 64 vs. 18 (p < 0.0001) | 10.4 vs. 5.1 (0.34 [0.23–0.49]; p < 0.0001) | 22.9 vs. 20.8 (0.93 [0.64–1.36]; p = 0.71) |
| LUX-Lung 3 [24,30] | Afatinib vs. Cis/Pem | 230 vs. 115 | Exon 18–21 | 56 vs. 23 (p = 0.001) | 11.1 vs. 6.9 (0.58 [0.43–0.78]; p < 0.0001) | 28.2 vs. 28.2 (0.88 [0.66–1.17]; p = 0.39) |
| LUX-Lung 6 [23,24] | Afatinib vs. Cis/Gem | 242 vs. 122 | Exon 18–21 | 67 vs. 23(p < 0.0001) | 11.0 vs. 5.6 (0.28 [0.20–0.39]; p < 0.0001) | 23.1 vs. 23.5 (0.93 [0.72–1.22]; p = 0.61) |
| ARCHER1050 [25,26] | Dacomitinib vs. Gefitinib | 227 vs. 225 | Exon 19 and 21 | 74.9 vs. 71.6 (p = 0.4234) | 14.7 vs. 9.2 (0·59[0.47–0.74]; p < 0·0001) | 34.1 vs. 26.8 (0.760 [0.58–0.99]; p = 0.044) |
| FLAURA [27] | Osimertinib vs. Gefitinib or Erlotinib | 279 vs. 277 | Exon 19 and 21 | 80 vs76 (p = 0.24) | 18.9 vs.10.2 (0.46 [0.37–0.57]; p < 0.001) | Not reached |
| Trial Name | PD-1 or PD-L1 Inhibitors | NSCLC Population | EGFR Mutation Rate (%) | Response Rate of ICIs (%) | Median OS, Months in ICI Treatment Group (HR [95%CI]) |
|---|---|---|---|---|---|
| KEYNOTE-010 [43] | Pembrolizumab 2 mg/kg and 10 mg/kg groups | PD-L1 positive (≥1% expression) | 6%–9% | 19% (2 mg/kg) 19% (10 mg/kg) | 10.4 (2 mg/kg) (0.71[0.58–0.88]; p = 0.0008) 12.7 (10 mg/kg) (0.61[0.49–0.75]; p < 0.0001) |
| KEYNOTE-024 [46] | Pembrolizumab 200mg | PD-L1 positive (≥50% expression); EGFR-wild-type ALK-wild-type | NA | 44.8% | 30.0 (0.49[0.34 to 0.69]; p = 0.002) |
| CheckMate 017 [47] | Nivolumab 3 mg/kg | Squamous NSCLC | NA | 20% | 9.2 (0.59 [0.44–0.79]; p < 0.001) |
| CheckMate 057 [48] | Nivolumab 3 mg/kg | Non-squamous NSCLC | 14% | 19% | 12.2 (0.73[0.59–0.89]; p = 0.002) |
| OAK [50] | Atezolizumab 1200 mg | PD-L1 positive and negative expression enrolled | 10% | 14% | 13.8 (0.73[0.62–0.87]; p = 0.0003) |
| PACIFIC [51] | Durvalumab 10 mg/kg | Stage III after chemoradiotherapy | 6% | 28.4 | Not reached 0.68 [0.47–0.997]; p = 0.0025 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, P.-C.; Jablons, D.M.; Yang, C.-T.; You, L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. https://doi.org/10.3390/ijms20153821
Hsu P-C, Jablons DM, Yang C-T, You L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). International Journal of Molecular Sciences. 2019; 20(15):3821. https://doi.org/10.3390/ijms20153821
Chicago/Turabian StyleHsu, Ping-Chih, David M. Jablons, Cheng-Ta Yang, and Liang You. 2019. "Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC)" International Journal of Molecular Sciences 20, no. 15: 3821. https://doi.org/10.3390/ijms20153821
APA StyleHsu, P.-C., Jablons, D. M., Yang, C.-T., & You, L. (2019). Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). International Journal of Molecular Sciences, 20(15), 3821. https://doi.org/10.3390/ijms20153821