High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. TMAO Induces Endothelial Dysfunction by Disrupting Endothelial Junction Proteins
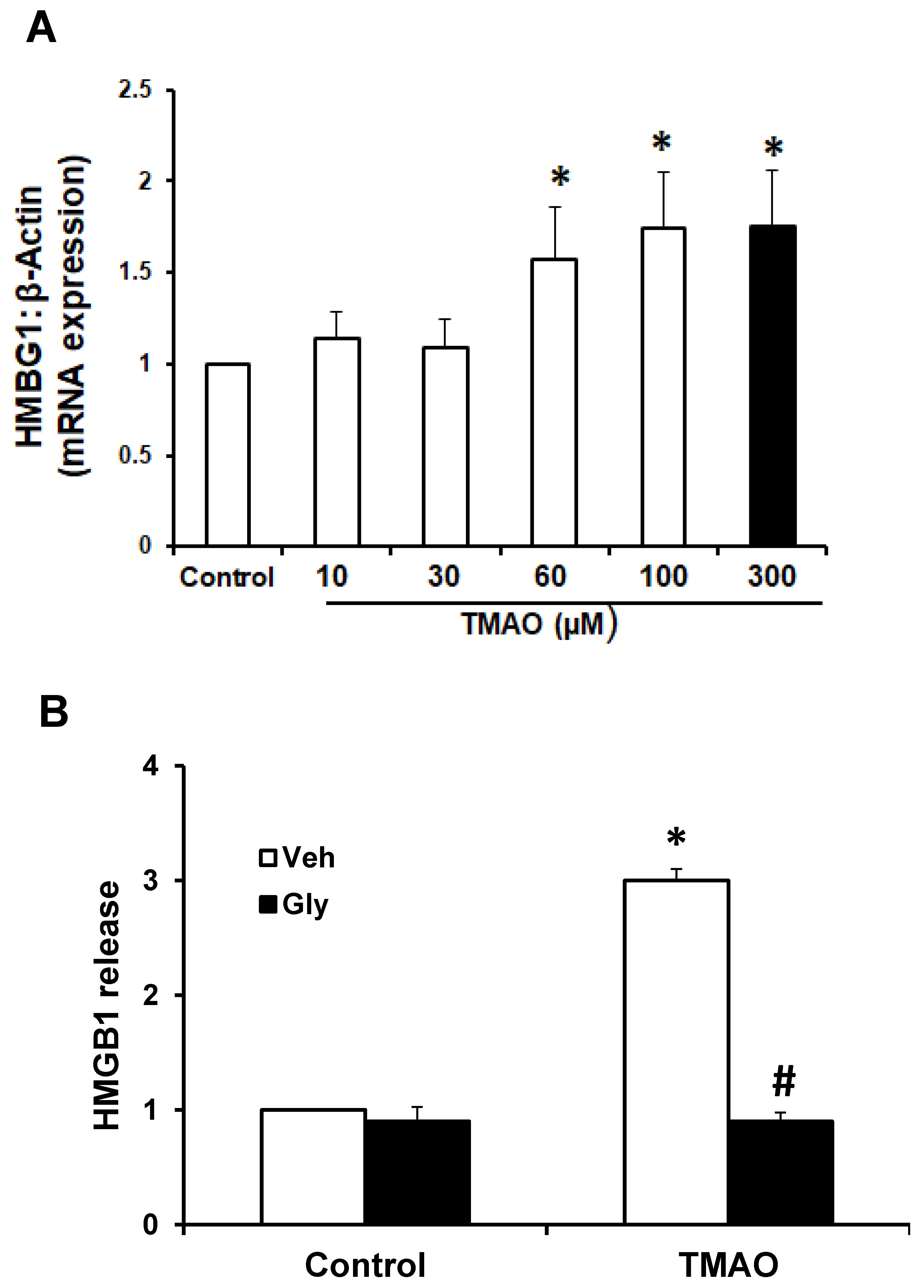
2.2. TMAO Upregulates the Expression of HMGB1, Accompanied by an Increase in Extracellular Release of HMGB1
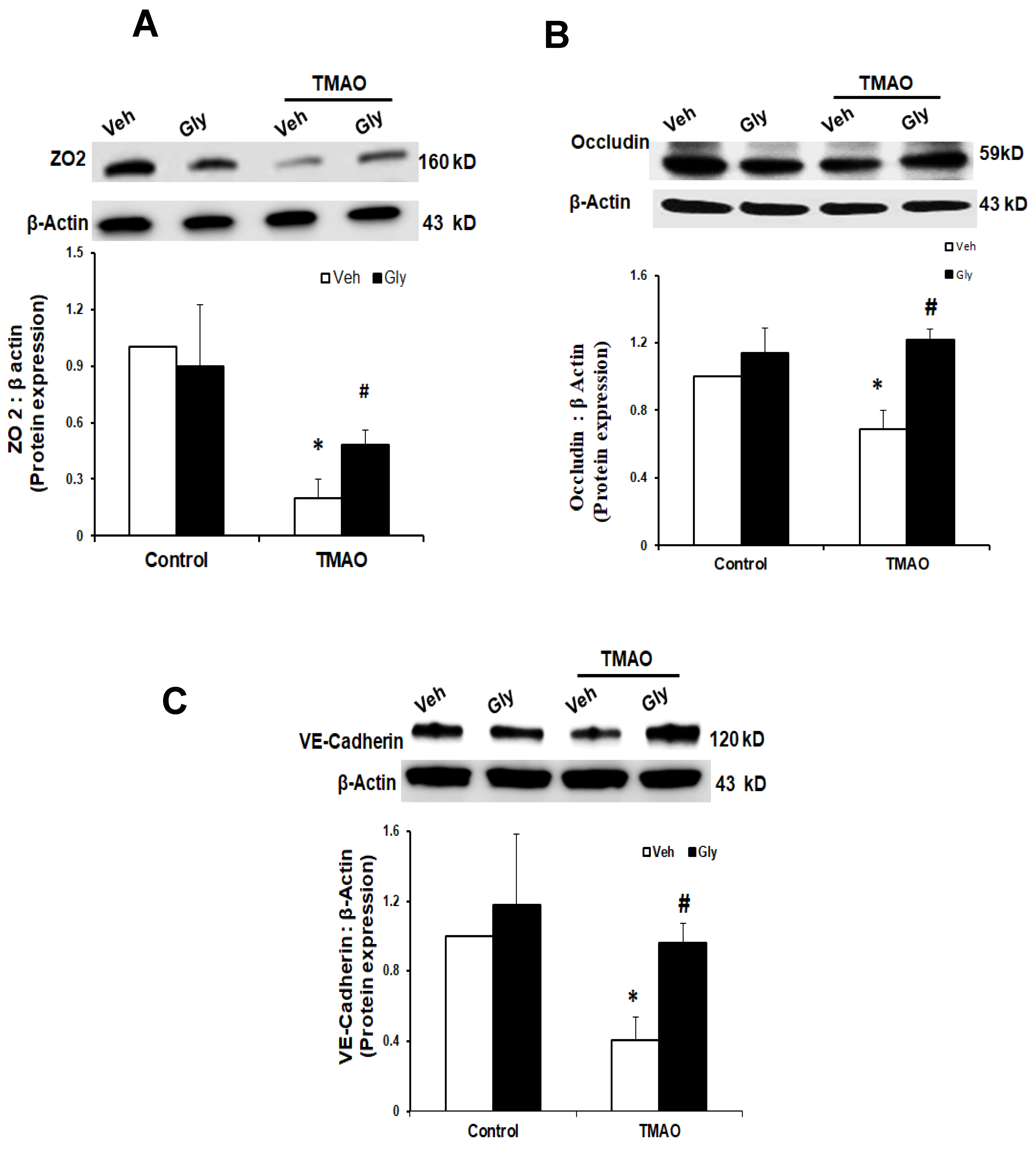
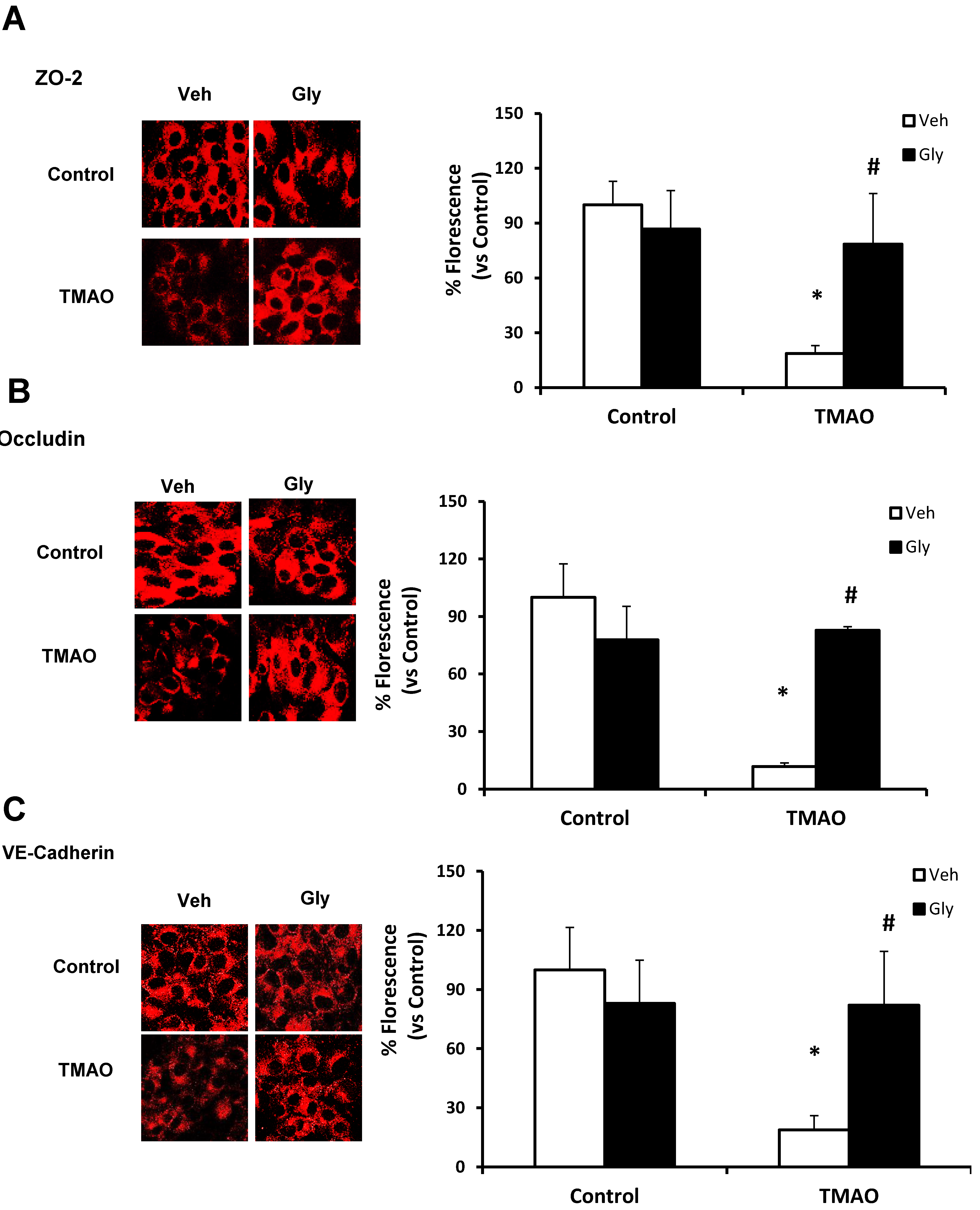
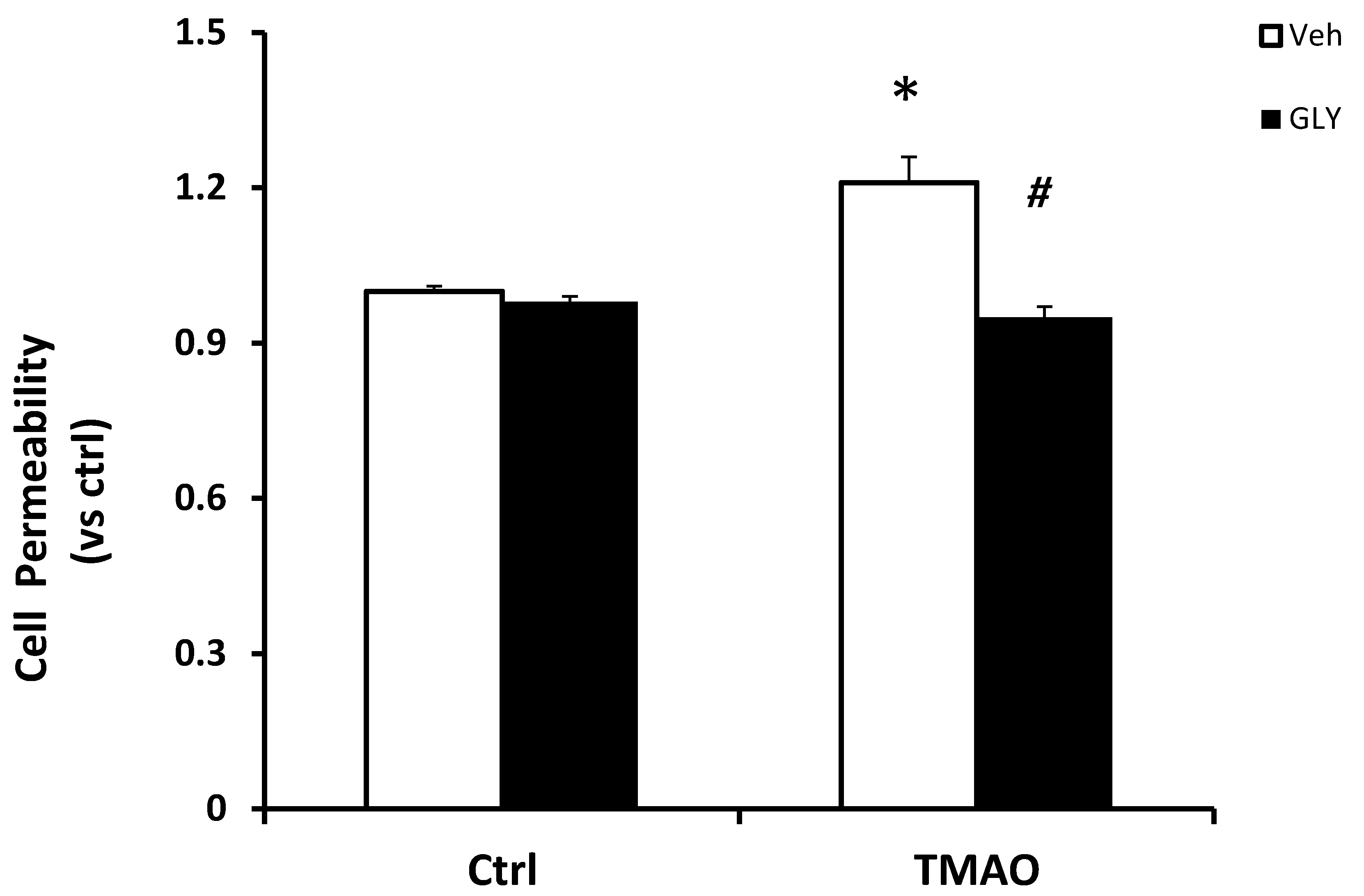
2.3. TMAO Causes Endothelial Cell Disjunction via HMGB1
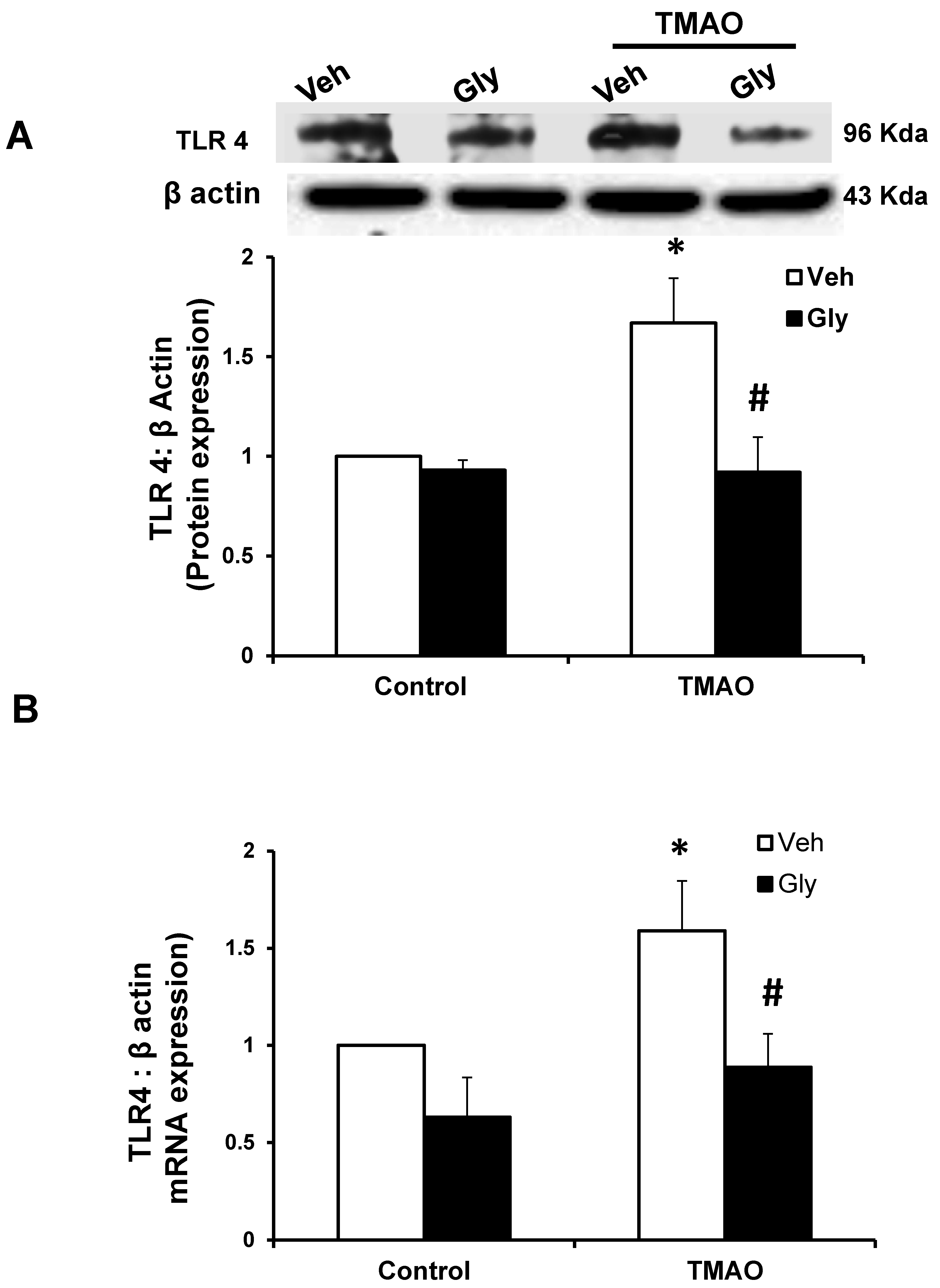
2.4. HMGB1 Acts in Synergy with TMAO to Upregulate the Expression of TLR4
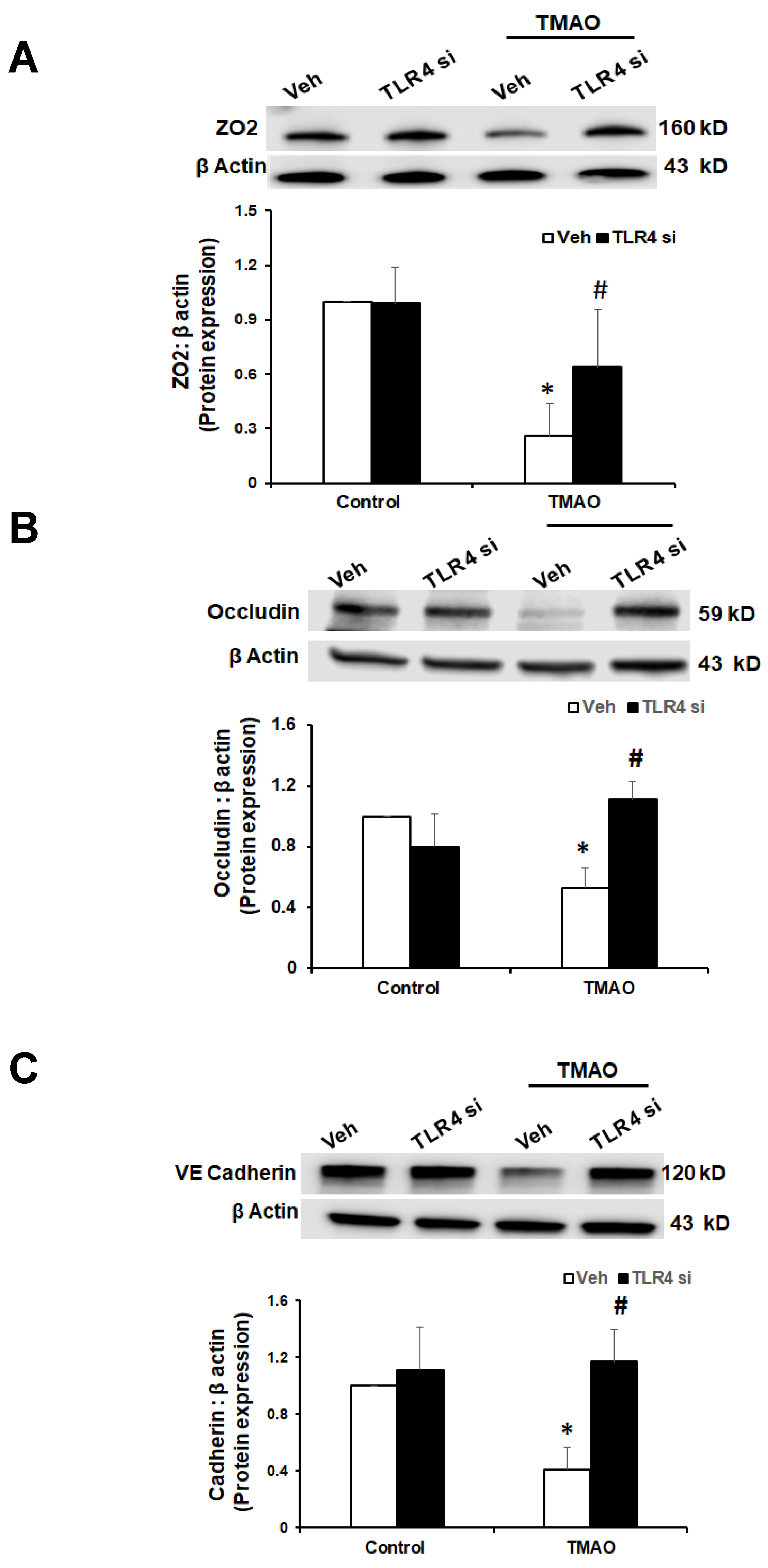
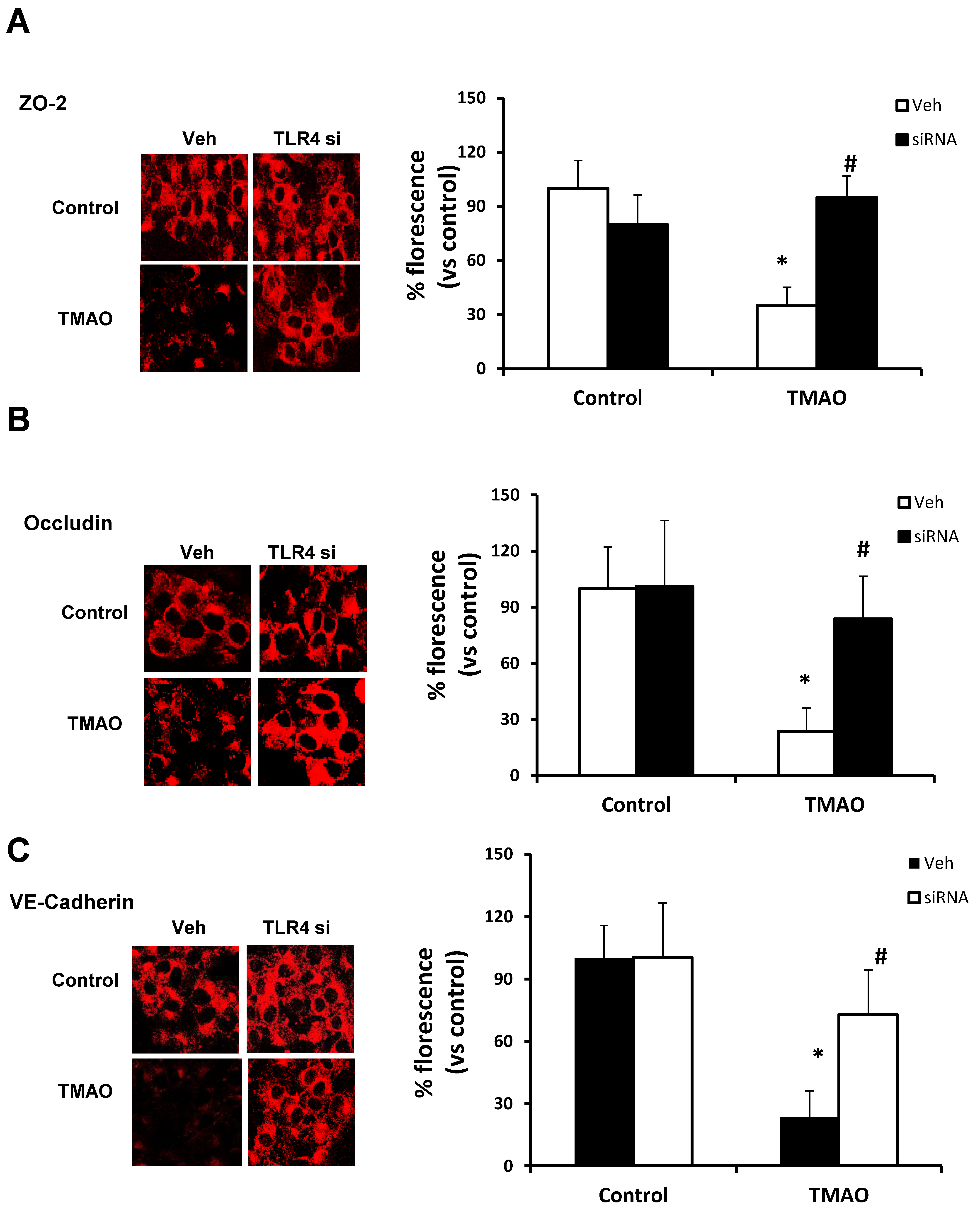
2.5. TMAO Causes Endothelial Dysfunction via HMGB1/TLR4 Axis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Immunofluorescence Analysis
4.3. Immunoblotting
4.4. qRT PCR
4.5. Detection of Extracellular Released HMGB1
4.6. RNA interference of TLR4
4.7. Endothelial Cell Permeability
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kitai, T.; Kirsop, J.; Tang, W.H. Exploring the Microbiome in Heart Failure. Curr. Heart Fail. Rep. 2016, 13, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; RaJapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Hazen, S.L. Microbiome, trimethylamine N-oxide, and cardiometabolic disease. Transl. Res. 2017, 179, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaide, M.A.I.; Singh, R.; Datta, P.; Rewers-Felkins, K.A.; Salguero, M.V.; Al-Obaidi, I.; Kottapalli, K.R.; Vasylyeva, T.L. Gut Microbiota-Dependent Trimethylamine-N-oxide and Serum Biomarkers in Patients with T2DM and Advanced CKD. J. Clin. Med. 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Miao, J.; Ling, A.V.; Manthena, P.V.; Gearing, M.E.; Graham, M.J.; Crooke, R.M.; Croce, K.J.; EsqueJo, R.M.; Clish, C.B.; Morbid Obesity Study Group; et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015, 6, 6498. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Miele, L.; Giorgio, V.; Alberelli, M.A.; De Candia, E.; Gasbarrini, A.; Grieco, A. Impact of Gut Microbiota on Obesity, Diabetes, and Cardiovascular Disease Risk. Curr. Cardiol. Rep. 2015, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Latkovskis, G.; Kuka, J.; Strele, I.; Konrade, I.; Grinberga, S.; Hartmane, D.; Pugovics, O.; Erglis, A.; Liepinsh, E. Diabetes is Associated with Higher Trimethylamine N-oxide Plasma Levels. Exp. Clin. Endocrinol. Diabetes 2016, 124, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37, e002767. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Kalinina, N.; Agrotis, A.; Antropova, Y.; DiVitto, G.; Kanellakis, P.; Kostolias, G.; Ilyinskaya, O.; Tararak, E.; Bobik, A. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: Role of activated macrophages and cytokines. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2320–2325. [Google Scholar] [CrossRef]
- Škrha, J., Jr.; Kalousová, M.; Švarcová, J.; Muravska, A.; Kvasnička, J.; Landova, L.; Zima, T.; Škrha, J. Relationship of soluble RAGE and RAGE ligands HMGB1 and EN-RAGE to endothelial dysfunction in type 1 and type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes. 2012, 120, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Zeh, H.; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 contributes to kidney ischemia reperfusion inury. Am. Soc. Nephrol. 2010, 21, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Han, N.J.; Kim, J.J.; Lee, M.J.; Park, S.K. TNF-alpha Activates High-Mobility Group Box 1-Toll-Like Receptor 4 Signaling Pathway in Human Aortic Endothelial Cells. Cell Physiol. Biochem. 2016, 38, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Den Dekker, W.K.; Cheng, C.; Pasterkamp, G.; Duckers, H. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis 2010, 209, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ialal, I.; Kaur, H.; Devara, S. Toll-like receptor status in obesity and metabolic syndrome: A translational perspective. Clin. Endocrinol. Metab. 2014, 99, 39–48. [Google Scholar]
- Bazzoni, G.; Deana, E. Endothelial cell-to-cell unctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef]
- Lu, B.; Wang, H.; Andersson, U.; Tracey, K. Regulation of HMGB1 release by inflammasomes. Protein Cell 2013, 4, 163–167. [Google Scholar] [CrossRef]
- Willingham, S.B.; Allen, I.C.; Bergstralh, D.T.; Brickey, W.J.; Huang, M.T.H.; Taxman, D.J.; Duncan, J.A.; Ting, J.P.Y. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. Immunol. 2009, 183, 2008–2015. [Google Scholar] [CrossRef]
- Chen, Y.; Pitzer, A.L.; Li, X.; Li, P.L.; Wang, L.; Zhang, Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial unction disruption: Role of HMGB1. Cel. Mol. Med. 2015, 19, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.L.; Zhang, Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 2016, 94, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Organ, C.L.; Otsuka, H.; Bhushan, S.; Wang, Z.; Bradley, J.; Trivedi, R.; Polhemus, D.J.; Tang, W.W.; Wu, Y.; Hazen, S.L.; et al. Choline Diet and Its Gut Microbe-Derived Metabolite, Trimethylamine N-Oxide, Exacerbate Pressure Overload-Induced Heart Failure. Circ. Heart Fail. 2016, 9, e002314. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cel. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Erlandsson-Harris, H.; Yang, H.; Tracey, K. HMGB1 as a DNA-binding cytokine. Leukoc. Biol. 2002, 72, 1084–1091. [Google Scholar]
- Dumitriu, I.E.; Baruah, P.; Manfredi, A.A.; Bianchi, M.E.; Rovere-Querini, P. HMGB1: Guiding immunity from within. Trends Immunol. 2005, 26, 381–387. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Mason, J.M.; Levine, J.; Yu, M.; Ulloa, L.; Czura, C.J.; Tracey, K.J.; Yang, H. Recombinant HMGB1 with cytokine-stimulating activity. Immunol. Methods 2004, 289, 211–223. [Google Scholar] [CrossRef]
- Yang, Z.; Deng, Y.; Su, D.; Tian, J.; Gao, Y.; He, Z.; Wang, X. TLR4 as receptor for HMGB1-mediated acute lung inury after liver ischemia/reperfusion inury. Lab. Investig. 2013, 93, 792–800. [Google Scholar] [CrossRef]
- Andrassy, M.; Volz, H.C.; Igwe, C.; Funke, B.; Eichberger, S.N.; Kaya, Z.; Buss, S.; Autschbach, F.; Pleger, S.T.; Lukic, I.K.; et al. High-mobility group box-1 in ischemia-reperfusion inury of the heart. Circulation 2008, 117, 3216–3226. [Google Scholar] [CrossRef]
- Andrassy, M.; Volz, H.C.; Maack, B.; Schuessler, A.; Gitsioudis, G.; Hofmann, N.; Laohachewin, D.; Wienbrandt, A.R.; Kaya, Z.; Bierhaus, A.; et al. HMGB1 is associated with atherosclerotic plaque composition and burden in patients with stable coronary artery disease. PLoS ONE 2012, 7, e52081. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.W.S.; Westra, J.; Limburg, P.C.; Bijl, M.; Kallenberg, C.G.M. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun. Rev. 2012, 11, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, S.; Strassheim, D.; Douglas, I.; Diaz del Valle, F.; Asehnoune, K.; Mitra, S.; Kwak, S.H.; Yamada, S.; Maruyama, I.; et al. HMGB1 contributes to the development of acute lung inury after hemorrhage. Am. Physiol. Lung Cell Mol. Physiol. 2005, 288, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Zeh, H.; Rubartelli, A.; Sparvero, L.; Amoscato, A.A.; Washburn, N.R.; Devera, M.E.; Liang, X.; Tor, M.; Billiar, T. The grateful dead: Damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev. 2007, 220, 60–81. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kawahara, K.; Biswas, K.K.; Ando, K.; Mitsudo, K.; Nobuyoshi, M.; Maruyama, I. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovasc. Pathol. 2007, 16, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.X.; Lu, L.; Peng, W.H.; Wang, L.; Zhang, Q.; Zhang, R.Y.; Chen, Q.; Shen, W.F. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 2009, 205, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, Y.; Li, L.; Zhang, R.; Liu, W.; Wu, J.; Mao, E.; Tang, Y. HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation 2012, 35, 350–362. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Deana, E. VE-cadherin and endothelial adherens unctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef]
- Lander, H.M.; Grant, A.M.; Albrecht, T.; Hill, T.; Peters, C. Endothelial cell permeability and adherens unction disruption induced by unin virus infection. Am. Trop. Med. Hyg. 2014, 90, 993–1002. [Google Scholar] [CrossRef]
- Runkle, E.A.; Mu, D. Tight unction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef]
- Wallez, Y.; Huber, P. Endothelial adherens and tight unctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Boini, K.M.; Pitzer, A.L.; Gulbins, E.; Zhang, Y.; Li, P.L. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim. Biophys. Acta 2015, 1853, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Tsan, M.F.; Gao, B. Endogenous ligands of Toll-like receptors. Leukoc. Biol. 2004, 76, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, G.F.; Dos Santos, R.A.; Oliveira, M.A.; Giachini, F.R.; Akamine, E.H.; Tostes, R.C.; Fortes, Z.B.; Webb, R.C.; Carvalho, M.H. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin. Sci. 2012, 122, 535–543. [Google Scholar] [CrossRef]
- De Batista, P.R.; Palacios, R.; Martin, A.; Hernanz, R.; Medici, C.T.; Silva, M.A.; Rossi, E.M.; Aguado, A.; Vassallo, D.V.; Salaices, M.; et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS ONE 2014, 9, e104020. [Google Scholar] [CrossRef]
- Eissler, R.; Schmaderer, C.; Rusai, K.; Kuhne, L.; Sollinger, D.; Lahmer, T.; Witzke, O.; Lutz, J.; Heemann, U.; Baumann, M. Hypertension augments cardiac Toll-like receptor 4 expression and activity. Hypertens. Res. 2011, 34, 551. [Google Scholar] [CrossRef]
- Marketou, M.E.; Kontaraki, E.; Zacharis, E.A.; Kochiadakis, G.E.; Giaouzaki, A.; Chlouverakis, G.; Vardas, P.E. TLR2 and TLR4 gene expression in peripheral monocytes in nondiabetic hypertensive patients: The effect of intensive blood pressure-lowering. Clin. Hypertens. 2012, 14, 330–335. [Google Scholar] [CrossRef]
- Sollinger, D.; Eißler, R.; Lorenz, S.; Strand, S.; Chmielewski, S.; Aoqui, C.; Schmaderer, C.; Bluyssen, H.; Zicha, J.; Witzke, O.; et al. Damage-associated molecular pattern activated Toll-like receptor 4 signalling modulates blood pressure in L-NAME-induced hypertension. Cardiovasc. Res. 2014, 101, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Pham, M.; Luttrell, I.; Bannerman, D.D.; Tupper, J.; Thaler, J.; Hawn, T.R.; Raines, E.W.; Schwartz, M.W. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ. Res. 2007, 100, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Katoh, S.; Honda, S.; Watanabe, T.; Suzuki, S.; Ishino, M.; Kitahara, T.; Funayama, A.; Netsu, S.; Sasaki, T.; Shishido, T.; et al. Atrial endothelial impairment through Toll-like receptor 4 signaling causes atrial thrombogenesis. Heart Vessels 2014, 29, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Yang, J.; Wang, W.; Zhao, W.; Peng, F.; Xiang, Y.; Chen, G.; Chen, T.; Chai, C.; Zheng, S.; et al. HMGB1-promoted and TLR2/4-dependent NK cell maturation and activation take part in rotavirus-induced murine biliary atresia. PLoS Pathog. 2014, 10, e1004011. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, G.B.; Zhang, Y.; Boini, K.M.; Koka, S. High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 3570. https://doi.org/10.3390/ijms20143570
Singh GB, Zhang Y, Boini KM, Koka S. High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction. International Journal of Molecular Sciences. 2019; 20(14):3570. https://doi.org/10.3390/ijms20143570
Chicago/Turabian StyleSingh, Gurinder Bir, Yang Zhang, Krishna M. Boini, and Saisudha Koka. 2019. "High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction" International Journal of Molecular Sciences 20, no. 14: 3570. https://doi.org/10.3390/ijms20143570
APA StyleSingh, G. B., Zhang, Y., Boini, K. M., & Koka, S. (2019). High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction. International Journal of Molecular Sciences, 20(14), 3570. https://doi.org/10.3390/ijms20143570