Mitochondrial Involvement in Cisplatin Resistance


Abstract
{kind=link}
{kind=link}
1. Introduction
2. Mitochondria
3. Mitochondria and Cancer
4. Mito-Nuclear Crosstalk
5. Intercellular Mitochondrial Transfer
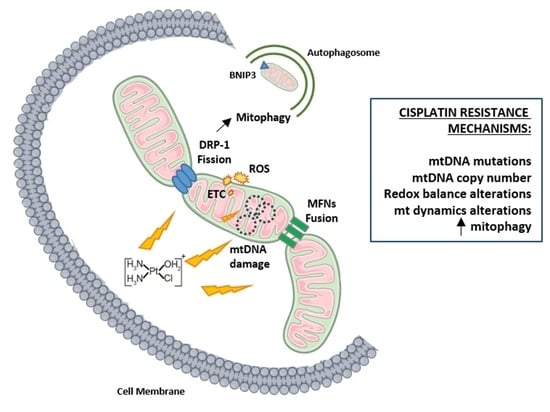
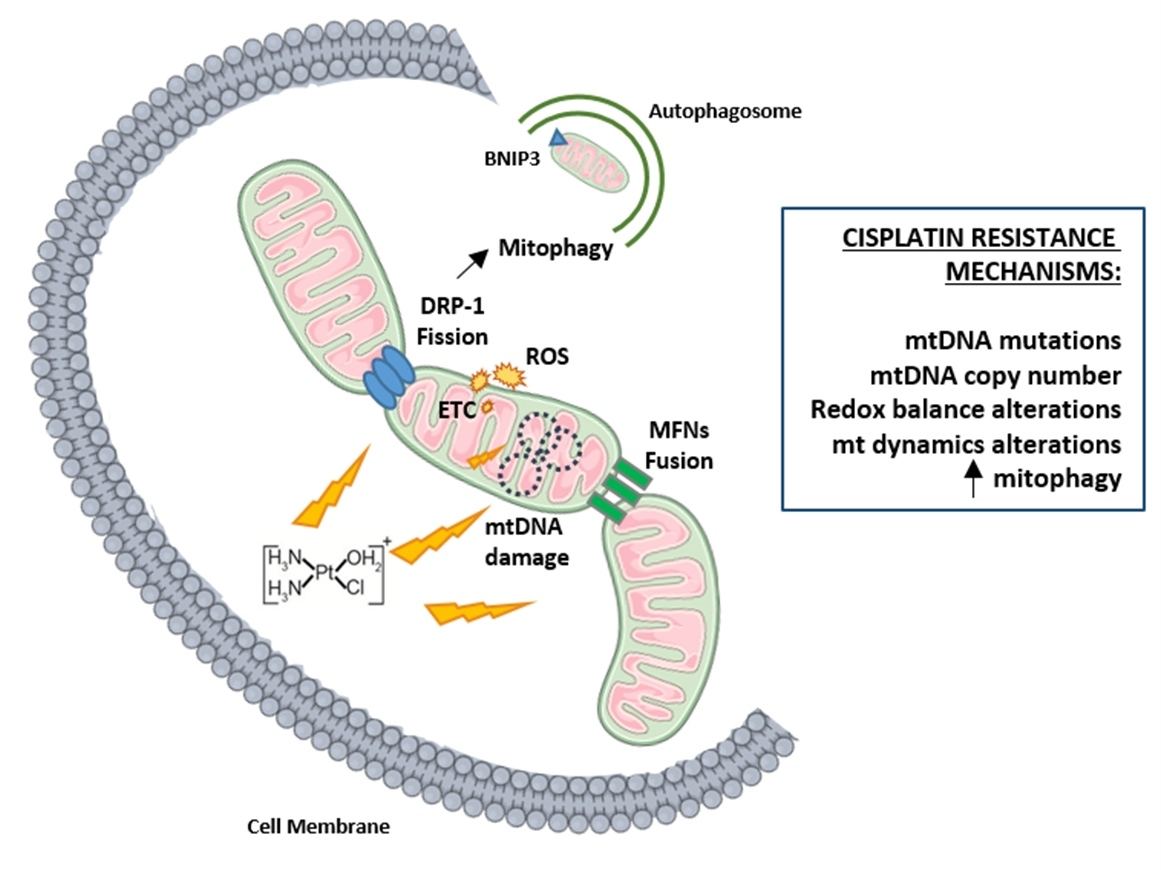
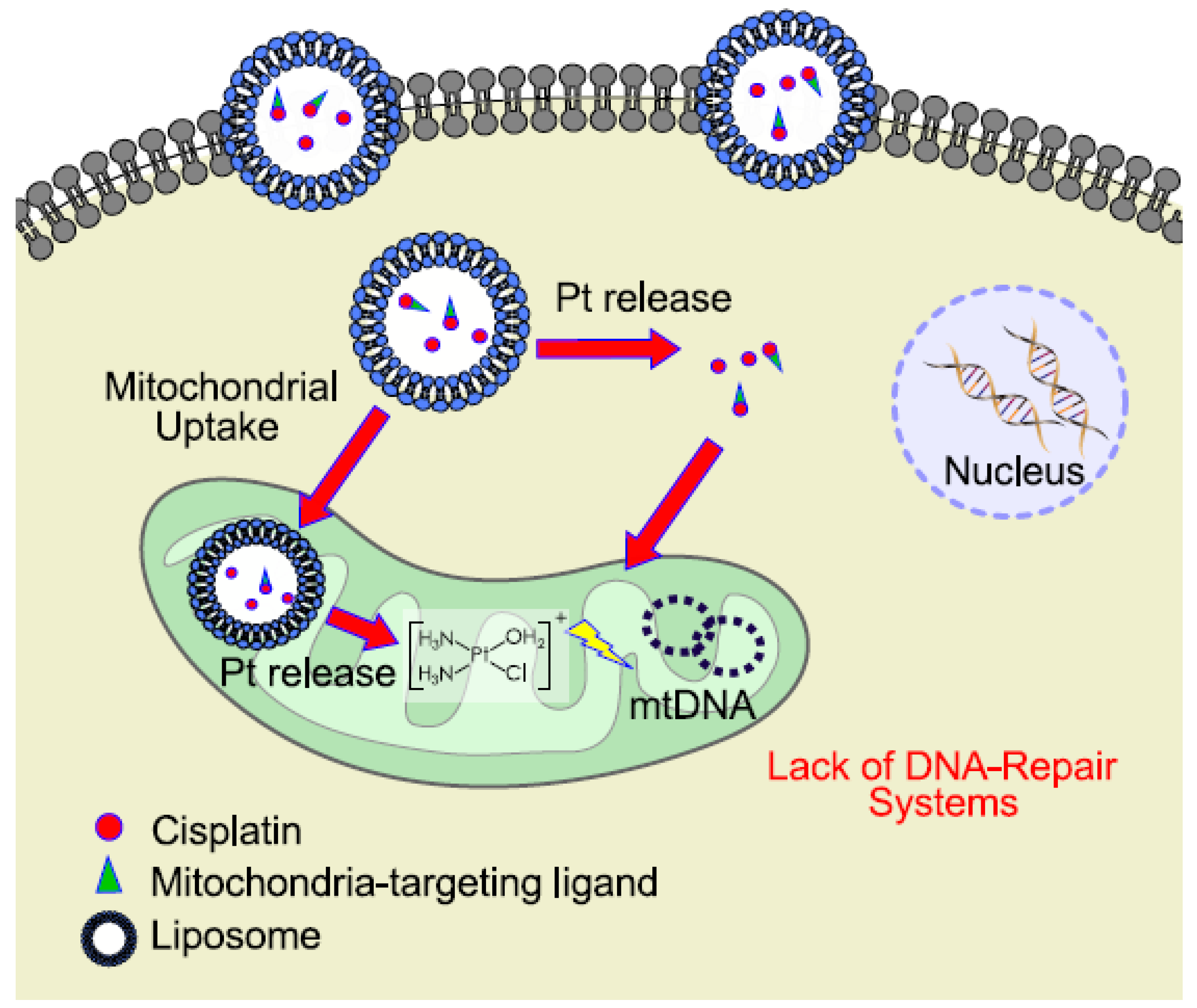
6. Mitochondrial DNA as a Target for Cisplatin
7. Mitochondrial Dynamics
8. Mitophagy and Chemoresistance
9. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Prestayko, A.W.; D’aoust, J.C.; Issell, B.F.; Crooke, S.T. Cispla.tin (cis-diamminedichloroplatinum II). Cancer Treat. Rev. 1979, 6, 17–39. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Galanski, M. Recent developments in the field of anticancer platinum complexes. Recent Pat. Anti-Cancer Drug Discov. 2006, 1, 285–295. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalt. Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F. Treatment goals in ovarian cancer. Int. J. Gynecol. Cancer 2005, 15 (Suppl. 1), 3–11. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. Clinical perspectives on platinum resistance. Drugs 2000, 59, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Köberle, B.; Tomicic, M.T.; Usanova, S.; Kaina, B. Cisplatin resistance: Preclinical findings and clinical implications. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2010, 1806, 172–182. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, N. Platinum resistance in breast and ovarian cancer cell lines. J. Exp. Clin. Cancer Res. 2011, 30, 91. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; Broggini, M. Platinum resistance in ovarian cancer: Role of DNA repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R. Preclinical perspectives on platinum resistance. Drugs 2000, 59, 1–8. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef]
- Bodiga, S.; Vemuri, P.K.; Bodiga, V.L. Low Ctr1p, due to lack of Sco1p results in lowered cisplatin uptake and mediates insensitivity of rho0 yeast to cisplatin. J. Inorg. Biochem. 2018, 187, 14–24. [Google Scholar] [CrossRef]
- Fichtinger-Schepman, A.M.J.; Van der Veer, J.L.; Den Hartog, J.H.J.; Lohman, P.H.M.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: Formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G.J.C.D. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy–an update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Structure of duplex DNA containing the cisplatin 1, 2-{Pt (NH3) 2} 2+-d (GpG) cross-link at 1.77 Å resolution. J. Inorg. Biochem. 2010, 104, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, J. Platinum anticancer coordination compounds: Study of DNA binding inspires new drug design. Eur. J. Inorg. Chem. 2009, 10, 1303–1312. [Google Scholar] [CrossRef]
- Koonin, E.V.; Aravind, L. Origin and evolution of eukaryotic apoptosis: The bacterial connection. Cell Death Differ. 2002, 9, 394. [Google Scholar] [CrossRef] [PubMed]
- Sickmann, A.; Reinders, J.; Wagner, Y.; Joppich, C.; Zahedi, R.; Meyer, H.E.; Schönfisch, B.; Perschil, I.; Chacinska, A.; Guiard, B.; et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 2003, 100, 13207–13212. [Google Scholar] [CrossRef] [PubMed]
- Forner, F.; Foster, L.J.; Campanaro, S.; Valle, G.; Mann, M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell. Proteom. 2006, 5, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Bustos, G.; Cruz, P.; Lovy, A.; Cárdenas, C. Endoplasmic reticulum–mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: A novel potential target. Front. Oncol. 2017, 7, 199. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Idelchik, M.D.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2017; Volume 47, pp. 57–66. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef]
- Okon, I.S.; Zou, M.H. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol. Res. 2015, 100, 170–174. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Gandin, V.; Fernandes, A. Metal-and semimetal-containing inhibitors of thioredoxin reductase as anticancer agents. Molecules 2015, 20, 12732–12756. [Google Scholar] [CrossRef]
- de Sá Junior, P.L.; Câmara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The roles of ROS in cancer heterogeneity and therapy. Oxidative Med. Cell. Longev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.W.; De Coo, I.F.M.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.M.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res./Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the center of cell signaling: Interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Tsuyoshi, H.; Wong, V.K.W.; Han, Y.; Orisaka, M.; Yoshida, Y.; Tsang, B.K. Saikosaponin-d, a calcium mobilizing agent, sensitizes chemoresistant ovarian cancer cells to cisplatin-induced apoptosis by facilitating mitochondrial fission and G2/M arrest. Oncotarget 2017, 8, 9982. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, H.; Wang, C.; Su, J.; Xie, Q.; Xu, L.; Yu, Y.; Liu, S.; Li, S.; Xu, Y.; et al. Failure of elevating calcium induces oxidative stress tolerance and imparts cisplatin resistance in ovarian Cancer cells. Aging Dis. 2016, 7, 254. [Google Scholar] [CrossRef]
- Corydon, T.J.; Wilsbech, M.; Jespersgaard, C.; Andresen, B.S.; Børglum, A.D.; Pedersen, S.; Bolund, L.; Gregersen, N.; Bross, P. Human and mouse mitochondrial orthologs of bacterial ClpX. Mamm. Genome 2000, 11, 899–905. [Google Scholar] [CrossRef]
- Zhang, Y.; Maurizi, M.R. Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 252–264. [Google Scholar] [CrossRef]
- Yang, D.; Kim, J. Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 2019, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Herrera, E.L.; Azzam, S.Z.; Berger, M.C.; Diaz-Martinez, L.A. Genomic heterogeneity meets cellular energetics: Crosstalk between the mitochondria and the cell cycle. Cancer Metastasis Treat 2018, 4, 42. [Google Scholar] [CrossRef][Green Version]
- Kleine, T.; Leister, D. Retrograde signaling: Organelles go networking. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- da Cunha, F.M.; Torelli, N.Q.; Kowaltowski, A.J. Mitochondrial retrograde signaling: Triggers, pathways, and outcomes. Oxidative Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Duncan, O.F.; Granat, L.; Ranganathan, R.; Singh, V.K.; Mazaud, D.; Fanto, M.; Chambers, D.; Ballard, C.G.; Bateman, J.M. Ras-ERK-ETS inhibition alleviates neuronal mitochondrial dysfunction by reprogramming mitochondrial retrograde signaling. PLoS Genet. 2018, 14, e1007567. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria retrograde signaling and the UPRmt: Where are we in mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef]
- Jazwinski, S.M.; Kriete, A. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front. Physiol. 2012, 3, 139. [Google Scholar] [CrossRef]
- Khurshed, M.; Aarnoudse, N.; Hulsbos, R.; Hira, V.V.; van Laarhoven, H.W.; Wilmink, J.W.; Molenaar, R.J.; van Noorden, C.J. IDH1-mutant cancer cells are sensitive to cisplatin and an IDH1-mutant inhibitor counteracts this sensitivity. FASEB J. 2018, 32, 6344–6352. [Google Scholar] [CrossRef]
- Wang, S.F.; Chen, M.S.; Chou, Y.C.; Ueng, Y.F.; Yin, P.H.; Yeh, T.S.; Lee, H.C. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway. Oncotarget 2016, 7, 74132. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Babenko, V.A.; Silachev, D.N.; Zorova, L.D.; Khryapenkova, T.G.; Savchenko, E.S.; SavchenkoI, B.; Zorov, D.B. Intercellular transfer of mitochondria. Biochemistry 2015, 80, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Crasso, C.; Neuzil, J. Mitochondrial genome transfer to tumor cells breaks the rules and establishes a new precedent in cancer biology. Mol. Cell. Oncol. 2018, 5, e1023929. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia–reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 2018, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Penta, J.S.; Johnson, F.M.; Wachsman, J.T.; Copeland, W.C. Mitochondrial DNA in human malignancy. Mutat. Res. /Rev. Mutat. Res. 2001, 488, 119–133. [Google Scholar] [CrossRef]
- Singh, K.K.; Russell, J.; Sigala, B.; Zhang, Y.; Williams, J.; Keshav, K.F. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene 1999, 18, 6641. [Google Scholar] [CrossRef]
- Guerra, F.; Perrone, A.M.; Kurelac, I.; Santini, D.; Ceccarelli, C.; Cricca, M.; Zamagni, C.; De Iaco, P.; Gasparre, G. Mitochondrial DNA mutation in serous ovarian cancer: Implications for mitochondria-coded genes in chemoresistance. J. Clin. Oncol. 2012, 30, e373–e378. [Google Scholar] [CrossRef] [PubMed]
- FanFan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA determines chemoresistance by reprogramming metabolism and regulating mitochondrial transcription. Cancer Res. 2019, 79, 1069–1084. [Google Scholar] [CrossRef]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction in cancer chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef]
- Qian, W.; Nishikawa, M.; Haque, A.M.; Hirose, M.; Mashimo, M.; Sato, E.; Inoue, M. Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am. J. Physiol. Cell Physiol. 2005, 289, C1466–C1475. [Google Scholar] [CrossRef]
- Park, S.Y.; Chang, I.; Kim, J.Y.; Kang, S.W.; Park, S.H.; Singh, K.; Lee, M.S. Resistance of mitochondrial DNA-depleted cells against cell death role of mitochondrial superoxide dismutase. J. Biol. Chem. 2004, 279, 7512–7520. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Marin, J.J.; Perez, M.J. The expression of genes involved in hepatocellular carcinoma chemoresistance is affected by mitochondrial genome depletion. Mol. Pharm. 2014, 11, 1856–1868. [Google Scholar] [CrossRef]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Sun, S.; Bai, Y.; Chen, Y.; Chai, R.; Li, H. Reduced mtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell Death Dis. 2015, 6, e1710. [Google Scholar] [CrossRef]
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. PNAS 2014, 111, 10444–10449. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Shaw, J.M. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005, 39, 503–536. [Google Scholar] [CrossRef] [PubMed]
- Bereiter-Hahn, J. Behavior of mitochondria in the living cell. In International Review of Cytology; Academic Press: Cambridge, MA, USA, 1990; Volume 122, pp. 1–63. [Google Scholar] [CrossRef]
- Westermann, B. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 2008, 283, 13501–13505. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, T.; Pendin, D.; Montagna, A.; Bova, S.; Ghelli, A.M.; Daga, A. Manipulation of mitochondria dynamics reveals separate roles for form and function in mitochondria distribution. Cell Rep. 2018, 23, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Mannella, C.A. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.J.; Pekkurnaz, G. Powerhouse of the mind: Mitochondrial plasticity at the synapse. Curr. Opin. Neurobiol. 2019, 57, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Arribat, Y.; Broskey, N.T.; Greggio, C.; Boutant, M.; Conde Alonso, S.; Kulkarni, S.S.; Lagarrigue, S.; Carnero, E.A.; Besson, C.; Cantó, C.; et al. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol. 2019, 225, e13179. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Ferree, A.; Shirihai, O. Mitochondrial dynamics: The intersection of form and function. In Mitochondrial Oxidative Phosphorylation; Springer: New York, NY, USA, 2012; pp. 13–40. [Google Scholar] [CrossRef]
- Sebastián, D.; Zorzano, A. Mitochondrial Dynamics: A Journey from Mitochondrial Morphology to Mitochondrial Function and Quality. In Mitochondrial Biology and Experimental Therapeutics; Springer: Cham, Switzerland, 2018; pp. 19–31. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vázquez, J.; Enríquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef]
- Hoppins, S.; Nunnari, J. The molecular mechanism of mitochondrial fusion. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Lenaers, G.; Hajnóczky, G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation–contraction coupling. J. Cell Biol. 2014, 205, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Boldogh, I.R.; Nowakowski, D.W.; Yang, H.C.; Chung, H.; Karmon, S.; Royes, P.; Pon, L.A. A protein complex containing Mdm10p, Mdm12p, and Mmm1p links mitochondrial membranes and DNA to the cytoskeleton-based segregation machinery. Mol. Biol. Cell 2003, 14, 4618–4627. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Grandemange, S.; Herzig, S.; Martinou, J.C. Mitochondrial dynamics and cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2009; Volume 19, pp. 50–56. [Google Scholar] [CrossRef]
- Wieder, S.Y.; Serasinghe, M.N.; Sung, J.C.; Choi, D.C.; Birge, M.B.; Yao, J.L.; Bernstein, E.; Celebi, J.T.; Chipuk, J.E. Activation of the mitochondrial fragmentation protein DRP1 correlates with BRAFV600E melanoma. J. Investig. Dermatol. 2015, 135, 2544. [Google Scholar] [CrossRef]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Yip, C.; Ahn, Y.R.; Ali, M.; Yllanes, A.P.; Chao, C.A.; McDonnell, D.P.; Wood, K.C. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G.; Ghiraldeli, L.P.; Pardee, T.S. Mitochondria in cancer metabolism, an organelle whose time has come? Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2018, 1870, 96–102. [Google Scholar] [CrossRef] [PubMed]
- von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-driven change in mitochondrial dynamics limits YAP/TAZ function in mammary epithelial cells and breast cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Chen, C.Y.; Chiou, S.H.; Wang, Y.T.; Lin, T.Y.; Chang, H.W.; Chiang, I.P.; Lan, K.J.; Chow, K.C. Overexpression of optic atrophy 1 protein increases cisplatin resistance via inactivation of caspase-dependent apoptosis in lung adenocarcinoma cells. Hum. Pathol. 2012, 43, 105–114. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Santin, G.; Piccolini, V.M.; Barni, S.; Veneroni, P.; Giansanti, V.; Dal Bo, V.; Bernocchi, G.; Bottone, M.G. Mitochondrial fusion: A mechanism of cisplatin-induced resistance in neuroblastoma cells? Neurotoxicology 2013, 34, 51–60. [Google Scholar] [CrossRef]
- Casinelli, G.; LaRosa, J.; Sharma, M.; Cherok, E.; Banerjee, S.; Branca, M.; Edmunds, L.; Wang, Y.; Sims-Lucas, S.; Churley, L.; et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2016, 2, 16082. [Google Scholar] [CrossRef]
- Han, X.J.; Shi, S.L.; Wei, Y.F.; Jiang, L.P.; Guo, M.Y.; Wu, H.L.; Wan, Y.Y. Involvement of mitochondrial dynamics in the antineoplastic activity of cisplatin in murine leukemia L1210 cells. Oncol. Rep. 2017, 38, 985–992. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Farrand, L.; Byun, S.; Kim, J.Y.; Im-Aram, A.; Lee, J.; Lim, S.; Lee, K.W.; Suh, J.Y.; Lee, H.J.; Tsang, B.K. Piceatannol enhances cisplatin sensitivity in ovarian cancer via modulation of p53, X-linked inhibitor of apoptosis protein (XIAP), and mitochondrial fission. J. Biol. Chem. 2013, 288, 23740–23750. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Wang, J.; Roginskaya, V.; McDermott, L.A.; Edwards, R.P.; Stolz, D.B.; Llambi, F.; Green, D.R.; Van Houten, B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43. [Google Scholar] [CrossRef] [PubMed]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting autophagy to overcome human diseases. Int. J. Mol. Sci. 2019, 20, 725. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Jahangiri, A.; DeLay, M.; Aghi, M.K. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012, 72, 4294–4299. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Yuan, Z.; Zhang, Q.; Long, Z.; Chen, J.; Tang, Z.; Zhu, Y.; Chen, S.; Xu, J.; Yan, M.; et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012, 8, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Masuda, N.; Kamigaki, S.; Morimoto, T.; Saji, S.; Imoto, S.; Sasano, H.; Toi, M. Differential Involvement of Autophagy and Apoptosis in Response to Chemoendocrine and Endocrine Therapy in Breast Cancer: JBCRG-07TR. Int. J. Mol. Sci. 2019, 20, 984. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Song, Z.; Qi, M.; Bai, X.; Wang, J.; Zhang, Y.; Zou, X.; Guo, Q.; Wan, P. Increased Autophagy Levels Mediate Cisplatin Resistance in Cisplatin-Resistant Cells While Also Rendering Them Vulnerable to Autophagy Induction. BioMed Res. Int. 2018. [Google Scholar] [CrossRef]
- Qiu, S.; Sun, L.; Jin, Y.; An, Q.; Weng, C.; Zheng, J. Silencing of BAG3 promotes the sensitivity of ovarian cancer cells to cisplatin via inhibition of autophagy. Oncol. Rep. 2017, 38, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; An, Y.; Wang, Y.; Zhang, C.; Zhang, H.; Huang, C.; Jiang, H.; Wang, X.; Li, X. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2013, 29, 2019–2024. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Um, J.H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299. [Google Scholar] [CrossRef]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK 1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.A.M.; Vieira, M.Q.; Ilha, M.; de Vasconcelos, M.; Biehl, H.B.; Lima, D.B.; Schein, V.; Barbé-Tuana, F.; Borojevic, R.; Guma, F.C.R. The interplay between apoptosis, mitophagy and mitochondrial biogenesis induced by resveratrol can determine activated hepatic stellate cells death or survival. Cell Biochem. Biophys. 2015, 71, 657–672. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, Z.; Qi, J.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zeng, M.; Zhang, B.; Wang, N.; et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 2017, 8, 20988. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.M.; Hu, X.; Ming, Q.L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef]
- Chen, L.; Yao, Y.; Sun, L.; Tang, J. Galectin-1 promotes tumor progression via NF-κB signaling pathway in epithelial ovarian cancer. J. Cancer 2017, 8, 3733. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Davuluri, G.V.N.; Chen, C.H.; Shiau, D.C.; Chen, C.C.; Chen, C.L.; Lin, Y.S.; Chang, C.P. Galectin-1-induced autophagy facilitates cisplatin resistance of hepatocellular carcinoma. PLoS ONE 2016, 11, e0148408. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Z.; Guo, J.; Wang, L.; Liu, X. Ambra1 induces autophagy and desensitizes human prostate cancer cells to cisplatin. Biosci. Rep. 2017, BSR20170770. [Google Scholar] [CrossRef] [PubMed]
- Abdrakhmanov, A.; Kulikov, A.V.; Luchkina, E.A.; Zhivotovsky, B.; Gogvadze, V. Involvement of mitophagy in cisplatin-induced cell death regulation. Biol. Chem. 2019, 400, 161–170. [Google Scholar] [CrossRef]
- Radogna, F.; Cerella, C.; Gaigneaux, A.; Christov, C.; Dicato, M.; Diederich, M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 2016, 35, 3839. [Google Scholar] [CrossRef]
- Wandee, J.; Prawan, A.; Senggunprai, L.; Kongpetch, S.; Kukongviriyapan, V. Metformin sensitizes cholangiocarcinoma cell to cisplatin-induced cytotoxicity through oxidative stress mediated mitochondrial pathway. Life Sci. 2019, 217, 155–163. [Google Scholar] [CrossRef]
- Fernandez-Gil, B.I.; Guerra-Librero, A.; Shen, Y.Q.; Florido, J.; Martínez-Ruiz, L.; García-López, S.; Adan, C.; Rodríguez-Santana, C.; Acuña-Castroviejo, D.; Quiñones-Hinojosa, A.; et al. Melatonin Enhances Cisplatin and Radiation Cytotoxicity in Head and Neck Squamous Cell Carcinoma by Stimulating Mitochondrial ROS Generation, Apoptosis, and Autophagy. Oxidative Med. Cell. Longev. 2019. [Google Scholar] [CrossRef]
- Stoyanova, E.; Petrov, P.; Karadjova, I.; Momekov, G.; Koseva, N. Cisplatin delivery vehicles based on stabilized polymeric aggregates comprising poly (acrylic acid) chains. Polym. J. 2017, 49, 607. [Google Scholar] [CrossRef][Green Version]
- Hang, Z.; Cooper, M.A.; Ziora, Z.M. Platinum-based anticancer drugs encapsulated liposome and polymeric micelle formulation in clinical trials. Biochem. Compd. 2016, 4, 2. [Google Scholar] [CrossRef]
- Duan, X.; He, C.; Kron, S.J.; Lin, W. Nanoparticle formulations of cisplatin for cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, Z.; Zhang, C.; Wang, Y.; Zhang, H.; Gan, Z.; Guo, Z.; Wang, X. Mitochondrion-targeted platinum complexes suppressing lung cancer through multiple pathways involving energy metabolism. Chem. Sci. 2019, 10, 3089–3095. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. https://doi.org/10.3390/ijms20143384
Cocetta V, Ragazzi E, Montopoli M. Mitochondrial Involvement in Cisplatin Resistance. International Journal of Molecular Sciences. 2019; 20(14):3384. https://doi.org/10.3390/ijms20143384
Chicago/Turabian StyleCocetta, Veronica, Eugenio Ragazzi, and Monica Montopoli. 2019. "Mitochondrial Involvement in Cisplatin Resistance" International Journal of Molecular Sciences 20, no. 14: 3384. https://doi.org/10.3390/ijms20143384
APA StyleCocetta, V., Ragazzi, E., & Montopoli, M. (2019). Mitochondrial Involvement in Cisplatin Resistance. International Journal of Molecular Sciences, 20(14), 3384. https://doi.org/10.3390/ijms20143384