The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications



 , , ,
, , ,
Abstract
1. Introduction
2. Restrictive Properties of the Blood Nerve Barrier (BNB)
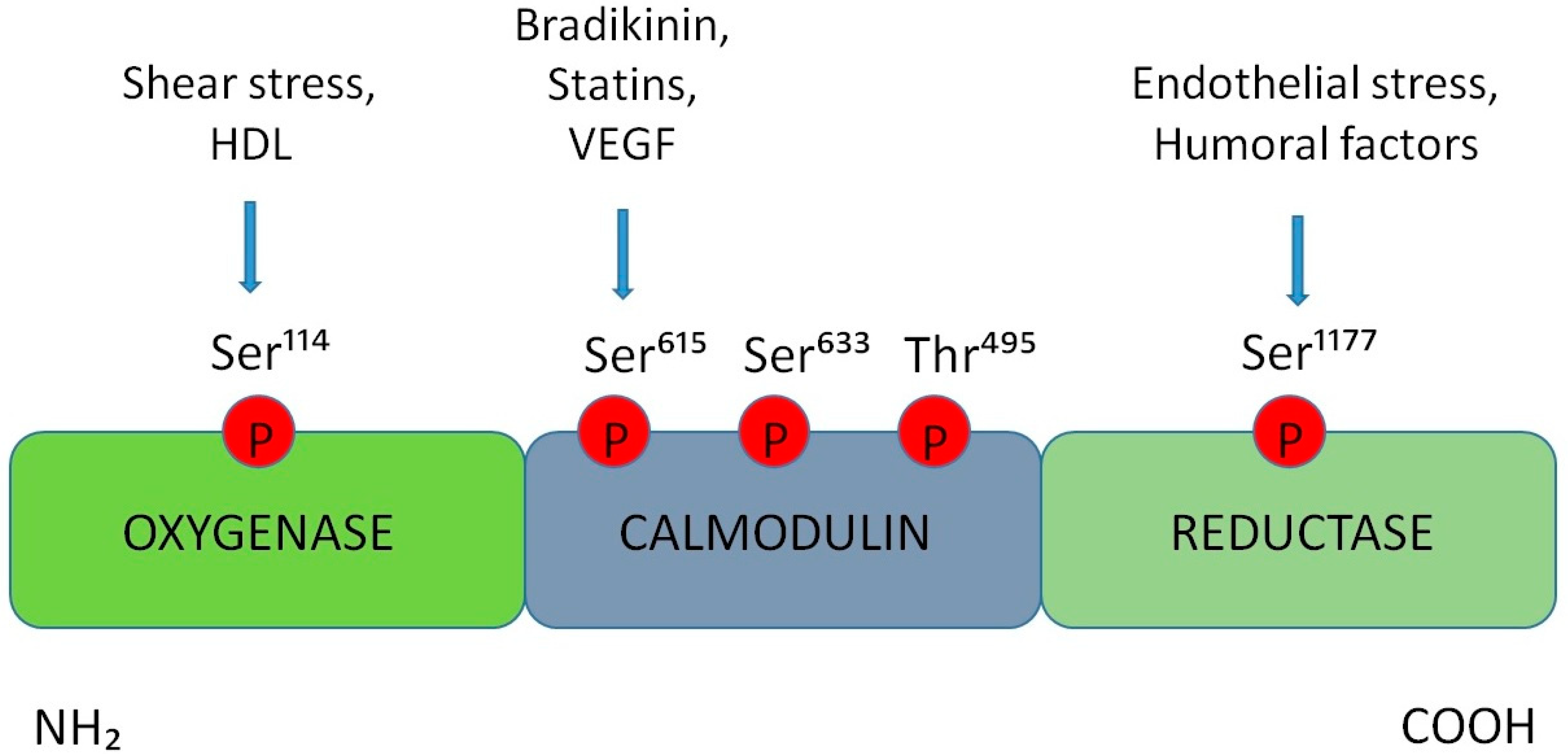
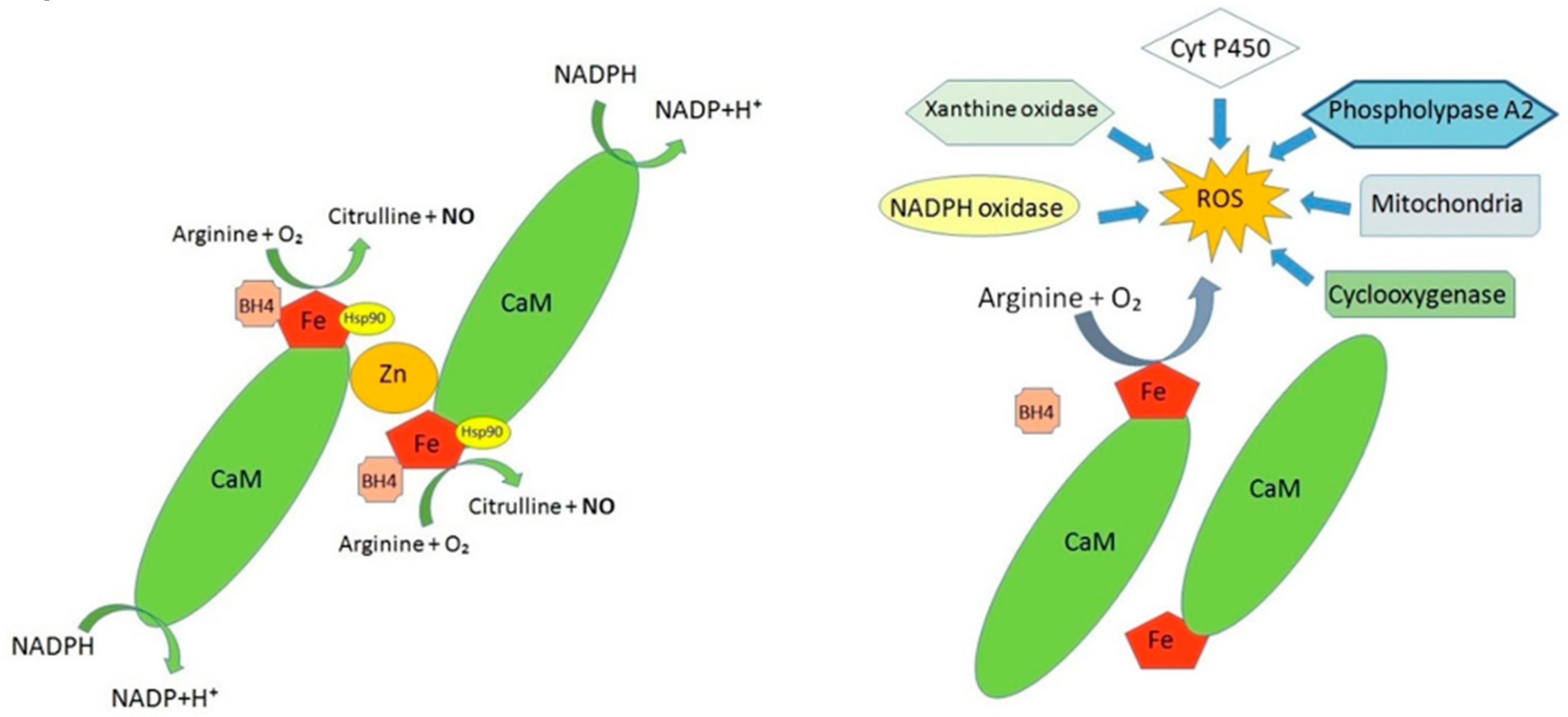
3. Alterations in the Production of Nitric Oxide at the BNB Level
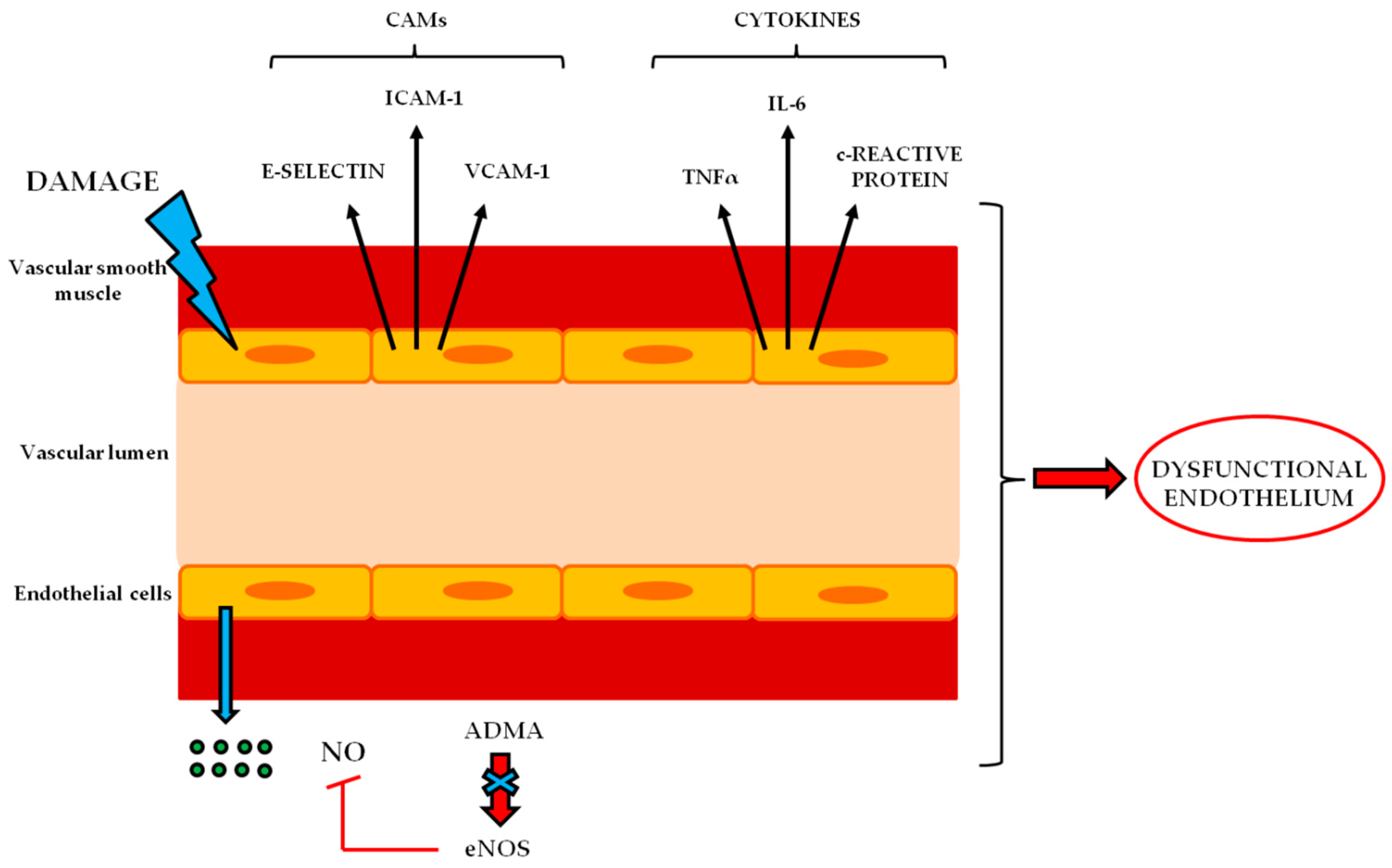
4. Endothelial Dysfunction in BNB-Related Disorders
5. BNB Dysfunction in Diabetic Neuropathy
6. BNB Involvement in Erectile Dysfunction
7. BNB and Neuropathic Pain
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef] [PubMed]
- Pelz, J.; Härtig, W.; Weise, C.; Hobohm, C.; Schneider, D.; Krueger, M.; Kacza, J.; Michalski, D. Endothelial Barrier Antigen immunoreactivity is Conversely Associated with Blood-Brain Barrier Dysfunction after Embolic Stroke in Rats. Eur J. Histochem. 2013, 57, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.D.; Ganz, P.; Hamburg, N.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The Assessment of Endothelial Function- From Research into Clinical Practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef]
- Cersosimo, E.; DeFronzo, R.A. Insulin resistance and endothelial dysfunction: The road map to cardiovascular diseases. Diabetes Metab Res. Rev. 2006, 22, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Reina, M.; López, A.; Villanueva, M.; De Andrés, J.; Machés, F. The blood-nerve barrier in peripheral nerves. Rev. Esp Anestesiol Reanim. 2003, 50, 80–86. [Google Scholar]
- Khazaei, M.; Moien-afshari, F.; Laher, I. Vascular endothelial function in health and diseases. Pathophysiology 2008, 15, 49–67. [Google Scholar] [CrossRef]
- Abe, M.; Sano, Y.; Maeda, T.; Shimizu, F.; Kashiwamura, Y.; Haruki, H.; Saito, K.; Tasaki, A.; Kawai, M.; Terasaki, T.; et al. Establishment and characterization of human peripheral nervemicrovascular endothelial cell lines: A new in vitro blood-nerve barrier (bnb) model. Cell Struct Funct. 2012, 37, 89–100. [Google Scholar] [CrossRef]
- Kanda, T.; Numata, Y.; Mizusawa, H. Chronic inflammatory demyelinating polyneuropathy: Decreased claudin-5 and relocated zo-1. J. Neurol. Neurosurg. Psychiatry 2004, 75, 765–769. [Google Scholar] [CrossRef]
- Petrova, N.L.; Shanahan, C.M. Neuropathy and the vascular-bone axis in diabetes: Lessons from Charcot osteoarthropathy. Osteoporos Int. 2014, 25, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Zenker, J.; Dan Ziegler, D.; Chrast, R. Novel pathogenic pathways in diabetic neuropathy. Trends Neurosci. 2013, 36, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.B.; Tsingalia, A.; Vidensky, S.; Lee, Y.; Jin, L.; Farah, M.H.; Lengacher, S.; Magistretti, P.J.; Pellerin, L.; Rothstein, J.D. Deficiency in Monocarboxylate Transporter 1 (MCT1) in Mice Delays Regeneration of Peripheral Nerves following Sciatic Nerve Crush. Exp. Neurol. 2015, 263, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Zhang, Z.J.; Ohtsu, T. Homozygous Disruption of the mdr1a P-glycoprotein Gene Affects Blood-Nerve Barrier Function in Mice Administered with Neurotoxic Drugs. Acta Oto-Laryngologica 2001, 121, 735–742. [Google Scholar] [PubMed]
- Yosef, N.; Ubogu, E.E. An immortalized human blood-nerve barrier endothelial cell line for in vitro permeability studies. Cell. Mol. Neurobiol. 2013, 33, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Tachikawa, M.; Takanaga, H.; Shimizu, H.; Watanabe, M.; Hosoya, K.; Terasaki, T. The blood-brain barrier creatine transporter is a major pathway for supplying creatine to the brain. J. Cereb Blood Flow Metab. 2002, 22, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The p-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.; Lerman, A. The endothelium: Dysfunction and beyond. J. Nucl Cardiol. 2001, 8, 197–206. [Google Scholar] [CrossRef]
- Gokce, N.; Keaney, J.F., Jr.; Hunter, L.M. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: A prospective study. Circulation 2002, 105, 1567–1572. [Google Scholar] [CrossRef]
- Schwarzmaier, S.M.; Terpolilli, N.A.; Dienel, A.; Gallozzi, M.; Schinzel, R.; Tegtmeier, F.; Plesnila, N. Endothelial nitric oxide synthase mediates arteriolar vasodilatation after traumatic brain injury in mice. J. Neurotrauma. 2015, 32, 731–738. [Google Scholar] [CrossRef]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, R.C.; Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M.; et al. Amyloid β peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflammation 2015, 3, 12–84. [Google Scholar] [CrossRef] [PubMed]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the NO/cGMP signaling pathway. Biochim. Biophys. Acta 1999, 1411, 334–3350. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from l-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Hai-na, Z.; Qiao-qiao, X.; Abhimanyu, T.; Martin Omondi, A.; Manas, C.; Arunima, G.; Xu-ben, Y. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar]
- Michel, T.; Feron, O. Nitric oxide synthases: Which, where, how, and why? J. Clin. Investig. 1997, 100, 2146–2152. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role invascular biology. Br. J. Pharmacol. 2006, 147, S193–S201. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowls, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Hideshi, I.; Atsushi, K.; Shingo, K.; Tomoaki, I.; Yuki, K.; Hiroyasu, T.; Tomohiro, S.; Yasuo, W.; Takaaki, A. Superoxide generation from nNOS splice variants and its potential involvement in redox signal regulation. Biochem. J. 2017, 474, 1149–1162. [Google Scholar]
- Mount, P.F.; Bruce, E.K.; Power, D.A. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. JMCC 2007, 42, 271–279. [Google Scholar] [CrossRef]
- Patel, C.; Rojas, M.; Narayanan, S.P.; Zhang, W.; Xu, Z.; Lemtalsi, T.; Jittiporn, K.; Caldwell, R.W.; Caldwell, R.B. Arginase as mediator of diabetic retinopathy. Front. Immunol. 2013, 4, 173. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Guo, Y.; Yang, X.; He, J.; Liu, J.; Yang, S.; Dong, H. Important roles of the Ca2+sensing receptor in vascular health and disease. Life Sci. 2018, 209, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Tat’yanenko, L.V.; Sanina, N.A.; Dobrokhotova, O.V.; Kotelnikov, A.I.; Goryachev, N.S.; Pihteleva, I.; Kozub, G.I.; Kondrateva, T.A. Action of Iron Nitrosyl Complexes, NO Donors, on the Activity of Sarcoplasmic Reticulum Ca2+-ATPase and Cyclic Guanosine Monophosphate Phosphodiesterase. Biochem. Biophys. Mol. Biol. 2018, 478, 8–13. [Google Scholar] [CrossRef]
- Quintana-Lopez, L.; Gonzalo Perez-Arana, M.; Cebada-Aleu, A.; Lechuga-Sancho, A.; Aguilar-Diosdado, M.; Segundo, C. Nitric Oxide Is a Mediator of Antiproliferative Effects Induced by Proinflammatory Cytokines on Pancreatic Beta Cells. Mediators Inflamm. 2013, 2013, 905175. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Mazumder, S.; Sinha, A.K. The role of inhibition of nitric oxide synthesis in the aggregation of platelets due to the stimulated production of thromboxane A2. Blood Coagul. Fibrinolysis 2014, 25, 585–591. [Google Scholar] [CrossRef]
- Gross, S.S.; Wolin, M.S. Nitric oxide: Pathophysiological mechanisms. Annu. Rev. Physiol. 1995, 57, 737–769. [Google Scholar] [CrossRef]
- Huang, A.L.; Silver, A.E.; Shvenke, E. Predictive value of reactive hyperemia for cardiovascular events in patients with peripheral arterial disease undergoing vascular surgery. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2113–2119. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. Endogenous nitric oxide: Physiology, pathology and clinical relevance. Eur. J. Clin Investig. 1991, 21, 361–374. [Google Scholar] [CrossRef]
- Pereira, M.L.; D’ancona, C.A.; Rojas-Moscoso, J.A.; Ramos, A.C.; Filho, M.F.Z.; Antunes, E. Effects of nitric oxide inhibitors in mice with bladder outlet obstruction. Int. Braz. J. Urol. 2017, 43, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Jagadapillai, R.; Vaishnav, R.A.; Friedland, R.P.; Drinovac, R.; Lin, X.; Gozal, E. Increased pulmonary arteriolar tone associated with lung oxidative stress and nitric oxide in a mouse model of Alzheimer’s disease. Physiol. Rep. 2016, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A CriticalMolecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol Sci. 2019, 20, 1658. [Google Scholar] [CrossRef]
- Maamoun, H.; Benameur, T.; Pintus, G.; Shankar Munusamy, S.; Agouni, A. Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 2008, 97, 2222–2229. [Google Scholar] [CrossRef]
- Taddei, S.; Salvetti, A. Endothelial dysfunction in essential hypertension: Clinical implications. J. Hypertens. 2002, 20, 1671–1674. [Google Scholar] [CrossRef]
- Sugamura, K.; Keaney, J.F. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef]
- Steyers, C.M.; Miller, F.J., Jr. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Singh, U.; Jialal, I. The Evolving Role of C-Reactive Protein in Atherothrombosis. Clin. Chem. 2009, 55, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Samarasekera, E.J.; Neilson, J.M.; Warren, R.B.; Parnham, J.; Smith, C.H. Incidence of cardiovascular disease in individuals with psoriasis: A systematic review and meta analysis. J. Investig. Dermatol. 2013, 133, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Ralevic, V. New insights into the local regulation of blood flow by perivascular nerves and endothelium. Br. J. Plasnc Surg. 1994, 47, 527–543. [Google Scholar] [CrossRef]
- Jaimes, L.; Vinet, R.; Knox, M.; Morales, B.; Benites, J.; Laurido, C.; Martínez, J.L. A Review of the Actions of Endogenous and Exogenous Vasoactive Substances during the Estrous Cycle and Pregnancy in Rats. Animals 2019, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Schmitz-Winnenthal, F.H.; Fe´ le´ tou, M.; Go¨ decke, A.; Huang, P.L.; Vanhoutte, P.M.; Fleming, I.; Busse, R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 9747–9752. [Google Scholar] [CrossRef]
- Kanda, T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 208–212. [Google Scholar] [CrossRef]
- Wälchli, T.; Wacker, A.; Frei, K.; Regli, L.; Schwab, M.E.; Hoerstrup, S.P.; Gerhardt, H.; Engelhardt, B. Wiring the Vascular Network with Neural Cues: A CNS Perspective. Neuron 2015, 87, 271–296. [Google Scholar] [CrossRef]
- Hackel, D.; Brack, A.; Fromm, M.; Rittner, H.L. Modulation of tight junction proteins in the perineurium for regional pain control. Ann. N. Y. Acad. Sci. 2012, 1257, 199–206. [Google Scholar] [CrossRef]
- Goethals, S.; Ydens, E.; Timmerman, V.; Janssens, S. Toll-like receptor expression in the peripheral nerve. Glia 2010, 58, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Freria, C.M.; Bernardes, D.; Almeida, G.L.; Simões, G.F.; Barbosa, G.O.; Oliveira, A.L.R. Impairment of toll-like receptors 2 and 4 leads to compensatory mechanisms after sciatic nerve axotomy. J. Neuroinflammation 2016, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Marchand, F.; Perretti, M.; McMahon, S.B. Role of the Immune system in chronic pain. Nat. Rev. Neurosci. 2005, 6, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc Immunol. 2015, 109, 1–10. [Google Scholar]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.K.Y.; Shi, X.Q.; Johnson, J.M.; Rone, M.B.; Antel, J.P.; David, S.; Zhang, J. Peripheral nerve injury induces persistent vascular dysfunction and endoneurial hypoxia, contributing to the genesis of neuropathic pain. J. Neurosci. 2015, 35, 3346–3359. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Jung, H.; Miller, R.J. Chemokines and the pathophysiology of neuropathic pain. Proc. Natl. Acad. Sci. USA 2007, 104, 20151–20158. [Google Scholar] [CrossRef]
- Wilmot, E.; Edwardson, C.; Achana, F.A.; Davies, M.J.; Gorely, T.; Gray, L.J.; Khunti, K.; Yates, T.; Biddle, S.J. Sedentary time in adults and the association with diabetes, cardiovascular disease and death: Systematic review and meta-analysis. Diabetologia 2012, 55, 2895–5905. [Google Scholar] [CrossRef]
- Feener, E.P.; King, G.L. Endothelial dysfunction in diabetes mellitus: Role in cardiovascular disease. Heart Fail. Monit. 2001, 1, 74–82. [Google Scholar]
- Callaghan, B.; Cheng, H.T.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef]
- Simmons, Z.; Feldman, E.L. Update on diabetic neuropathy. Curr. Opin. Neurol. 2002, 15, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.L.; Galea, M.P.; O’Neal, D.N.; Dodson, M.J. Peripheral neuropathy in the hands of people with diabetes mellitus. Diabetes Res. Clin. Pract. 2016, 119, 23–31. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic neuropathy: A position statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Basha, B.; Samuel, S.M.; Triggle, C.R.; Ding, H. Endothelial dysfunction in diabetes mellitus: Possible involvement of endoplasmic reticulum stress? Exp. Diabetes Res. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Oates, P.J. Aldose reductase, still a compelling target for diabetic neuropathy. Curr. Drug Targets 2008, 9, 14–36. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, K.; Kaufman, J. The unfolded protein response- A stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Bishara, N.B.; Ding, H. Glucose enhances expression of TRPC1 and calcium entry in endothelial cells. Am. J. Physiol. 2010, 298, H171–H178. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.W.; Fugl-Meyer, K.S.; Corona, G.; Hayes, R.D.; Laumann, E.O. Definitions/epidemiology/risk factors for sexual dysfunction. J. Sex. Med. 2010, 7, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Sopko, N.A.; Hannan, J.L.; Bivalacqua, T.J. Pathophysiology of Erectile Dysfunction. Curr. Drug Targets 2015, 16, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Malara, N.; Gratteri, S.; Palma, E.; Zappia, L.; Costa, N.; Rosano, G.; Paone, S. Bergamot polyphenolic fraction counteracts erectile dysfunction occurring in patients suffering from type 2 diabetes. Pharma Nutr. 2016, 4S, S41–S46. [Google Scholar] [CrossRef]
- De Berardis, G.; Pellegrini, F.; Franciosi, M.; Belfiglio, M.; Di Nardo, B.; Greenfield, S. Identifying patients with type 2 diabetes with a higher likelihood of erectile dysfunction: The role of the interaction between clinical and psychological factors. J. Urol. 2003, 169, 1422–1428. [Google Scholar] [CrossRef]
- Kirby, M.; Jackson, G.; Betteridge, J.; Friedli, K. Is erectile dysfunction a marker for cardiovascular disease? Int. J. Clin. Pract. 2001, 55, 614–618. [Google Scholar]
- Maiorino, M.I.; Bellastella, G.; Esposito, K. Lifestyle modifications and erectile dysfunction: What can be expected? Asian J. Androl. 2015, 17, 5–10. [Google Scholar]
- Cao, S.; Yin, X.; Wang, Y.; Zhou, H.; Song, F.; Lu, Z. Smoking and risk of erectile dysfunction: Systematic review of observational studies with meta-analysis. PLoS ONE 2013, 8, e60443. [Google Scholar] [CrossRef]
- Pourmand, G.; Alidaee, M.R.; Rasuli, S.; Maleki, A.; Mehrsai, A. Do cigarette smokers with erectile dysfunction benefit from stopping? A prospective study. B.J.U. Int. 2004, 94, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Ponholzer, A.; Temml, C.; Mock, K.; Marszalek, M.; Obermayr, R. Prevalence and risk factors for erectile dysfunction in 2869 men using a validated questionnaire. Eur. Urol. 2005, 47, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.H.; Wagner, G.; Heitmann, B.L. Sexual function and obesity. Int. J. Obes. 2007, 31, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Dai, S.; Wang, M.; Morrison, H. Erectile dysfunction and fruit/vegetable consumption among diabetic Canadian men. Urology 2013, 82, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Atlantis, E.; Lange, K.; Taylor, A.W.; O’Loughlin, P. Predictors of sexual dysfunction incidence and remission in men. J. Sex. Med. 2014, 11, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Leite, L.N.; Gabriel, T.; do Vale, G.T.; Simplicio, J.A.; De Martinis, B.F.; Carneiro, F.S.; Tirapelli, C.R. Ethanol-induced erectile dysfunction and increased expression of proinflammatory proteins in the rat cavernosal smooth muscle are mediated by NADPH oxidase-derived reactive oxygen species. Eur. J. Pharmacol. 2017, 804, 82–93. [Google Scholar] [CrossRef]
- Blick, C.; Ritchie, R.W.; Sullivan, M.E. Is Erectile Dysfunction an Example of Abnormal Endothelial Function? Curr. Vasc. Pharmacol. 2016, 14, 163–167. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular noxs, and hypertension: Focus on translational and clinical research. Antioxid Redox Signal. 2014, 20, 164–182. [Google Scholar] [CrossRef]
- Tuncayengin, A.; Biri, H.; Onaran, M.; Sen, I.; Tuncayengin, O.; Polat, F. Cavernosal tissue nitrite, nitrate, malondialdehyde and glutathione levels in diabetic and non-diabetic erectile dysfunction. Int. J. Androl. 2003, 26, 250–254. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Silva, F.H.; Mónica, F.Z.; Báu, F.R.; Brugnerotto, A.F.; Priviero, F.B.; Toque, H.A.; Antunes, E. Superoxide anion production by NADPH oxidase plays a major role in erectile dysfunction in middle-aged rats: Prevention by antioxidant therapy. J. Sex. Med. 2013, 10, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Yafi, F.A.; Jenkins, L.; Albersen, M.; Corona, G.; Isidori, A.M.; Goldfarb, S.; Maggi, M.; Nelson, C.J.; Parish, S.; Salonia, A.; et al. Erectile dysfunction. Nat. Rev. Dis. Prim. 2016, 2, 16003. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.L.; Fais, R.S.; Tostes, R.C.; Carneiro, F.S. There is a Link Between Erectile Dysfunction and Heart Failure: It could be Inflammation. Curr. Drug Targets 2015, 16, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, L.V.; Schneider, A.P.; Barbosa, S.; Ilha, J.; Faccioni-Heuser, M.C. Balance and coordination training and endurance training after nerve injury. Muscle Nerve 2015, 51, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef]
- Challa, S.R. Surgical animal models of neuropathic pain: Pros and Cons. Int. J. Neurosci. 2015, 125, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Savastano, L.E.; Laurito, S.R.; Fitt, M.R.; Rasmussen, J.A.; Gonzalez Polo, V.; Patterson, S.I. Sciatic nerve injury: A simple and subtle model for investigating many aspects of nervous system damage and recovery. J. Neurosci. Methods 2014, 227, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.M.; Shu, Y.H.; Qiu, C.H.; Chen, K.T.; Wang, Y.T. Protective effects and anti-apoptotic role of nerve growth factor on spinal cord neurons in sciatic nerve-injured rats. Neurol. Res. 2014, 36, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Karsidag, S.; Akcal, A.; Sahin, S.; Karsidag, S.; Kabukcuoglu, F.; Ugurlu, K. Neurophysiological and morphological responses to treatment with acetyl-l-carnitine in a sciatic nerve injury model: Preliminary data. J. Hand Surg. 2012, 37, 529–536. [Google Scholar] [CrossRef]
- Blom, C.L.; Martensson, L.B.; Dahlin, L.B. Nerve injury-induced c-Jun activation in Schwann cells is JNK independent. Biomed. Res. Int. 2014, 2014, 392971. [Google Scholar]
- Pelletier, J.; Fromy, B.; Morel, G.; Roquelaure, Y.; Saumet, J.L. Sigaudo Roussel, D. Chronic sciatic nerve injury impairs the local cutaneous neurovascular interaction in rats. Pain 2012, 153, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Khoa, P.; Nima, N.; Ranjan, G. c-Jun, krox-20, and integrin β4 expression following chronic nerve compression injury. Neurosci Lett. 2009, 465, 194–198. [Google Scholar]
- Qinwen, L.I.; Jianghai, C.; Yanhua, C.; Xiaobin, C.; Zhenbing, C. Chronic sciatic nerve compression induces fibrosis in dorsal root ganglia. Mol. Med. Rep. 2016, 13, 2393–2400. [Google Scholar]
- Khan, J.; Hassun, H.; Zusman, T.; Korczeniewska, O.; Eliav, E. Interleukin-8 levels in rat models of nerve damage and neuropathic pain. Neurosci. Lett. 2017, 657, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Lauro, F.; Ilari, S.; Giancotti, L.A.; Ventura, C.A.; Morabito, C.; Gliozzi, M.; Malafoglia, V.; Palma, E.; Paolino, D.; Mollace, V.; et al. Pharmacological effect of a new idebenone formulation in a model of carrageenan-induced inflammatory pain. Pharmacol. Res. 2016, 111, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Guedes, R.P.; Dal Bosco, L.; da Rosa Araújo, A.S.; Belló-Klein, A.; Marques Ribeiro, M.F.; Aparecida Partata, W. Sciatic nerve transection increases gluthatione antioxidant system activity and neuronal nitric oxide synthase expression in the spinal cord. Brain Res. Bull. 2009, 80, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.F.; Yan, N.; Xu, H.; Sun, J.H.; Xiong, Y.C.; Deng, X.M. Edaravone, a free radical scavenger, is effective on neuropathic pain in rats. Brain Res. 2009, 1248, 68–75. [Google Scholar] [CrossRef]
- Liu, T.; Knight, K.R.; Tracey, D.J. Hyperalgesia due to nerve injury-role of peroxynitrite. Neuroscience 2000, 97, 125–131. [Google Scholar] [CrossRef]
- Menorca, B.S.; Fussell, T.S.; Elfar, M.D. Peripheral Nerve Trauma: Mechanisms of Injury and Recovery. Hand Clin. 2013, 29, 317–330. [Google Scholar] [CrossRef]
- Geuna, S.; Raimondo, S.; Ronchi, G.; Di Scipio, F.; Tos, P.; Czaja, K. Histology of theperipheral nerve and changes occurring during nerve regeneration. Int. Rev. Neu-robiol 2009, 87, 27–46. [Google Scholar]
- Bazzoni, G.; Dejana, E. Pores in the sieve and channels in the wall: Control of paracellular permeability by junctional proteins in endothelial cells. Microcirculation 2001, 8, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Moreau, N.; Mauborgne, A.; Bourgoin, S.; Couraud, P.O.; Romero, I.A.; Weksler, B.B.; Villanueva, L.; Pohl, M.; Boucher, Y. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation and neuropathic pain development. Pain 2016, 157, 827–839. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BNB Components | Role in BNB | Expression in BNB-Related Disorders |
|---|---|---|
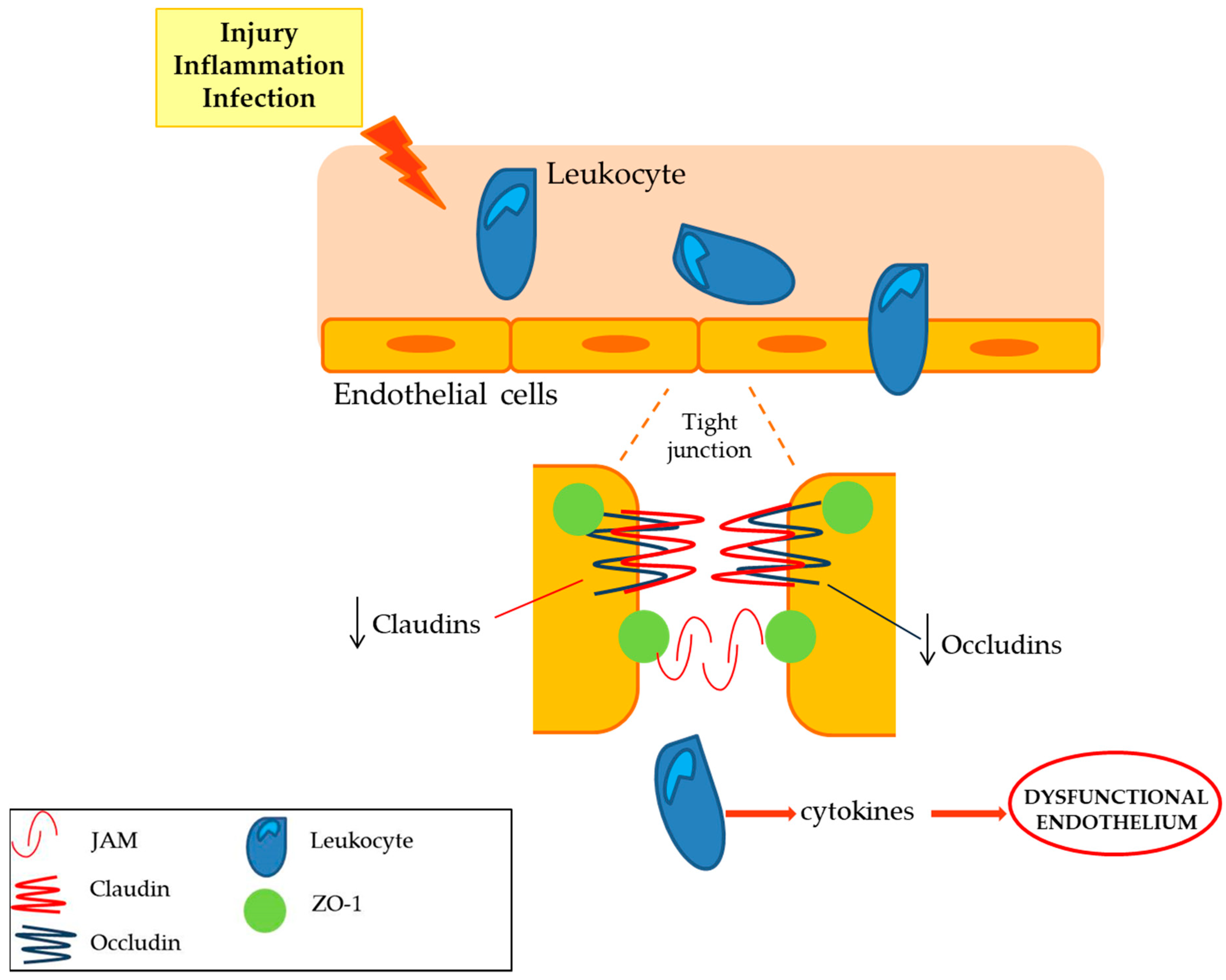
| Claudins (claudin 1, claudin 2, claudin 5 and claudin 19) | Tight junction that limits the cellular permeability. They modulates the passage of leukocyte, regulating the immunosurveillance of the tissues [10]. | The expression of claudin 5 is decreased while the claudin 1, 2 expression is not affected in peripheral nerve inflammation. Deficient claudin 19 mice showed a deficit in PNS [10,11]. |
| Occludins (ZO-1, ZO-2) | The expression of occludins is decreased in peripheral nerve inflammation, whilst ZO-1 and ZO-2 localization is altered [10,11]. | |
| Cell adhesion molecules | The expression of intercellular adhesion molecules, such as ICAM -1, VCAM-1 and selectin E, is up-regulated in peripheral neuroinflammatory disease [12,13]. |
| BNB Transportes | Role in BNB | Expression in BNB-Related Disorders |
|---|---|---|
| Alkaline phosphatase, AP | Ionic transporter of the capillary endothelium transferring phosphate groups and preserving ionic concentrations [14]. | AP has been linked to the degradation of the calcification inhibitor pyrophosphate to promote VSMC calcification [14]. |
| Glucose transporter-1, GLUT-1 | Transporter of D-glucose. It facilitates its passage into the endothelium as source of energy [15]. | GLUT-1 expression in diabetic sensorimotor polyneuropathy don’t change significantly, but it is possible that diabetic condition leads to an alteration in their localization or in post-translational modification [16]. |
| Monocarboxylate transporter 1, MCT-1 | Transporter of monocarboxylic acids such as L-lactate. Under anaerobic conditions or starvation, it provides lactate as source of energy [15]. | The expression levels of MCT-1 is reduced after sciatic nerve injury [17]. |
| Creatine transporter, CRT | Transporter for creatinine that is necessary to supplies high-energy phosphate groups for the production of ATP [18]. | Not so far investigated. |
| ABC transporters (ATP bond box), MDR-1 | Efflux transporter that guarantee the outflow of xenobiotics and toxic tissue metabolism intermediates. It is fundamental for the protection of peripheral nerves from external factors [19]. | Lack of MDR-1 expression leads to an increased toxicity drugs induced in BNB related disorders [20]. |
| Toll Like Receptors (TLRs) on BNB | Role | TLRs in BNB-Related Disorders |
|---|---|---|
| TLR-1 | Toll like receptors are transmembrane receptors able to recognize pathogens or microbes that activate the sentinel cells of the immune system. These receptors are involved in the immune response during neurodegeneration [62,63,64]. | TLR-1 is strongly induced in neurodegeneration in the sciatic nerve after injury [62]. |
| TLR-2 | TLR-2 knockout mice showed an increased rate of degenerated axons. However its absence does not influence the overall functional recovery [65]. | |
| TLR-3 | TLR-3 is modestly induced in neurodegeneration in the sciatic nerve after injury [62]. | |
| TLR-4 | TLR-4 is linked to neuropathic pain. In TLR-4 ko mice a decresed level of proinflammatory interleuchine 1β, interferon-γ and TNFalpha, has been showed, without a mechanical allodynia after peripheral nerve injury [66]. | |
| TRL-6 | TLR-6 is modestly induced in neurodegeneration in the sciatic nerve after injury [62]. | |
| TLR-7 | TLR-7 and TLR-9 are not affected in neurodegenertion in the sciatic nerve after injury [62]. | |
| TLR-9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. https://doi.org/10.3390/ijms20123022
Maiuolo J, Gliozzi M, Musolino V, Carresi C, Nucera S, Macrì R, Scicchitano M, Bosco F, Scarano F, Ruga S, et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. International Journal of Molecular Sciences. 2019; 20(12):3022. https://doi.org/10.3390/ijms20123022
Chicago/Turabian StyleMaiuolo, Jessica, Micaela Gliozzi, Vincenzo Musolino, Cristina Carresi, Saverio Nucera, Roberta Macrì, Miriam Scicchitano, Francesca Bosco, Federica Scarano, Stefano Ruga, and et al. 2019. "The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications" International Journal of Molecular Sciences 20, no. 12: 3022. https://doi.org/10.3390/ijms20123022
APA StyleMaiuolo, J., Gliozzi, M., Musolino, V., Carresi, C., Nucera, S., Macrì, R., Scicchitano, M., Bosco, F., Scarano, F., Ruga, S., Zito, M. C., Oppedisano, F., Mollace, R., Paone, S., Palma, E., Muscoli, C., & Mollace, V. (2019). The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. International Journal of Molecular Sciences, 20(12), 3022. https://doi.org/10.3390/ijms20123022