Experimental Approaches and Computational Modeling of Rat Serum Albumin and Its Interaction with Piperine

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
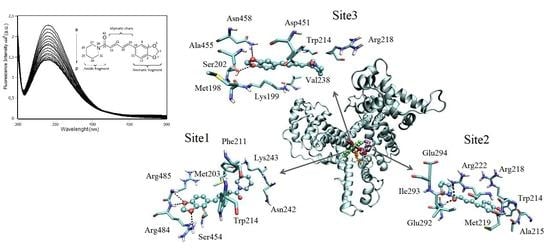
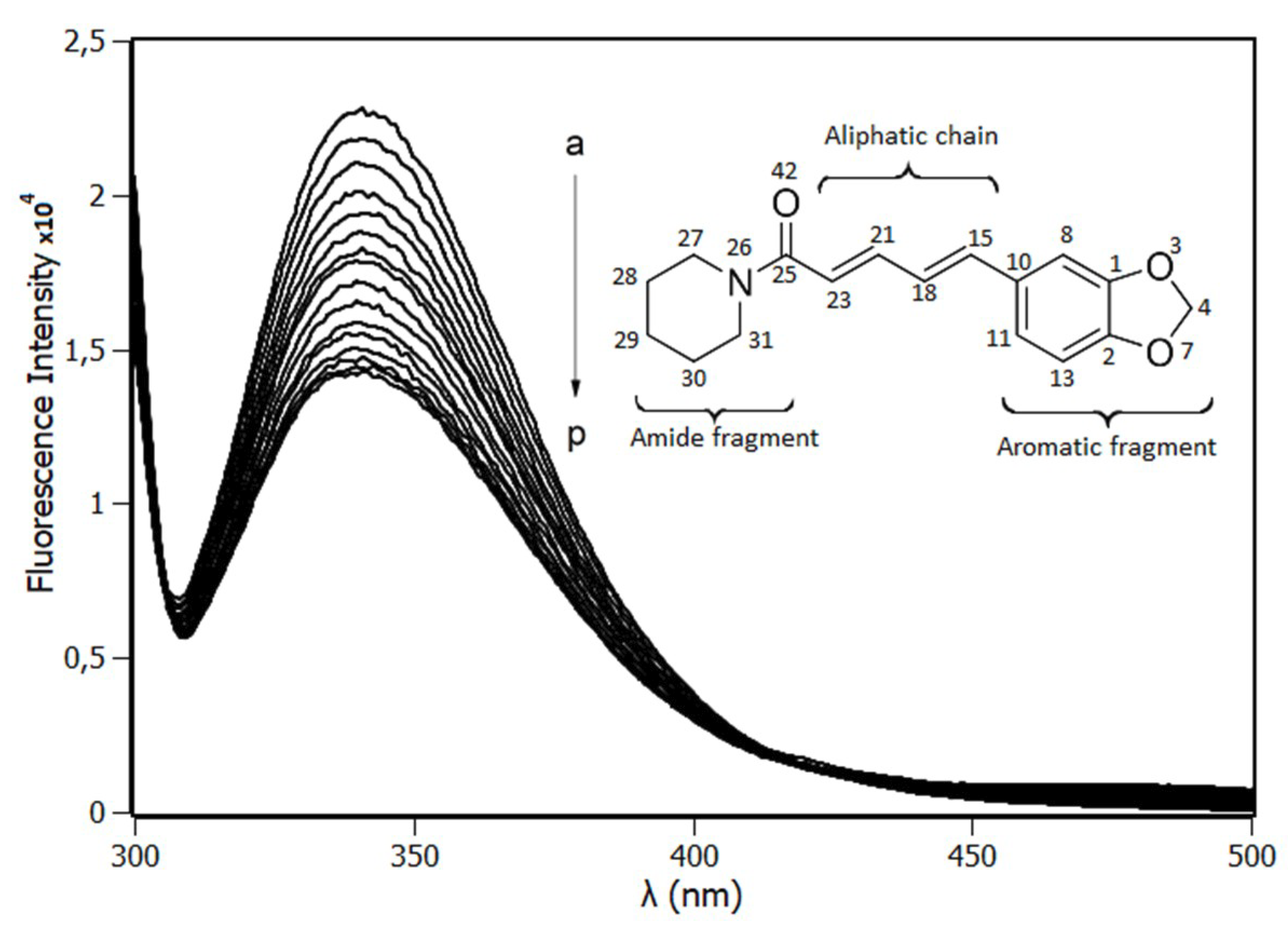
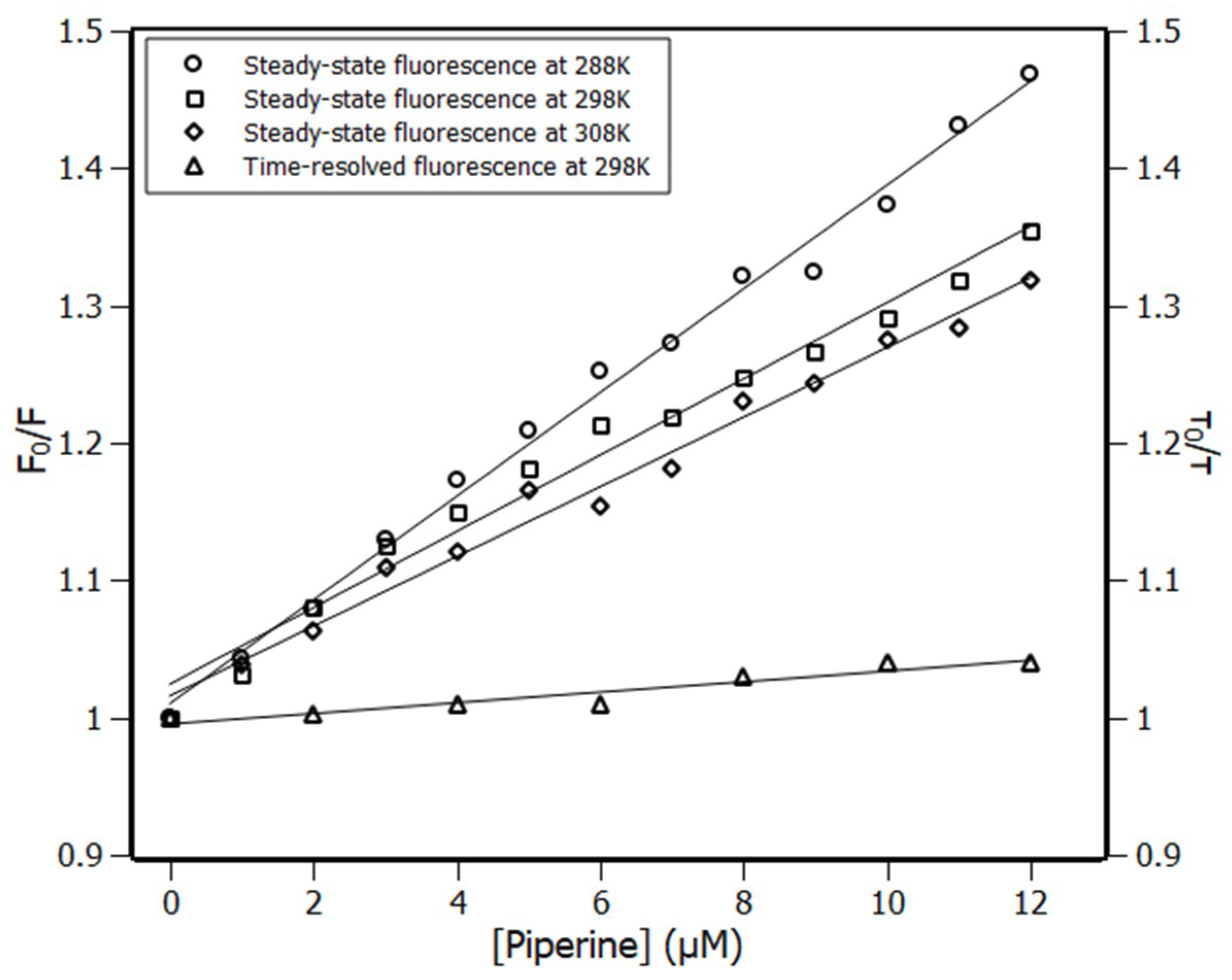
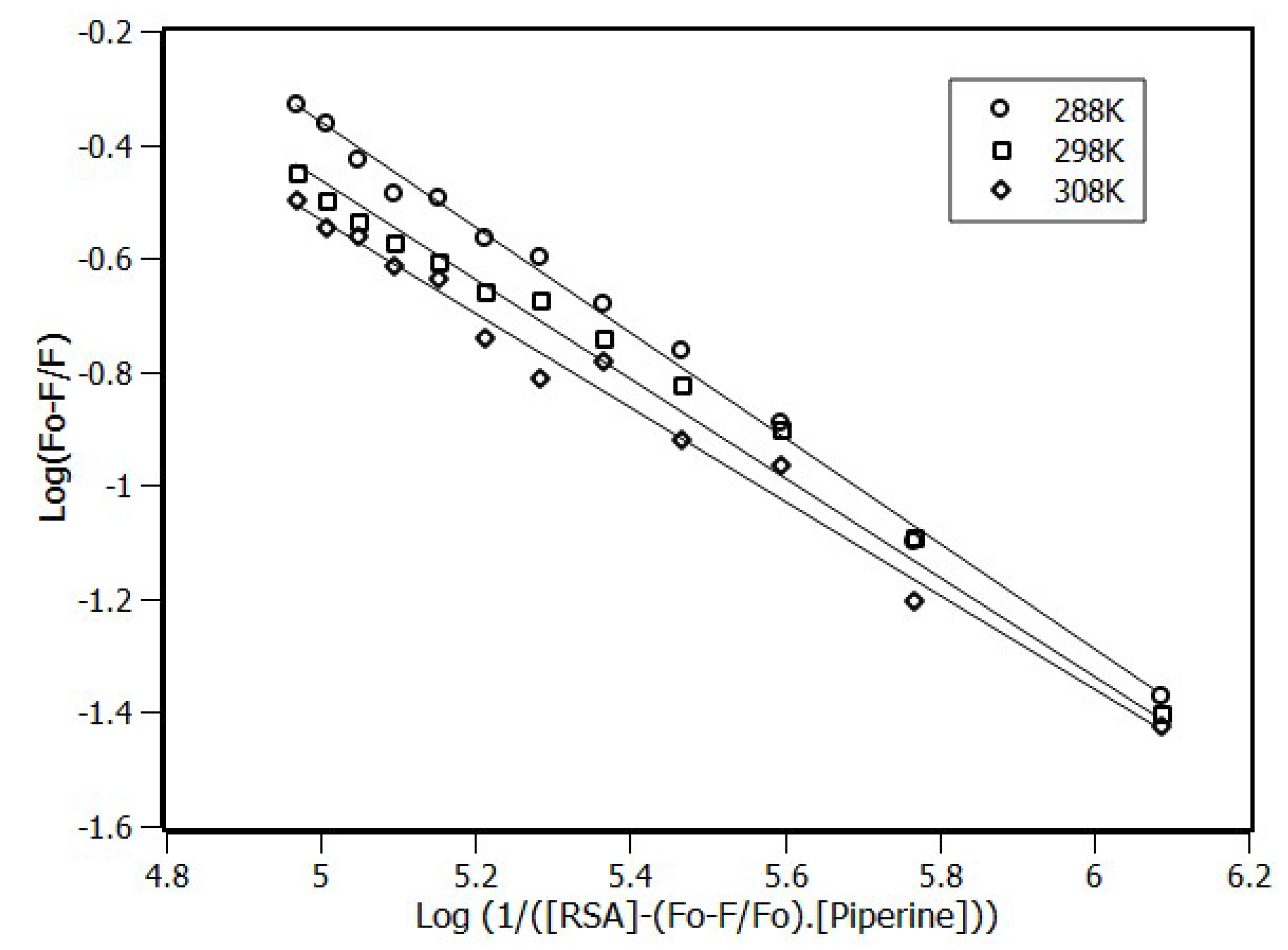
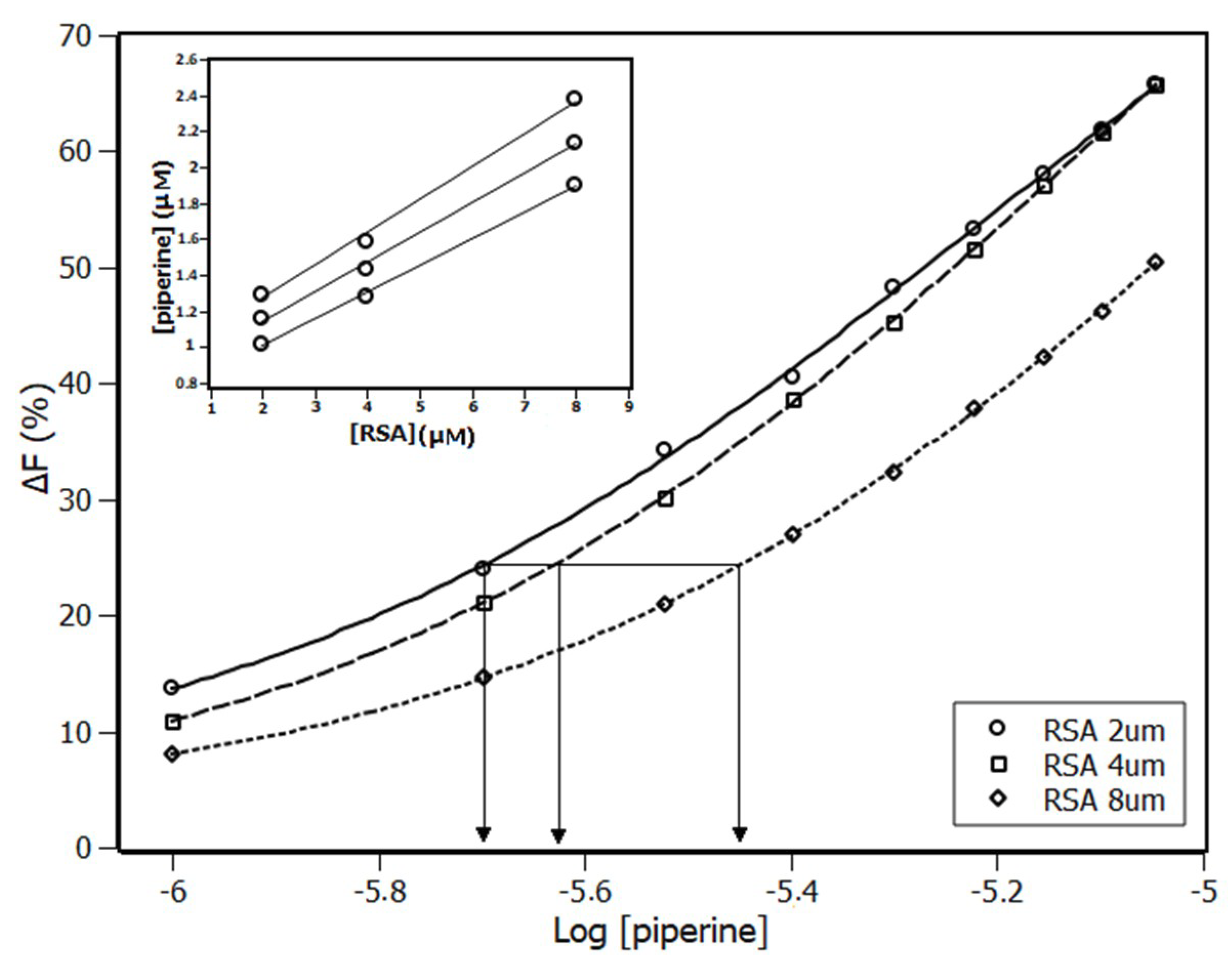
2.1. Fluorescence Spectroscopy
2.2. Thermodynamic Parameters
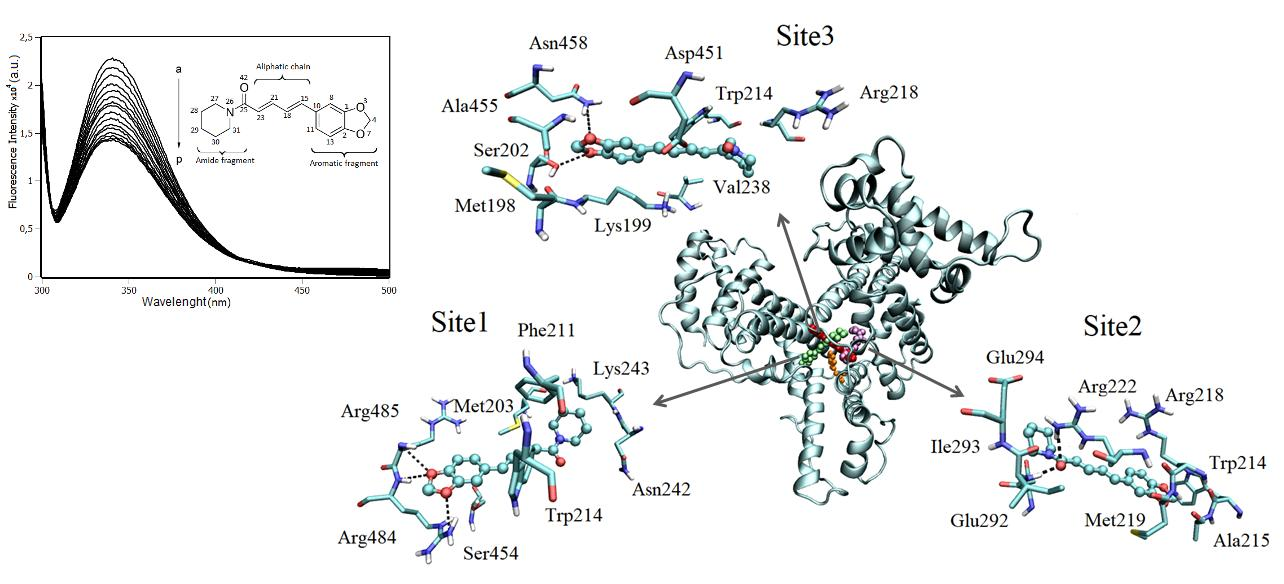
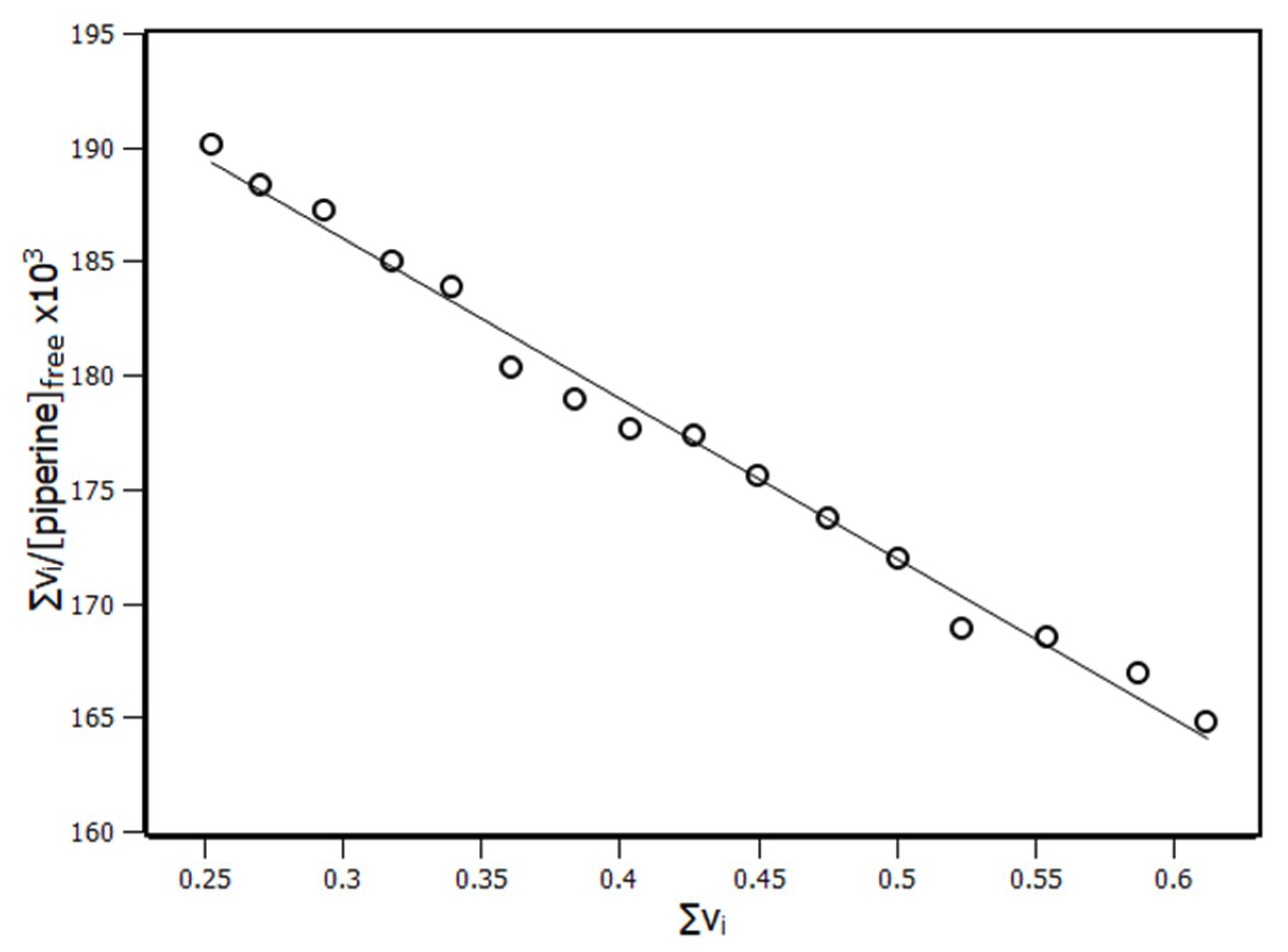
2.3. Interaction Density Function (IDF)
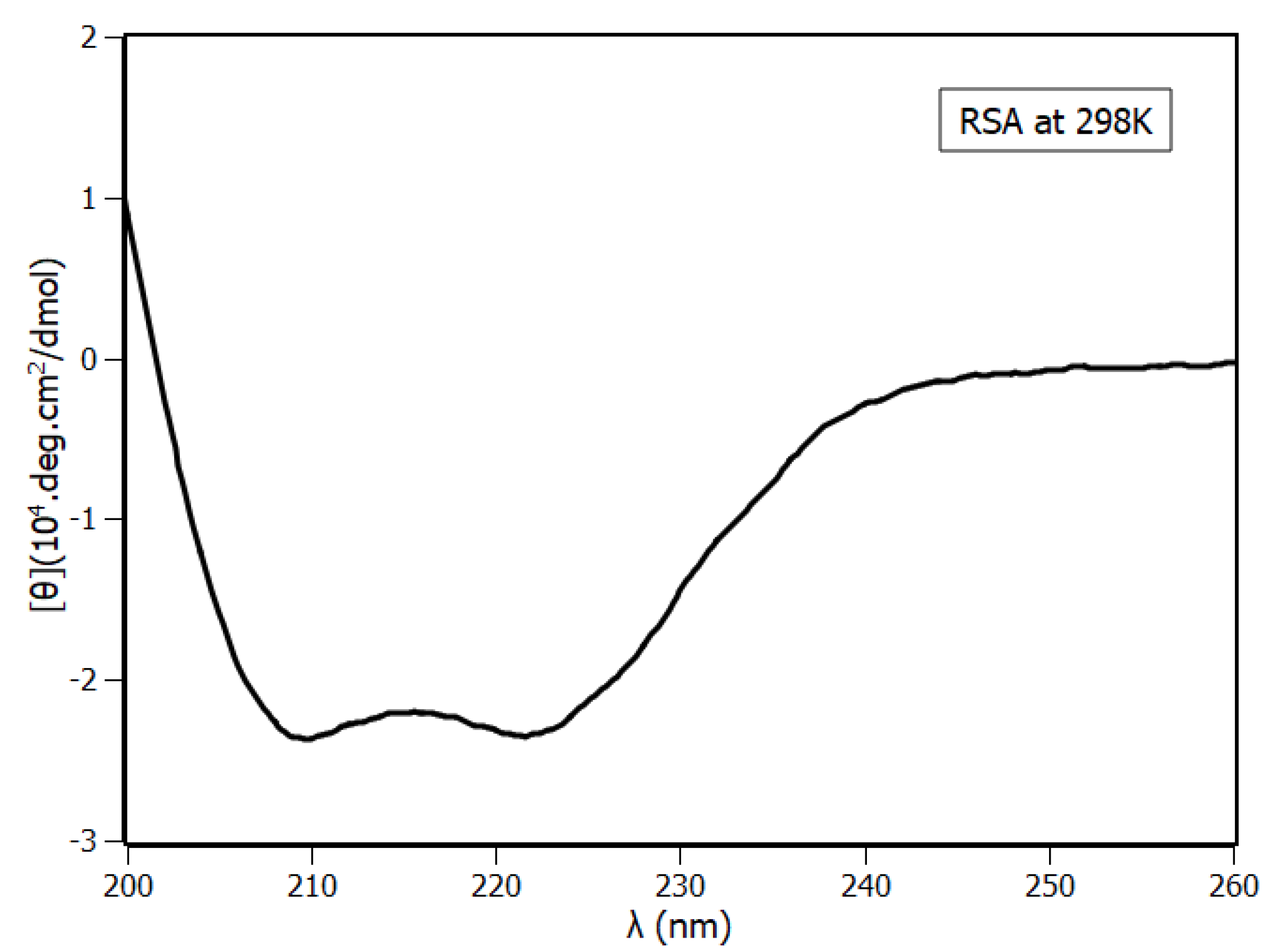
2.4. Circular Dichroism Spectroscopy
2.5. Protein Comparative Modeling
2.6. Ab Initio Parametrization of Piperine
2.7. Molecular Docking
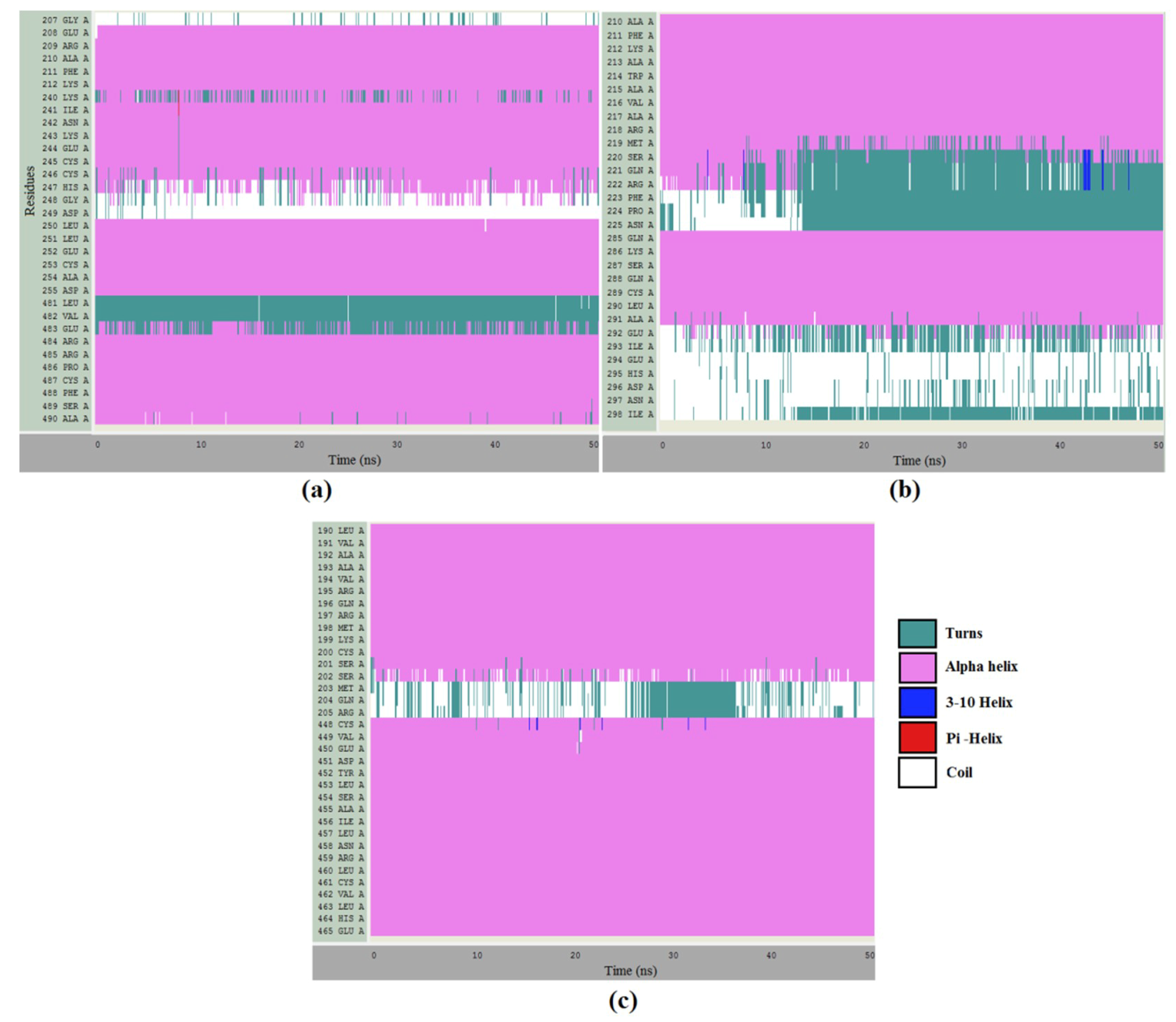
2.8. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Reagents
3.2. Steady-State Fluorescence Spectroscopy
3.3. Time-Resolved Fluorescence
3.4. Circular Dichroism Spectroscopy
3.5. Piperine Optimization by Ab Initio Calculations
3.6. RSA Modeling
3.7. Molecular Docking
3.8. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RSA | Rat Serum Albumin |
| PDB | Protein Data Bank |
| IDF | Interaction Density Function |
| CD | Circular Dichroism |
| λexc | Excitation wavelenght |
| ΔH | Enthalpy variation |
| ΔS | Entropy variation |
| ΔG | Gibbs free energy |
| MEP | Potential electrostatic map |
| MD | Molecular Dynamics |
| VMD | Visual Molecular Dynamics software |
References
- Kotte, S.C.B.; Dubey, P.K.; Murali, P.M. Identification and characterization of stress degradation products of piperine and profiling of a black pepper (Piper nigrum L.) extract using LC/Q-TOF-dual ESI-MS. Anal. Methods 2014, 6, 8022–8029. [Google Scholar] [CrossRef]
- Li, C. A quantitative method using one marker for simultaneous assay of gallic acid, hydroxysafflor yellow A, cinnamic aldehyde and piperine in Tibetan medicine Dangzuo. Chin. J. Pharm. Anal. 2011, 31, 1658–1663. [Google Scholar]
- Liu, H.-L.; Luo, R.; Chen, X.-Q.; Ba, Y.-Y.; Zheng, L.; Guo, W.-W.; Wu, X. Identification and simultaneous quantification of five alkaloids in Piper longum L. by HPLC–ESI-MSn and UFLC–ESI-MS/MS and their application to Piper nigrum L. Food Chem. 2015, 177, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Yuan, E.L.; Zhang, J.; Yan, C.; Jin, Q.A.; Peng, Z.Q. Isolation and identification of piperine from Piper sarmentosum and its insecticidal activity. Chin. J. Trop. Crop. 2013, 34, 2246–2250. [Google Scholar]
- He, H.; Guo, W.W.; Chen, X.Q.; Zhao, H.Y.; Wu, X. Comparative metabolism of three amide alkaloids from Piper longum in five different species of liver microsomes. China J. Chin. Mater. Med. 2016, 41, 3084–3089. [Google Scholar]
- Shang, Z.; Cai, W.; Cao, Y.; Wang, F.; Wang, Z.; Lu, J.; Zhang, J. An integrated strategy for rapid discovery and identification of the sequential piperine metabolites in rats using ultra high-performance liquid chromatography/high resolution mass spectrometery. J. Pharm. Biomed. Anal. 2017, 146, 387–401. [Google Scholar] [CrossRef]
- Tripathi, S.M.; Sharma, R.J.; Bansal, A.K.; Bhutani, K.K.; Singh, I.P. Development of chewable tablet of Trikatu churna and standardization by densitometry. Indian J. Tradit. Knowl. 2017, 16, 256–262. [Google Scholar]
- Bang, J.S.; Oh, D.H.; Choi, H.M.; Sur, B.-J.; Lim, S.-J.; Kim, J.Y.; Yang, H.-I.; Yoo, M.C.; Hahm, D.-H.; Kim, K.S. Anti-inflammatory and antiarthritic effects of piperine in human interleukin 1β-stimulated fibroblast-like synoviocytes and in rat arthritis models. Arthritis Res. Ther. 2009, 11, R49. [Google Scholar] [CrossRef]
- Ying, X.; Chen, X.; Cheng, S.; Shen, Y.; Peng, L.; Xu, H. Piperine inhibits IL-β induced expression of inflammatory mediators in human osteoarthritis chondrocyte. Int. Immunopharmacol. 2013, 17, 293–299. [Google Scholar] [CrossRef]
- Ying, X.; Yu, K.; Chen, X.; Chen, H.; Hong, J.; Cheng, S.; Peng, L. Piperine inhibits LPS induced expression of inflammatory mediators in RAW 264.7 cells. Cell. Immunol. 2013, 285, 49–54. [Google Scholar] [CrossRef]
- Wang-sheng, C.; Jie, A.; Jian-jun, L.; Lan, H.; Zeng-bao, X.; Chang-qing, L. Piperine attenuates lipopolysaccharide (LPS)-induced inflammatory responses in BV2 microglia. Int. Immunopharmacol. 2017, 42, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Manayi, A.; Nabavi, S.M.; Setzer, W.N.; Jafari, S. Piperine as a potential anti-cancer agent: A Review on Preclinical studies. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Ahamad, M.S.; Jafri, A.; Afzal, M.; Arshad, M. Piperine triggers apoptosis of human oral squamous carcinoma through cell cycle arrest and mitochondrial oxidative stress. Nutr. Cancer 2017, 69, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Choo, G.S.; Yoo, E.S.; Kim, S.H.; Woo, J.S.; Kim, H.J.; Park, Y.S.; Kim, B.S.; Kim, S.K.; Park, B.K.; Cho, S.D.; et al. Antitumor and apoprosis-inducing effects of piperine in human melanoma cells. Anticancer Res. 2019, 39, 1883–1892. [Google Scholar]
- Ding, Y.; Wang, C.; Wang, Y.; Xu, Y.; Zhao, J.; Gao, M.; Ding, Y.; Peng, J.; Li, L. Development and evaluation of a novel drug delivery: Soluplus®/TPGS mixed micelles loaded with piperine in vitro and in vivo. Drug Dev. Ind. Pharm. 2018, 44, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Prashant, A.; Rangaswamy, C.; Yadav, A.K.; Reddy, V.; Sowmya, M.N.; Madhunapantula, S. In Vitro anticancer activity of ethanolic extracts of Piper nigrum against colorectal carcinoma cell lines. Int. J. Appl. Basic Med. Res. 2017, 7, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.V.; Srinivas, P.V.; Praveen, B.; Kishore, K.H.; Raju, B.C.; Murthy, U.S.; Rao, J.M. Antibacterial constituents from the berries of Piper nigrum. Phytomedicine 2004, 11, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.C.; Haque, M.E.; Rahman, S.; Al-Mansur, M.A. Piperine and isoflavan-4-one from the stems of piper chaba hunter and their in vitro antimicrobial activities. J. Pharmacogn. Phytochem. 2018, 7, 2653–2662. [Google Scholar]
- Morsy, N.F.S.; El-Salam, E.A.A. Antimicrobial and Antiproliferative Activities of Black Pepper (Piper nigrum L.) Essential Oil and Oleoresin. J. Essent. Oil Bear. Plants 2017, 20, 779–790. [Google Scholar] [CrossRef]
- Ribeiro, T.S.; Freire-de-Lima, L.; Previato, J.O.; Mendonça-Previato, L.; Heise, N.; de Lima, M.E.F. Toxic effects of natural piperine and its derivatives on epimastigotes and amastigotes of Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 2004, 14, 3555–3558. [Google Scholar] [CrossRef]
- Romero-Benavides, J.C.; Ruano, A.L.; Silva-Rivas, R.; Castillo-Veintimilla, P.; Vivanco-Jaramillo, S.; Bailon-Moscoso, N. Medicinal plants used as anthelmintics: Ethnomedical, pharmacological, and phytochemical studies. Eur. J. Med. Chem. 2017, 129, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; Koul, J.L.; Taneja, S.C.; Dhar, K.L.; Jamwal, D.S.; Singh, K.; Reen, R.K.; Singh, J. Structure–activity relationship of piperine and its synthetic analogues for their inhibitory potentials of rat hepatic microsomal constitutive and inducible cytochrome P450 activities. Bioorg. Med. Chem. 2000, 8, 251–268. [Google Scholar] [CrossRef]
- Suresh, D.; Srinivasan, K. Tissue distribution & elimination of capsaicin, piperine & curcumin following oral intake in rats. Indian J. Med. Res. 2010, 131, 682–691. [Google Scholar] [PubMed]
- Qu, Y.; Harte, F.M.; Elias, R.J.; Coupland, J.N. Effect of ethanol on the solubilization of hydrophobic molecules by sodium caseinate. Food Hydrocoll. 2018, 77, 454–459. [Google Scholar] [CrossRef]
- Rothschild, M.A.; Oratz, M.; Schreiber, S.S. Serum albumin. Hepatology 1988, 8, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Jagodzinski, L.L.; Sargent, T.D.; Yang, M.; Glackin, C.; Bonner, J. Sequence homology between RNAs encoding rat alpha-fetoprotein and rat serum albumin. Proc. Natl. Acad. Sci. USA 1981, 78, 3521–3525. [Google Scholar] [CrossRef] [PubMed]
- Kosa, T.; Maruyama, T.; Otagiri, M. Species differences of serum albumins: II. Chemical and thermal stability. Pharm. Res. 1998, 15, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Q.Y.; Wang, Y.P.; Liu, Y.M.; Liu, B.; Liu, M.M.; Liu, B.M. Spectroscopic investigation of the anticancer alkaloid piperlongumine binding to human serum albumin from the viewpoint of drug delivery. Luminescence 2018, 33, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Povinelli, A.P.R.; Zazeri, G.; Cornélio, M.L. Molecular Mechanism of Flavonoids Using Fluorescence Spectroscopy and Computational Tools. In Flavonoids-A Coloring Model For Cheering up Life; IntechOpen: London, UK, 2019. [Google Scholar]
- Bi, S.; Ding, L.; Tian, Y.; Song, D.; Zhou, X.; Liu, X.; Zhang, H. Investigation of the interaction between flavonoids and human serum albumin. J. Mol. Struct. 2004, 703, 37–45. [Google Scholar] [CrossRef]
- Otagiri, M. A molecular functional study on the interactions of drugs with plasma proteins. Drug Metab. Pharmacokinet. 2005, 20, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Dangles, O. Flavonoid–serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Camargo, C.R.; Caruso, Í.P.; Gutierrez, S.J.C.; Fossey, M.A.; Barbosa Filho, J.M.; Cornélio, M.L. Spectral and computational features of the binding between riparins and human serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 190, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Caruso, Í.P.; Vilegas, W.; de Souza, F.P.; Fossey, M.A.; Cornélio, M.L. Binding of antioxidant flavone isovitexin to human serum albumin investigated by experimental and computational assays. J. Pharm. Biomed. Anal. 2014, 98, 100–106. [Google Scholar] [CrossRef] [PubMed]
- de Alcântara-Contessoto, N.S.; Caruso, Í.P.; Bezerra, D.P.; Barbosa Filho, J.M.; Cornélio, M.L. An investigation into the interaction between piplartine (piperlongumine) and human serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 220, 117084. [Google Scholar] [CrossRef]
- Beckett, D. Measurement and analysis of equilibrium binding titrations: A beginner’s guide. Methods Enzymol. 2011, 488, 1–16. [Google Scholar]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- Van Holde, K.E.; Johnson, W.C.; Ho, P.S. Principles of Physical Biochemistry; Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, 2006. [Google Scholar]
- Baig, M.H.; Rahman, S.; Rabbani, G.; Imran, M.; Ahmad, K.; Choi, I. Multi-Spectroscopic Characterization of Human Serum Albumin Binding with Cyclobenzaprine Hydrochloride: Insights from Biophysical and In Silico Approaches. Int. J. Mol. Sci. 2019, 20, 662. [Google Scholar] [CrossRef]
- Schulz, H.; Baranska, M.; Quilitzsch, R.; Schütze, W.; Lösing, G. Characterization of peppercorn, pepper oil, and pepper oleoresin by vibrational spectroscopy methods. J. Agric. Food Chem. 2005, 53, 3358–3363. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Slater, J.C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385. [Google Scholar] [CrossRef]
- Parr, R.G. Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry; Springer: Berlin/Heidelberg, Germany, 1980; pp. 5–15. [Google Scholar]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. Potential derived charges using a geodesic point selection scheme. J. Comput. Chem. 1996, 17, 1–18. [Google Scholar] [CrossRef]
- Bode, B.M.; Gordon, M.S. MacMolPlt: A graphical user interface for GAMESS. J. Mol. Graph. Model. 1998, 16, 133–138. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Eswar, N.; John, B.; Mirkovic, N.; Fiser, A.; Ilyin, V.A.; Pieper, U.; Stuart, A.C.; Marti-Renom, M.A.; Madhusudhan, M.S.; Yerkovich, B.; et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003, 31, 3375–3380. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (K) | KSV (×104 M−1) | Ka (×104 M−1) | n |
|---|---|---|---|
| 288 | 3.77 ± 0.09 | 3.90 ± 0.67 | 0.93 |
| 298 | 2.78 ± 0.12 | 2.77 ± 0.54 | 0.88 |
| 308 | 2.54 ± 0.09 | 2.06 ± 0.65 | 0.83 |
| T (K) | ∆G (kJ/mol) | ∆H (kJ/mol) | ∆S (J/mol.K) | T. ∆S (kJ/mol) |
|---|---|---|---|---|
| 288 | −25.29 ± 0.95 | −23.54 ± 0.52 | 6.1 ± 1.5 | 1.76 ± 0.43 |
| 298 | −25.35 ± 0.96 | 1.82 ± 0.44 | ||
| 308 | −25.41 ± 0.98 | 1.88 ± 0.46 |
| Methods | α-Helix (%) | Turns (%) | Coil (%) |
|---|---|---|---|
| Circular Dichroism | 63 | 17 | 17 |
| RSA model | 75 | 11 | 11 |
| RSA model with MD | 65 | 17 | 16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zazeri, G.; Povinelli, A.P.R.; Lima, M.d.F.; Cornélio, M.L. Experimental Approaches and Computational Modeling of Rat Serum Albumin and Its Interaction with Piperine. Int. J. Mol. Sci. 2019, 20, 2856. https://doi.org/10.3390/ijms20122856
Zazeri G, Povinelli APR, Lima MdF, Cornélio ML. Experimental Approaches and Computational Modeling of Rat Serum Albumin and Its Interaction with Piperine. International Journal of Molecular Sciences. 2019; 20(12):2856. https://doi.org/10.3390/ijms20122856
Chicago/Turabian StyleZazeri, Gabriel, Ana Paula Ribeiro Povinelli, Marcelo de Freitas Lima, and Marinônio Lopes Cornélio. 2019. "Experimental Approaches and Computational Modeling of Rat Serum Albumin and Its Interaction with Piperine" International Journal of Molecular Sciences 20, no. 12: 2856. https://doi.org/10.3390/ijms20122856
APA StyleZazeri, G., Povinelli, A. P. R., Lima, M. d. F., & Cornélio, M. L. (2019). Experimental Approaches and Computational Modeling of Rat Serum Albumin and Its Interaction with Piperine. International Journal of Molecular Sciences, 20(12), 2856. https://doi.org/10.3390/ijms20122856