Chemokines in COPD: From Implication to Therapeutic Use


Abstract
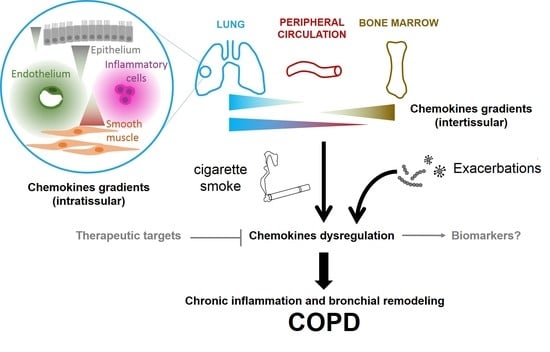
1. Introduction
2. General Considerations on Chemokines in COPD
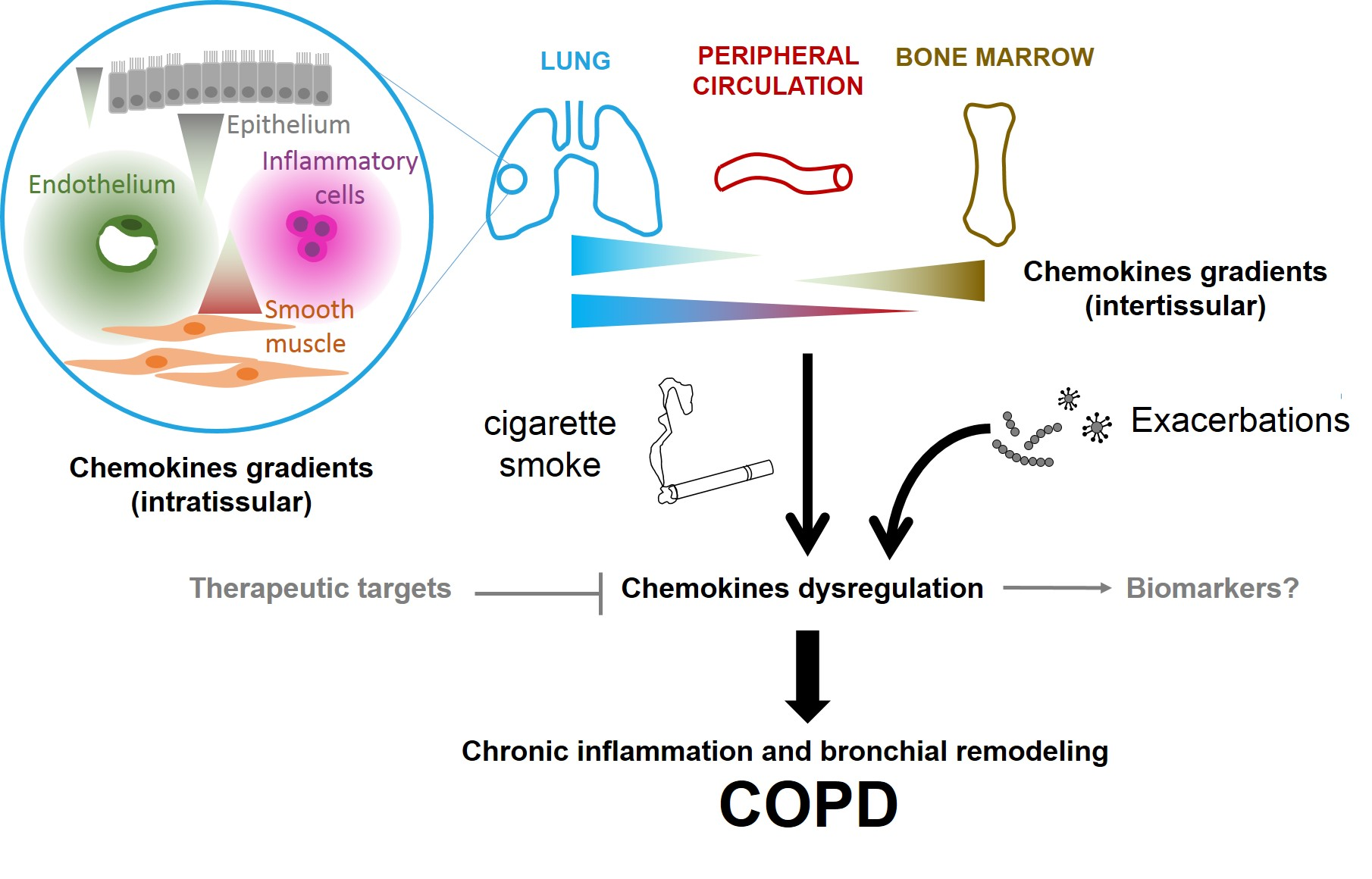
2.1. Chemokines and their Receptors: Definition and Properties, in Relation with COPD
2.2. Chemokines Gradients in COPD
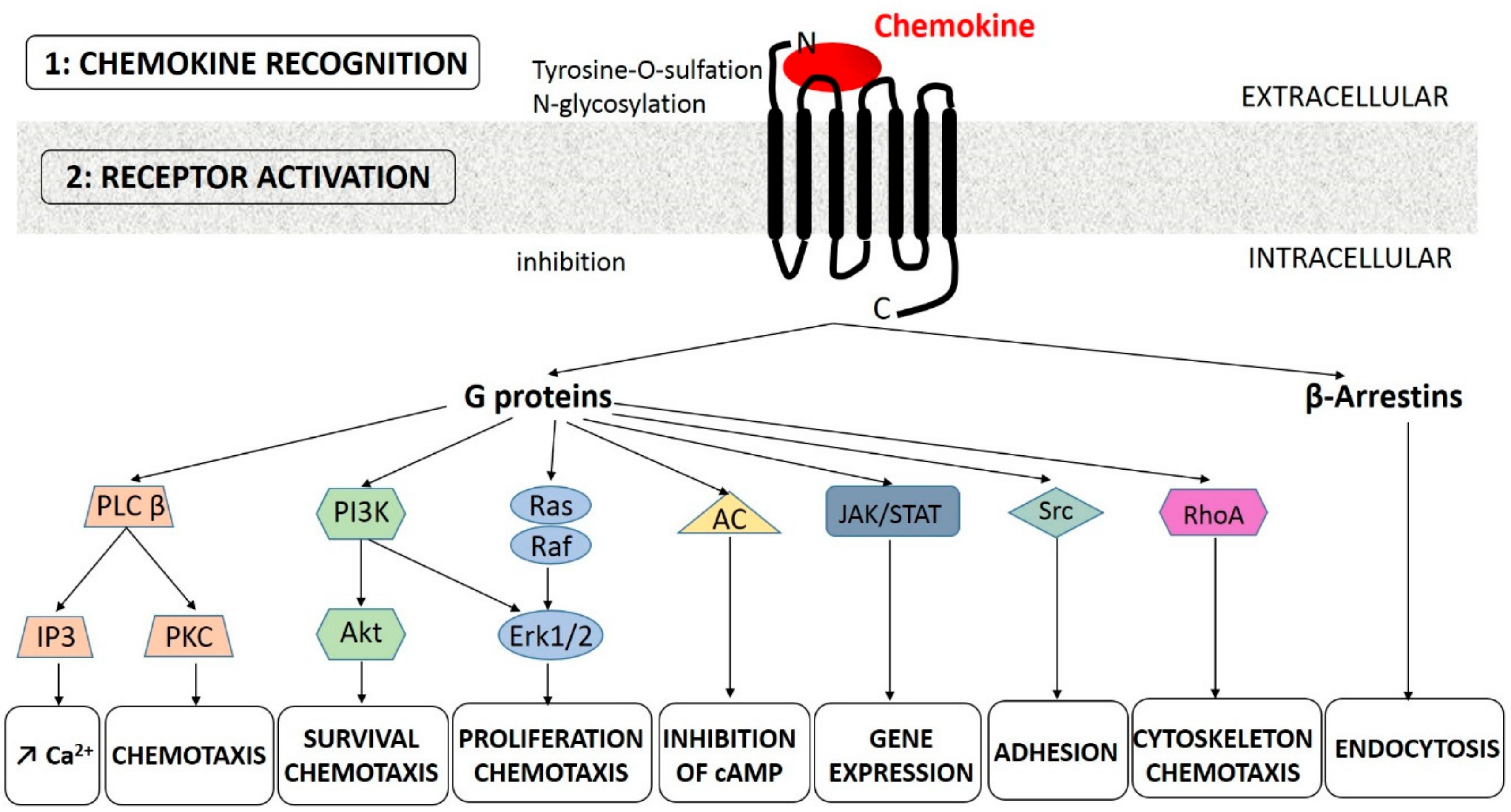
3. Chemokines at the Stable State and During Exacerbation
3.1. The CXCL8-CXCR1/2 Axis
3.1.1. At the Stable State
3.1.2. During Exacerbation
3.2. The CXCL9/10/11-CXCR3 Axis
3.2.1. At the Stable State
3.2.2. During Exacerbation
3.3. The CCL2-CCR2 Axis
3.3.1. At the Stable State
3.3.2. During an Exacerbation
3.4. The CXCL12-CXCR4 Axis
3.4.1. At the Stable State
3.4.2. During Exacerbation
4. Chemokines in COPD: Therapeutic Implications
4.1. Chemokine Levels as Biomarkers in COPD
4.1.1. CXCL8 as a Biomarker of Disease Severity
4.1.2. CXCL10 as a Genetic Biomarker of Disease Susceptibility
4.2. Chemokines or Chemokine Receptors Neutralization in COPD
4.2.1. The CXCL8-CXCR1/2 Axis as Therapeutic Target
4.2.2. The CXCL9/10/11-CXCR3 Axis as a Therapeutic Target
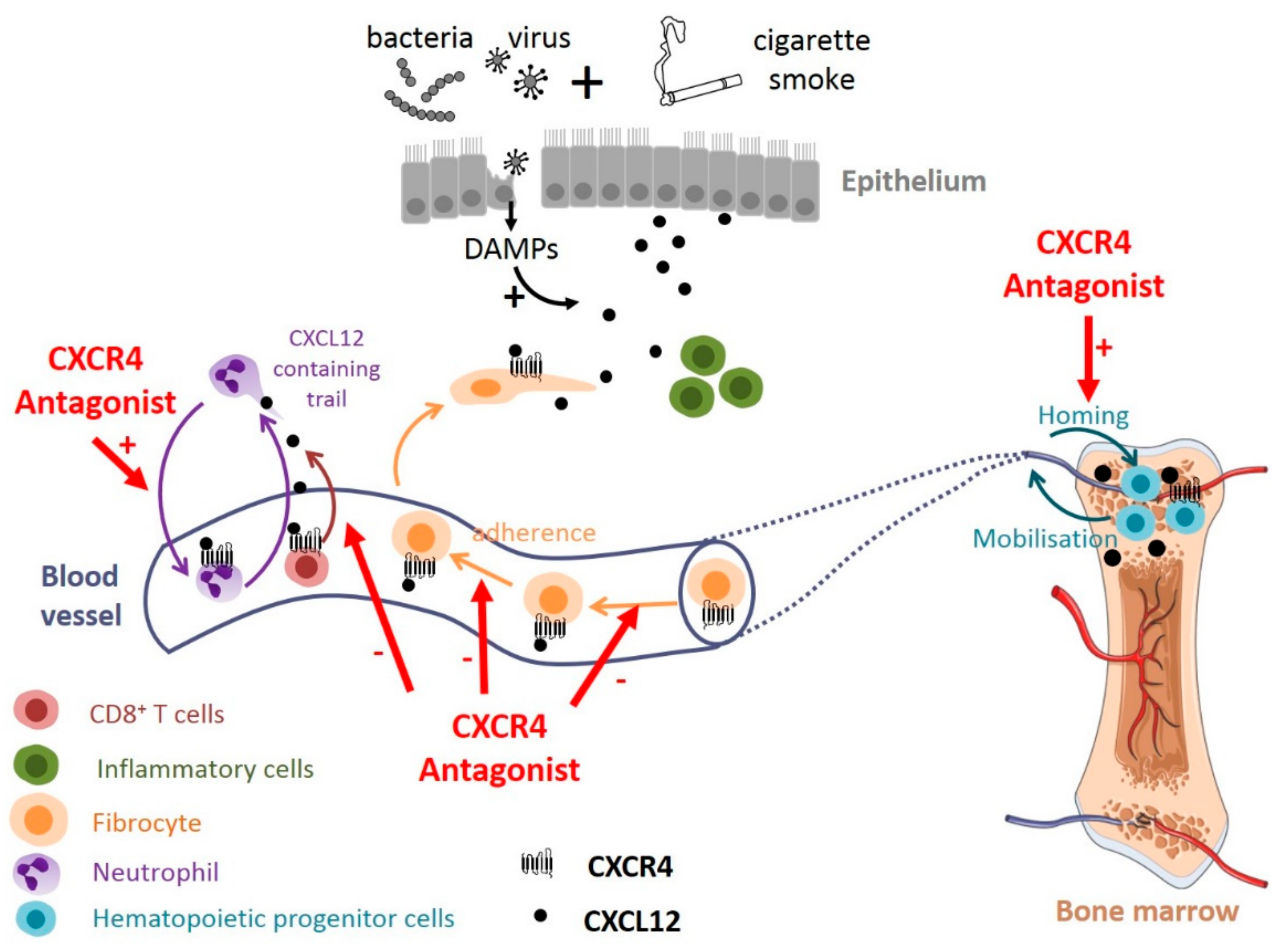
4.2.3. The CXCL12-CXCR4 Axis as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| AIDS | Acquired immune deficiency syndrome |
| AC | Adenylate cyclase |
| BALF | Broncho-alveolar lung fluid |
| BM-MSCs | Bone marrow-mesenchymal stem cells |
| CC16 | Club cell-16 |
| CDK9 | Cyclin-dependent kinase 9 |
| COPD | Chronic obstructive pulmonary disease |
| CS | Cigarette smoke |
| CSE | Cigarette smoke extract |
| CT | Computed tomography |
| DAMPs | Damage-associated molecular patterns |
| DPPIV | Dipeptidyl peptidase-4 |
| ECLIPSE | Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points |
| ECM | Extracellular matrix |
| EPCs | Endothelial progenitor cells |
| FDA | Food and drug administration |
| FEV1 | Forced expiratory volume in one second |
| FEV1/FVC FLAME | Forced expiratory volume in one second/forced volume vital capacity EFfect of Indacaterol Glycopyronium Vs Fluticasone Salmeterol on COPD Exacerbations |
| GAGs | Glycosaminoglycans |
| GDP | Guanosine-5′-diphosphate |
| GPCRs | G-protein coupled receptors |
| GTP | Guanosine-5′-triphosphate |
| 16-HBE | 16-human bronchial epithelial cells |
| HMGB1 | High mobility group box 1 protein |
| HIV | Human immunodeficiency virus |
| HPCs | Hematopoietic progenitor cells |
| IFN-γ IMPACT | Interferon-γ InforMing the PAthway of COPD Treatment |
| IRF3 | IFN regulatory factor III |
| IL | Interleukine |
| IP3 | Inositol trisphosphate |
| JAK/STAT | Janus kinase/signaling transducer and activator of transcription |
| kDa | kilo Daltons |
| LPS | Lipopolysaccharide |
| MAPK/Erk | Mitogen activated protein kinase/extracellular signal-regulated kinase |
| MCP-1 | Monocyte chemotactic protein 1 |
| MMP | Matrix metalloproteinase |
| PBMC | Peripheral blood mononucleated cells |
| PI3K | Phosphoinositide 3-kinase |
| PKC | Protein kinase C |
| PLC | Phosphoinositide-specific phospholipase C |
| PPE | Porcine pancreatic elastase |
| PRRs | Pattern recognition receptors |
| SNP | Single nucleotide polymorphism |
| SP-D | Surfactant protein D |
| sRAGE SUMMIT | Soluble receptor for advanced glycation end-products Study to Understand Mortality and Morbidity in COPD |
| TNF-α TORCH | Tumor necrosis factor-α TOwards a Revolution in COPD Health |
| TSG-6 UPLIFT | TNF-stimulated gene/protein-6 Understanding Potential Long-term Impacts on Function with Tiotropium |
| VEGF | Vascular endothelial growth factor |
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet Lond. Engl. 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Lond. Engl. 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Terzikhan, N.; Verhamme, K.M.C.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and incidence of COPD in smokers and non-smokers: The Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Aboussouan, L.S. Alpha1-antitrypsin deficiency. Lancet Lond. Engl. 2005, 365, 2225–2236. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Altose, M.D.; Bleecker, E.R.; Connett, J.E.; Kanner, R.E.; Lee, W.W.; Wise, R. The lung health study: Airway responsiveness to inhaled methacholine in smokers with mild to moderate airflow limitation. The Lung Health Study Research Group. Am. Rev. Respir. Dis. 1992, 145, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.A.; Morgan, W.J.; Wright, A.L.; Guerra, S.; Martinez, F.D. Poor airway function in early infancy and lung function by age 22 years: A non-selective longitudinal cohort study. Lancet Lond. Engl. 2007, 370, 758–764. [Google Scholar] [CrossRef]
- Lange, P.; Celli, B.; Agustí, A.; Boje Jensen, G.; Divo, M.; Faner, R.; Guerra, S.; Marott, J.L.; Martinez, F.D.; Martinez-Camblor, P.; et al. Lung-Function Trajectories Leading to Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2015, 373, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ko, F.W.; Chan, K.P.; Hui, D.S.; Goddard, J.R.; Shaw, J.G.; Reid, D.W.; Yang, I.A. Acute exacerbation of COPD. Respirol. Carlton VIC 2016, 21, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Wedzicha, J.A.; Brill, S.E.; Allinson, J.P.; Donaldson, G.C. Mechanisms and impact of the frequent exacerbator phenotype in chronic obstructive pulmonary disease. BMC Med. 2013, 11, 181. [Google Scholar] [CrossRef]
- Calverley, P.M.A.; Anderson, J.A.; Celli, B.; Ferguson, G.T.; Jenkins, C.; Jones, P.W.; Yates, J.C.; Vestbo, J. TORCH investigators Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N. Engl. J. Med. 2007, 356, 775–789. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Celli, B.; Senn, S.; Burkhart, D.; Kesten, S.; Menjoge, S.; Decramer, M. UPLIFT Study Investigators A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N. Engl. J. Med. 2008, 359, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Vestbo, J.; Anderson, J.A.; Brook, R.D.; Calverley, P.M.A.; Celli, B.R.; Crim, C.; Martinez, F.; Yates, J.; Newby, D.E. SUMMIT Investigators Fluticasone furoate and vilanterol and survival in chronic obstructive pulmonary disease with heightened cardiovascular risk (SUMMIT): A double-blind randomised controlled trial. Lancet Lond. Engl. 2016, 387, 1817–1826. [Google Scholar] [CrossRef]
- Wedzicha, J.A.; Banerji, D.; Chapman, K.R.; Vestbo, J.; Roche, N.; Ayers, R.T.; Thach, C.; Fogel, R.; Patalano, F.; Vogelmeier, C.F.; et al. Indacaterol-Glycopyrronium versus Salmeterol-Fluticasone for COPD. N. Engl. J. Med. 2016, 374, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.A.; Barnhart, F.; Brealey, N.; Brooks, J.; Criner, G.J.; Day, N.C.; Dransfield, M.T.; Halpin, D.M.G.; Han, M.K.; Jones, C.E.; et al. Once-Daily Single-Inhaler Triple versus Dual Therapy in Patients with COPD. N. Engl. J. Med. 2018, 378, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef]
- Dupin, I.; Contin-Bordes, C.; Berger, P. Fibrocytes in Asthma and Chronic Obstructive Pulmonary Disease: Variations on the Same Theme. Am. J. Respir. Cell Mol. Biol. 2018, 58, 288–298. [Google Scholar] [CrossRef]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef]
- Mackay, C.R. Chemokines: immunology’s high impact factors. Nat. Immunol. 2001, 2, 95–101. [Google Scholar] [CrossRef]
- Youn, B.S.; Mantel, C.; Broxmeyer, H.E. Chemokines, chemokine receptors and hematopoiesis. Immunol. Rev. 2000, 177, 150–174. [Google Scholar] [CrossRef]
- Belperio, J.A.; Keane, M.P.; Arenberg, D.A.; Addison, C.L.; Ehlert, J.E.; Burdick, M.D.; Strieter, R.M. CXC chemokines in angiogenesis. J. Leukoc. Biol. 2000, 68, 1–8. [Google Scholar]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.; Arthos, J.; Cicala, C.; Fauci, A.S. Chemokines, cytokines and HIV: A complex network of interactions that influence HIV pathogenesis. Immunol. Rev. 2000, 177, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R.; Weber, C. Therapeutic targeting of chemokine interactions in atherosclerosis. Nat. Rev. Drug Discov. 2010, 9, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.B.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; Van Damme, J.; Walz, A.; Marriott, D. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef]
- Ludeman, J.P.; Stone, M.J. The structural role of receptor tyrosine sulfation in chemokine recognition. Br. J. Pharmacol. 2014, 171, 1167–1179. [Google Scholar] [CrossRef]
- Veldkamp, C.T.; Seibert, C.; Peterson, F.C.; De la Cruz, N.B.; Haugner, J.C.; Basnet, H.; Sakmar, T.P.; Volkman, B.F. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci. Signal. 2008, 1, ra4. [Google Scholar] [CrossRef]
- Legler, D.F.; Thelen, M. New insights in chemokine signaling. F1000Research 2018, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M. Dancing to the tune of chemokines. Nat. Immunol. 2001, 2, 129–134. [Google Scholar] [CrossRef]
- Colamussi, M.L.; Secchiero, P.; Gonelli, A.; Marchisio, M.; Zauli, G.; Capitani, S. Stromal derived factor-1 alpha (SDF-1 alpha) induces CD4+ T cell apoptosis via the functional up-regulation of the Fas (CD95)/Fas ligand (CD95L) pathway. J. Leukoc. Biol. 2001, 69, 263–270. [Google Scholar] [PubMed]
- Drury, L.J.; Wendt, M.K.; Dwinell, M.B. CXCL12 chemokine expression and secretion regulates colorectal carcinoma cell anoikis through Bim-mediated intrinsic apoptosis. PLoS ONE 2010, 5, e12895. [Google Scholar] [CrossRef] [PubMed]
- Kremer, K.N.; Peterson, K.L.; Schneider, P.A.; Meng, X.W.; Dai, H.; Hess, A.D.; Smith, B.D.; Rodriguez-Ramirez, C.; Karp, J.E.; Kaufmann, S.H.; et al. CXCR4 chemokine receptor signaling induces apoptosis in acute myeloid leukemia cells via regulation of the Bcl-2 family members Bcl-XL, Noxa, and Bak. J. Biol. Chem. 2013, 288, 22899–22914. [Google Scholar] [CrossRef] [PubMed]
- Ravi, A.K.; Plumb, J.; Gaskell, R.; Mason, S.; Broome, C.S.; Booth, G.; Catley, M.; Vestbo, J.; Singh, D. COPD monocytes demonstrate impaired migratory ability. Respir. Res. 2017, 18, 90. [Google Scholar] [CrossRef]
- Kufareva, I.; Gustavsson, M.; Zheng, Y.; Stephens, B.S.; Handel, T.M. What Do Structures Tell Us About Chemokine Receptor Function and Antagonism? Annu. Rev. Biophys. 2017, 46, 175–198. [Google Scholar] [CrossRef]
- Arimont, M.; Sun, S.-L.; Leurs, R.; Smit, M.; de Esch, I.J.P.; de Graaf, C. Structural Analysis of Chemokine Receptor-Ligand Interactions. J. Med. Chem. 2017, 60, 4735–4779. [Google Scholar] [CrossRef]
- Proudfoot, A.E.I.; Uguccioni, M. Modulation of Chemokine Responses: Synergy and Cooperativity. Front. Immunol. 2016, 7, 183. [Google Scholar] [CrossRef]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef]
- Campana, L.; Bosurgi, L.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J. Leukoc. Biol. 2009, 86, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I. Chemokines and Glycosaminoglycans. Front. Immunol. 2015, 6, 246. [Google Scholar] [CrossRef] [PubMed]
- Drury, L.J.; Ziarek, J.J.; Gravel, S.; Veldkamp, C.T.; Takekoshi, T.; Hwang, S.T.; Heveker, N.; Volkman, B.F.; Dwinell, M.B. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 17655–17660. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.C.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Oehrle, B.; Gerckens, M.; White, E.S.; Schiller, H.B.; Eickelberg, O. The instructive extracellular matrix of the lung: Basic composition and alterations in chronic lung disease. Eur. Respir. J. 2017, 50, 1601805. [Google Scholar] [CrossRef]
- Sand, J.M.B.; Knox, A.J.; Lange, P.; Sun, S.; Kristensen, J.H.; Leeming, D.J.; Karsdal, M.A.; Bolton, C.E.; Johnson, S.R. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir. Res. 2015, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Deng, W.; Wang, Z.; Ning, M.; Zhang, W.; Zhou, Y.; Lo, E.H.; Xing, C. Differential subnetwork of chemokines/cytokines in human, mouse, and rat brain cells after oxygen-glucose deprivation. J. Cereb. Blood Flow Metab. 2017, 37, 1425–1434. [Google Scholar] [CrossRef]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef]
- Gangavarapu, P.; Rajagopalan, L.; Kolli, D.; Guerrero-Plata, A.; Garofalo, R.P.; Rajarathnam, K. The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 2012, 91, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Tanino, Y.; Coombe, D.R.; Gill, S.E.; Kett, W.C.; Kajikawa, O.; Proudfoot, A.E.I.; Wells, T.N.C.; Parks, W.C.; Wight, T.N.; Martin, T.R.; et al. Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J. Immunol. 2010, 184, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- GOLD 1998. Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease. NIH Publication (Updated 2019). Available online: http://www.goldcopd.org (accessed on 1 April 2019).
- Pauwels, R.A.; Buist, A.S.; Calverley, P.M.; Jenkins, C.R.; Hurd, S.S. GOLD Scientific Committee Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am. J. Respir. Crit. Care Med. 2001, 163, 1256–1276. [Google Scholar] [CrossRef] [PubMed]
- Inui, T.; Watanabe, M.; Nakamoto, K.; Sada, M.; Hirata, A.; Nakamura, M.; Honda, K.; Ogawa, Y.; Takata, S.; Yokoyama, T.; et al. Bronchial epithelial cells produce CXCL1 in response to LPS and TNFα: A potential role in the pathogenesis of COPD. Exp. Lung Res. 2019, 44, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Traves, S.L.; Culpitt, S.V.; Russell, R.E.K.; Barnes, P.J.; Donnelly, L.E. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax 2002, 57, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Beeh, K.M.; Kornmann, O.; Buhl, R.; Culpitt, S.V.; Giembycz, M.A.; Barnes, P.J. Neutrophil chemotactic activity of sputum from patients with COPD: Role of interleukin 8 and leukotriene B4. Chest 2003, 123, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhu, J.; Bandi, V.; Atmar, R.L.; Hattotuwa, K.; Guntupalli, K.K.; Jeffery, P.K. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 168, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Stefano, A.D.; Caramori, G.; Gnemmi, I.; Contoli, M.; Bristot, L.; Capelli, A.; Ricciardolo, F.L.M.; Magno, F.; D’Anna, S.E.; Zanini, A.; et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 2009, 64, 968–975. [Google Scholar] [CrossRef]
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [Google Scholar] [CrossRef]
- Osei, E.T.; Noordhoek, J.A.; Hackett, T.L.; Spanjer, A.I.R.; Postma, D.S.; Timens, W.; Brandsma, C.-A.; Heijink, I.H. Interleukin-1α drives the dysfunctional cross-talk of the airway epithelium and lung fibroblasts in COPD. Eur. Respir. J. 2016, 48, 359–369. [Google Scholar] [CrossRef]
- Patel, B.S.; Kugel, M.J.; Baehring, G.; Ammit, A.J. Doxofylline does not increase formoterol-induced cAMP nor MKP-1 expression in ASM cells resulting in lack of anti-inflammatory effect. Pulm. Pharmacol. Ther. 2017, 45, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Ikari, J.; Nelson, A.J.; Obaid, J.; Giron-Martinez, A.; Ikari, K.; Makino, F.; Iwasawa, S.; Gunji, Y.; Farid, M.; Wang, X.; et al. Reduced microRNA-503 expression augments lung fibroblast VEGF production in chronic obstructive pulmonary disease. PLoS ONE 2017, 12, e0184039. [Google Scholar] [CrossRef] [PubMed]
- De Llano, L.P.; Cosío, B.G.; Iglesias, A.; de Las Cuevas, N.; Soler-Cataluña, J.J.; Izquierdo, J.L.; López-Campos, J.L.; Calero, C.; Plaza, V.; Miravitlles, M.; et al. Mixed Th2 and non-Th2 inflammatory pattern in the asthma-COPD overlap: A network approach. Int. J. Chronic Obstruct. Pulm. Dis. 2018, 13, 591–601. [Google Scholar] [CrossRef] [PubMed]
- De Boer, W.I.; Sont, J.K.; van Schadewijk, A.; Stolk, J.; van Krieken, J.H.; Hiemstra, P.S. Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPD. J. Pathol. 2000, 190, 619–626. [Google Scholar] [CrossRef]
- Belchamber, K.B.R.; Donnelly, L.E. Macrophage Dysfunction in Respiratory Disease. Results Probl. Cell Differ. 2017, 62, 299–313. [Google Scholar] [PubMed]
- Huang, Z.-W.; Lien, G.-S.; Lin, C.-H.; Jiang, C.-P.; Chen, B.-C. p300 and C/EBPβ-regulated IKKβ expression are involved in thrombin-induced IL-8/CXCL8 expression in human lung epithelial cells. Pharmacol. Res. 2017, 121, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Rufino, R.; Traves, S.L.; Lapa E Silva, J.R.; Barnes, P.J.; Donnelly, L.E. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest 2008, 133, 26–33. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Aksoy, M.O.; Georgy, M.; Hershman, R.; Ji, R.; Li, X.; Hurford, M.; Solomides, C.; Chatila, W.; Kim, V. Lymphoid Follicle Cells in Chronic Obstructive Pulmonary Disease Overexpress the Chemokine Receptor CXCR3. Am. J. Respir. Crit. Care Med. 2009, 179, 799–805. [Google Scholar] [CrossRef]
- Tura-Ceide, O.; Lobo, B.; Paul, T.; Puig-Pey, R.; Coll-Bonfill, N.; García-Lucio, J.; Smolders, V.; Blanco, I.; Barberà, J.A.; Peinado, V.I. Cigarette smoke challenges bone marrow mesenchymal stem cell capacities in guinea pig. Respir. Res. 2017, 18, 50. [Google Scholar] [CrossRef]
- Hardaker, E.L.; Bacon, A.M.; Carlson, K.; Roshak, A.K.; Foley, J.J.; Schmidt, D.B.; Buckley, P.T.; Comegys, M.; Panettieri, R.A.; Sarau, H.M.; et al. Regulation of TNF-alpha- and IFN-gamma-induced CXCL10 expression: Participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 191–193. [Google Scholar]
- Saetta, M.; Mariani, M.; Panina-Bordignon, P.; Turato, G.; Buonsanti, C.; Baraldo, S.; Bellettato, C.M.; Papi, A.; Corbetta, L.; Zuin, R.; et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 165, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Quint, J.K.; Donaldson, G.C.; Goldring, J.J.P.; Baghai-Ravary, R.; Hurst, J.R.; Wedzicha, J.A. Serum IP-10 as a biomarker of human rhinovirus infection at exacerbation of COPD. Chest 2010, 137, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Barwinska, D.; Oueini, H.; Poirier, C.; Albrecht, M.E.; Bogatcheva, N.V.; Justice, M.J.; Saliba, J.; Schweitzer, K.S.; Broxmeyer, H.E.; March, K.L.; et al. AMD3100 ameliorates cigarette smoke-induced emphysema-like manifestations in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L382–L386. [Google Scholar] [CrossRef]
- Karagiannis, K.; Proklou, A.; Tsitoura, E.; Lasithiotaki, I.; Kalpadaki, C.; Moraitaki, D.; Sperelakis, I.; Kontakis, G.; Antoniou, K.M.; Tzanakis, N. Impaired mRNA Expression of the Migration Related Chemokine Receptor CXCR4 in Mesenchymal Stem Cells of COPD Patients. Int. J. Inflamm. 2017, 2017, 6089425. [Google Scholar] [CrossRef]
- Dupin, I.; Allard, B.; Ozier, A.; Maurat, E.; Ousova, O.; Delbrel, E.; Trian, T.; Bui, H.-N.; Dromer, C.; Guisset, O.; et al. Blood fibrocytes are recruited during acute exacerbations of chronic obstructive pulmonary disease through a CXCR4-dependent pathway. J. Allergy Clin. Immunol. 2016, 137, 1036.e7–1042.e7. [Google Scholar] [CrossRef] [PubMed]
- Vishweswaraiah, S.; Thimraj, T.A.; George, L.; Krishnarao, C.S.; Lokesh, K.S.; Siddaiah, J.B.; Larsson, K.; Upadhyay, S.; Palmberg, L.; Anand, M.P.; et al. Putative Systemic Biomarkers of Biomass Smoke-Induced Chronic Obstructive Pulmonary Disease among Women in a Rural South Indian Population. Dis. Mark. 2018, 2018, 4949175. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.; Collado, A.; Escudero, P.; Rius, C.; González, C.; Servera, E.; Piqueras, L.; Sanz, M.-J. Cigarette Smoke Increases Endothelial CXCL16-Leukocyte CXCR6 Adhesion In Vitro and In Vivo. Potential Consequences in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2017, 8, 1766. [Google Scholar] [CrossRef]
- Jing, H.; Liu, L.; Zhou, J.; Yao, H. Inhibition of C-X-C Motif Chemokine 10 (CXCL10) Protects Mice from Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 5748–5753. [Google Scholar] [CrossRef]
- Di Stefano, A.; Coccini, T.; Roda, E.; Signorini, C.; Balbi, B.; Brunetti, G.; Ceriana, P. Blood MCP-1 levels are increased in chronic obstructive pulmonary disease patients with prevalent emphysema. Int. J. Chronic Obstruct. Pulm. Dis. 2018, 13, 1691–1700. [Google Scholar] [CrossRef]
- Ravi, A.K.; Khurana, S.; Lemon, J.; Plumb, J.; Booth, G.; Healy, L.; Catley, M.; Vestbo, J.; Singh, D. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir. Res. 2014, 15, 103. [Google Scholar] [CrossRef]
- Bradford, E.; Jacobson, S.; Varasteh, J.; Comellas, A.P.; Woodruff, P.; O’Neal, W.; DeMeo, D.L.; Li, X.; Kim, V.; Cho, M.; et al. The value of blood cytokines and chemokines in assessing COPD. Respir. Res. 2017, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- D’Armiento, J.M.; Scharf, S.M.; Roth, M.D.; Connett, J.E.; Ghio, A.; Sternberg, D.; Goldin, J.G.; Louis, T.A.; Mao, J.T.; O’Connor, G.T.; et al. Eosinophil and T cell markers predict functional decline in COPD patients. Respir. Res. 2009, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Bocchino, V.; Bertorelli, G.; Bertrand, C.P.; Ponath, P.D.; Newman, W.; Franco, C.; Marruchella, A.; Merlini, S.; Del Donno, M.; Zhuo, X.; et al. Eotaxin and CCR3 are up-regulated in exacerbations of chronic bronchitis. Allergy 2002, 57, 17–22. [Google Scholar] [PubMed]
- Miller, M.; Ramsdell, J.; Friedman, P.J.; Cho, J.Y.; Renvall, M.; Broide, D.H. Computed tomographic scan-diagnosed chronic obstructive pulmonary disease-emphysema: Eotaxin-1 is associated with bronchodilator response and extent of emphysema. J. Allergy Clin. Immunol. 2007, 120, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Miller, B.E.; Duvoix, A.; Man, S.F.P.; Zhang, X.; Silverman, E.K.; Connett, J.E.; Anthonisen, N.A.; Wise, R.A.; Tashkin, D.; et al. Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Esquerre, M.; Aliagas, E.; López-Sánchez, M.; Escobar, I.; Huertas, D.; Penín, R.; Dorca, J.; Santos, S. Vascular disease in COPD: Systemic and pulmonary expression of PARC (Pulmonary and Activation-Regulated Chemokine). PLoS ONE 2017, 12, e0177218. [Google Scholar] [CrossRef] [PubMed]
- Demedts, I.K.; Bracke, K.R.; Van Pottelberge, G.; Testelmans, D.; Verleden, G.M.; Vermassen, F.E.; Joos, G.F.; Brusselle, G.G. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 998–1005. [Google Scholar] [CrossRef]
- Hao, W.; Li, M.; Zhang, C.; Zhang, Y.; Xue, Y. High Serum Fractalkine/CX3CL1 in Patients with Chronic Obstructive Pulmonary Disease: Relationship with Emphysema Severity and Frequent Exacerbation. Lung 2019, 197, 29–35. [Google Scholar] [CrossRef]
- Barnes, P.J. The Cytokine Network in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef]
- Kim, V.; Cornwell, W.D.; Oros, M.; Durra, H.; Criner, G.J.; Rogers, T.J. Plasma Chemokine signature correlates with lung goblet cell hyperplasia in smokers with and without chronic obstructive pulmonary disease. BMC Pulm. Med. 2015, 15, 111. [Google Scholar] [CrossRef]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Fang, C.; Cousins, D.; Zhang, G.; Gu, S.; Gao, Z.; Shamji, B.; et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J. Immunol. 2008, 181, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Tanino, M.; Betsuyaku, T.; Takeyabu, K.; Tanino, Y.; Yamaguchi, E.; Miyamoto, K.; Nishimura, M. Increased levels of interleukin-8 in BAL fluid from smokers susceptible to pulmonary emphysema. Thorax 2002, 57, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, C.; Yoneda, T.; Yoshikawa, M.; Fu, A.; Tokuyama, T.; Tsukaguchi, K.; Narita, N. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest 1997, 112, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, I.S.; Bayley, D.L.; Stockley, R.A. Sputum chemotactic activity in chronic obstructive pulmonary disease: Effect of alpha(1)-antitrypsin deficiency and the role of leukotriene B(4) and interleukin 8. Thorax 2002, 57, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Peveri, P.; Walz, A.; Dewald, B.; Baggiolini, M. A novel neutrophil-activating factor produced by human mononuclear phagocytes. J. Exp. Med. 1988, 167, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Zarcone, M.C.; Duistermaat, E.; Alblas, M.J.; van Schadewijk, A.; Ninaber, D.K.; Clarijs, V.; Moerman, M.M.; Vaessen, D.; Hiemstra, P.S.; Kooter, I.M. Effect of diesel exhaust generated by a city bus engine on stress responses and innate immunity in primary bronchial epithelial cell cultures. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2018, 48, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wan, C.; Zhang, W.; Guan, L.; Tian, G.; Zhang, F.; Ding, W. MiR-146a regulates PM1-induced inflammation via NF-κB signaling pathway in BEAS-2B cells. Environ. Toxicol. 2018, 33, 743–751. [Google Scholar] [CrossRef]
- Sangiorgi, C.; Vallese, D.; Gnemmi, I.; Bucchieri, F.; Balbi, B.; Brun, P.; Leone, A.; Giordano, A.; Conway de Macario, E.; Macario, A.J.; et al. HSP60 activity on human bronchial epithelial cells. Int. J. Immunopathol. Pharmacol. 2017, 30, 333–340. [Google Scholar] [CrossRef]
- Reutershan, J.; Morris, M.A.; Burcin, T.L.; Smith, D.F.; Chang, D.; Saprito, M.S.; Ley, K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Investig. 2006, 116, 695–702. [Google Scholar] [CrossRef]
- Jiang, J.-X.; Zhang, S.-J.; Shen, H.-J.; Guan, Y.; Liu, Q.; Zhao, W.; Jia, Y.-L.; Shen, J.; Yan, X.-F.; Xie, Q.-M. Rac1 signaling regulates cigarette smoke-induced inflammation in the lung via the Erk1/2 MAPK and STAT3 pathways. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1778–1788. [Google Scholar] [CrossRef]
- Aaron, S.D.; Angel, J.B.; Lunau, M.; Wright, K.; Fex, C.; Le Saux, N.; Dales, R.E. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Mikami, M.; Llewellyn-Jones, C.G.; Bayley, D.; Hill, S.L.; Stockley, R.A. The chemotactic activity of sputum from patients with bronchiectasis. Am. J. Respir. Crit. Care Med. 1998, 157, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bai, C. The Significance of Serum Interleukin-8 in Acute Exacerbations of Chronic Obstructive Pulmonary Disease. Tanaffos 2018, 17, 13–21. [Google Scholar] [PubMed]
- Han, M.K.; Quibrera, P.M.; Carretta, E.E.; Barr, R.G.; Bleecker, E.R.; Bowler, R.P.; Cooper, C.B.; Comellas, A.; Couper, D.J.; Curtis, J.L.; et al. Frequency of exacerbations in patients with chronic obstructive pulmonary disease: An analysis of the SPIROMICS cohort. Lancet Respir. Med. 2017, 5, 619–626. [Google Scholar] [CrossRef]
- Soler, N.; Ewig, S.; Torres, A.; Filella, X.; Gonzalez, J.; Zaubet, A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur. Respir. J. 1999, 14, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Kawaguchi, M.; Matsuyama, M.; Ota, K.; Fujita, J.; Matsukura, S.; Huang, S.-K.; Morishima, Y.; Ishii, Y.; Satoh, H.; et al. Transcription Elongation Factor P-TEFb Is Involved in IL-17F Signaling in Airway Smooth Muscle Cells. Int. Arch. Allergy Immunol. 2018, 176, 83–90. [Google Scholar] [CrossRef]
- Donnelly, L.E.; Barnes, P.J. Chemokine receptors as therapeutic targets in chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 2006, 27, 546–553. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Goldmann, T.; Ludwig, C.; Prasse, A.; Vollmer, E.; Müller-Quernheim, J.; Zissel, G. CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir. Res. 2005, 6, 75. [Google Scholar] [CrossRef]
- Sauty, A.; Dziejman, M.; Taha, R.A.; Iarossi, A.S.; Neote, K.; Garcia-Zepeda, E.A.; Hamid, Q.; Luster, A.D. The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J. Immunol. 1999, 162, 3549–3558. [Google Scholar]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. Lond. Engl. 2017, 131, 1541–1558. [Google Scholar] [CrossRef]
- Costa, C.; Traves, S.L.; Tudhope, S.J.; Fenwick, P.S.; Belchamber, K.B.R.; Russell, R.E.K.; Barnes, P.J.; Donnelly, L.E. Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD. Eur. Respir. J. 2016, 47, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.E.; Strick, C.A.; Paradis, T.J.; Ogborne, K.T.; Loetscher, M.; Gladue, R.P.; Lin, W.; Boyd, J.G.; Moser, B.; Wood, D.E.; et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): A novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998, 187, 2009–2021. [Google Scholar] [CrossRef] [PubMed]
- Grumelli, S.; Corry, D.B.; Song, L.-Z.; Song, L.; Green, L.; Huh, J.; Hacken, J.; Espada, R.; Bag, R.; Lewis, D.E.; et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004, 1, e8. [Google Scholar] [CrossRef] [PubMed]
- Cosio, M.G.; Majo, J.; Cosio, M.G. Inflammation of the airways and lung parenchyma in COPD: Role of T cells. Chest 2002, 121, 160S–165S. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.C.; Falzon, M.; Hall, A. Polarized localization of epithelial CXCL11 in chronic obstructive pulmonary disease and mechanisms of T cell egression. J. Immunol. 2008, 180, 1866–1877. [Google Scholar] [CrossRef] [PubMed]
- Warwick, G.; Thomas, P.S.; Yates, D.H. Non-invasive biomarkers in exacerbations of obstructive lung disease. Respirol. Carlton VIC 2013, 18, 874–884. [Google Scholar] [CrossRef]
- Bafadhel, M.; McKenna, S.; Terry, S.; Mistry, V.; Reid, C.; Haldar, P.; McCormick, M.; Haldar, K.; Kebadze, T.; Duvoix, A.; et al. Acute exacerbations of chronic obstructive pulmonary disease: Identification of biologic clusters and their biomarkers. Am. J. Respir. Crit. Care Med. 2011, 184, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.L.; Clifford, R.L.; Jindarat, S.; Proud, D.; Pang, L.; Belvisi, M.; Knox, A.J. TNFα and IFNγ synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-κB, and the transcriptional coactivator CREB-binding protein. J. Biol. Chem. 2010, 285, 29101–29110. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Ganz, T.; Liese, A.M.; Burdick, M.D.; Liu, L.; Strieter, R.M. Cutting edge: IFN-inducible ELR- CXC chemokines display defensin-like antimicrobial activity. J. Immunol. 2001, 167, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Song, H.; Cai, C.; Zhang, M.; Xu, S.; Tan, J. The association of monocyte chemotactic protein-1 and CC chemokine receptor 2 gene variants with chronic obstructive pulmonary disease. DNA Cell Biol. 2012, 31, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shi, Y.; Xiong, L.; Zhang, W.; Li, Y.; Gibson, P.G.; Simpson, J.L.; Zhang, C.; Lu, J.; Sai, J.; et al. The Expression of IL-6, TNF-α, and MCP-1 in Respiratory Viral Infection in Acute Exacerbations of Chronic Obstructive Pulmonary Disease. J. Immunol. Res. 2017, 2017, 8539294. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Hosoki, K.; Nikura, Y.; Yamashita, N.; Nagase, T.; Yamashita, N. IFN Regulatory Factor 3 Potentiates Emphysematous Aggravation by Lipopolysaccharide. J. Immunol. 2017, 198, 3637–3649. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, B.N.; Belperio, J.A.; Rao, P.N.; Randell, S.H.; Fishbein, M.C.; Burdick, M.D.; Strieter, R.M. Circulating progenitor epithelial cells traffic via CXCR4/CXCL12 in response to airway injury. J. Immunol. 2006, 176, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Heissig, B.; Tashiro, K.; Honjo, T.; Tateno, M.; Shieh, J.H.; Hackett, N.R.; Quitoriano, M.S.; Crystal, R.G.; Rafii, S.; et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood 2001, 97, 3354–3360. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Oppenheim, J.J. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation 2003, 10, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.C.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef]
- Dupin, I.; Thumerel, M.; Maurat, E.; Coste, F.; Eyraud, E.; Begueret, H.; Trian, T.; Montaudon, M.; Marthan, R.; Girodet, P.-O.; et al. Fibrocyte accumulation in the airway walls of COPD patients. Eur. Respir. J. in press.
- Liu, X.; Xie, C. Human endothelial progenitor cells isolated from COPD patients are dysfunctional. Mol. Cell. Biochem. 2012, 363, 53–63. [Google Scholar] [CrossRef]
- Lim, K.; Hyun, Y.-M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Tybjaerg-Hansen, A.; Vestbo, J.; Lange, P.; Nordestgaard, B.G. Elevated plasma fibrinogen associated with reduced pulmonary function and increased risk of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Vestbo, J.; Lange, P.; Bojesen, S.E.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. C-reactive protein as a predictor of prognosis in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.T.; Kim, D.K.; Cockayne, D.A.; Belousov, A.; Bitter, H.; Cho, M.H.; Duvoix, A.; Edwards, L.D.; Lomas, D.A.; Miller, B.E.; et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 188, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Atochina-Vasserman, E.N.; Holz, O.; Beers, M.F.; Erpenbeck, V.J.; Krug, N.; Roepcke, S.; Lauer, G.; Elmlinger, M.; Hohlfeld, J.M. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir. Res. 2011, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Coxson, H.O.; Dirksen, A.; Edwards, L.D.; Yates, J.C.; Agusti, A.; Bakke, P.; Calverley, P.M.; Celli, B.; Crim, C.; Duvoix, A.; et al. The presence and progression of emphysema in COPD as determined by CT scanning and biomarker expression: A prospective analysis from the ECLIPSE study. Lancet Respir. Med. 2013, 1, 129–136. [Google Scholar] [CrossRef]
- Lomas, D.A.; Silverman, E.K.; Edwards, L.D.; Miller, B.E.; Coxson, H.O.; Tal-Singer, R. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) investigators Evaluation of serum CC-16 as a biomarker for COPD in the ECLIPSE cohort. Thorax 2008, 63, 1058–1063. [Google Scholar] [CrossRef]
- Faner, R.; Tal-Singer, R.; Riley, J.H.; Celli, B.; Vestbo, J.; MacNee, W.; Bakke, P.; Calverley, P.M.A.; Coxson, H.; Crim, C.; et al. Lessons from ECLIPSE: A review of COPD biomarkers. Thorax 2014, 69, 666–672. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Hassibi, S.; Fenwick, P.S.; Donnelly, L.E.; Ito, K.; Barnes, P.J. MicroRNA-570 is a novel regulator of cellular senescence and inflammaging. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 33, 1605–1616. [Google Scholar] [CrossRef]
- De Novaes Rocha, N.; de Oliveira, M.V.; Braga, C.L.; Guimarães, G.; de Albuquerque Maia, L.; de Araújo Padilha, G.; Silva, J.D.; Takiya, C.M.; Capelozzi, V.L.; Silva, P.L.; et al. Ghrelin therapy improves lung and cardiovascular function in experimental emphysema. Respir. Res. 2017, 18, 185. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Q.; Dong, L.; Xiong, M.; Jiang, H.; Guo, M.; Zhao, L.; Yuan, L.; Li, Z.; Liu, H.; et al. The effects of CXCL10 polymorphisms on COPD susceptibility. Mol. Genet. Genom. MGG 2018, 293, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Devine, S.M.; Flomenberg, N.; Vesole, D.H.; Liesveld, J.; Weisdorf, D.; Badel, K.; Calandra, G.; DiPersio, J.F. Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non-Hodgkin’s lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Hitchinson, B.; Eby, J.M.; Gao, X.; Guite-Vinet, F.; Ziarek, J.J.; Abdelkarim, H.; Lee, Y.; Okamoto, Y.; Shikano, S.; Majetschak, M.; et al. Biased antagonism of CXCR4 avoids antagonist tolerance. Sci. Signal. 2018, 11, eaat2214. [Google Scholar] [CrossRef] [PubMed]
- Galzi, J.-L.; Hachet-Haas, M.; Bonnet, D.; Daubeuf, F.; Lecat, S.; Hibert, M.; Haiech, J.; Frossard, N. Neutralizing endogenous chemokines with small molecules. Principles and potential therapeutic applications. Pharmacol. Ther. 2010, 126, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Agten, S.M.; Eckardt, V.; Blanchet, X.; Schmitt, M.M.; Ippel, H.; Neideck, C.; Bidzhekov, K.; Leberzammer, J.; Wichapong, K.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9, eaah6650. [Google Scholar] [CrossRef] [PubMed]
- Mahler, D.A.; Huang, S.; Tabrizi, M.; Bell, G.M. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: A pilot study. Chest 2004, 126, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Thomson, J.M.; Hermant, A.; Jowitt, T.A.; Handel, T.M.; Proudfoot, A.E.I.; Day, A.J.; Milner, C.M. TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J. Immunol. 2014, 192, 2177–2185. [Google Scholar] [CrossRef]
- Stevenson, C.S.; Coote, K.; Webster, R.; Johnston, H.; Atherton, H.C.; Nicholls, A.; Giddings, J.; Sugar, R.; Jackson, A.; Press, N.J.; et al. Characterization of cigarette smoke-induced inflammatory and mucus hypersecretory changes in rat lung and the role of CXCR2 ligands in mediating this effect. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L514–L522. [Google Scholar] [CrossRef]
- Thatcher, T.H.; McHugh, N.A.; Egan, R.W.; Chapman, R.W.; Hey, J.A.; Turner, C.K.; Redonnet, M.R.; Seweryniak, K.E.; Sime, P.J.; Phipps, R.P. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L322–L328. [Google Scholar] [CrossRef]
- Rennard, S.I.; Dale, D.C.; Donohue, J.F.; Kanniess, F.; Magnussen, H.; Sutherland, E.R.; Watz, H.; Lu, S.; Stryszak, P.; Rosenberg, E.; et al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Lazaar, A.L.; Miller, B.E.; Tabberer, M.; Yonchuk, J.; Leidy, N.; Ambery, C.; Bloomer, J.; Watz, H.; Tal-Singer, R. Effect of the CXCR2 antagonist danirixin on symptoms and health status in COPD. Eur. Respir. J. 2018, 52, 1801020. [Google Scholar] [CrossRef] [PubMed]
- Planagumà, A.; Domènech, T.; Pont, M.; Calama, E.; García-González, V.; López, R.; Aulí, M.; López, M.; Fonquerna, S.; Ramos, I.; et al. Combined anti CXC receptors 1 and 2 therapy is a promising anti-inflammatory treatment for respiratory diseases by reducing neutrophil migration and activation. Pulm. Pharmacol. Ther. 2015, 34, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Leaker, B.R.; Barnes, P.J.; O’Connor, B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir. Res. 2013, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.P.; Cox, R.J. Small Molecule CXCR3 Antagonists. J. Med. Chem. 2016, 59, 2894–2917. [Google Scholar] [CrossRef] [PubMed]
- Takekoshi, T.; Ziarek, J.J.; Volkman, B.F.; Hwang, S.T. A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol. Cancer Ther. 2012, 11, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.W.; Liu, Y.; Getschman, A.E.; Peterson, F.C.; Ziarek, J.J.; Li, R.; Volkman, B.F.; Chen, Y. Structural analysis of a novel small molecule ligand bound to the CXCL12 chemokine. J. Med. Chem. 2014, 57, 9693–9699. [Google Scholar] [CrossRef] [PubMed]
- Veldkamp, C.T.; Ziarek, J.J.; Peterson, F.C.; Chen, Y.; Volkman, B.F. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J. Am. Chem. Soc. 2010, 132, 7242–7243. [Google Scholar] [CrossRef] [PubMed]
- Hachet-Haas, M.; Balabanian, K.; Rohmer, F.; Pons, F.; Franchet, C.; Lecat, S.; Chow, K.Y.C.; Dagher, R.; Gizzi, P.; Didier, B.; et al. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J. Biol. Chem. 2008, 283, 23189–23199. [Google Scholar] [CrossRef] [PubMed]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Peng, D.; Cao, B.; Zhou, Y.-J.; Long, Y.-Q. The chemical diversity and structure-based evolution of non-peptide CXCR4 antagonists with diverse therapeutic potential. Eur. J. Med. Chem. 2018, 149, 148–169. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Pastrana, D.V.; Calvo, K.R.; Pittaluga, S.; Velez, D.; Cho, E.; Liu, Q.; Trout, H.H.; Neves, J.F.; Gardner, P.J.; et al. Plerixafor for the Treatment of WHIM Syndrome. N. Engl. J. Med. 2019, 380, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Bonig, H.; Chudziak, D.; Priestley, G.; Papayannopoulou, T. Insights into the biology of mobilized hematopoietic stem/progenitor cells through innovative treatment schedules of the CXCR4 antagonist AMD3100. Exp. Hematol. 2009, 37, 402–415.e1. [Google Scholar] [CrossRef] [PubMed]
- Cardones, A.R.; Murakami, T.; Hwang, S.T. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via beta(1) integrin. Cancer Res. 2003, 63, 6751–6757. [Google Scholar] [PubMed]
- Devi, S.; Wang, Y.; Chew, W.K.; Lima, R.; A-González, N.; Mattar, C.N.Z.; Chong, S.Z.; Schlitzer, A.; Bakocevic, N.; Chew, S.; et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J. Exp. Med. 2013, 210, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Jujo, K.; Hamada, H.; Iwakura, A.; Thorne, T.; Sekiguchi, H.; Clarke, T.; Ito, A.; Misener, S.; Tanaka, T.; Klyachko, E.; et al. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc. Natl. Acad. Sci. USA 2010, 107, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, X.; Zhou, X.; Ji, W.; Zhang, L.; Luo, T.; Liu, H.; Huang, T.; Jiang, T.; Li, Y. Short-term intermittent administration of CXCR4 antagonist AMD3100 facilitates myocardial repair in experimental myocardial infarction. Acta Biochim. Biophys. Sin. 2013, 45, 561–569. [Google Scholar] [CrossRef] [PubMed][Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attracting Cells | Expressing Cells | Chemokine | Receptor | Studied Site/Organ | Model | Result | Ref. |
|---|---|---|---|---|---|---|---|
| Neutrophils > monocytes | Macrophages, mast cells | CXCL1 (GROalpha) | CXCR2 > CXCR1 | Sputum | Human | Increased in COPD compared to IP | [55,56] |
| Neutrophils > monocytes | Macrophages, mast cells | CXCL2 (MIP-2, GRObeta) | CXCR2 | BALF | Mouse | Increased in COPD compared to control | [57] |
| Neutrophils > monocytes | Alveolar macrophages, epithelial cells, platelets | CXCL5 (epithelial neutrophil-activating peptide 78) | CXCR2 | Bronchial epithelium | Human | Increased at the mRNA level in severe exacerbatory COPD patients compared to stable COPD patients and controls | [58] |
| Neutrophils > monocytes | Inflammatory cells, fibroblasts, endothelial cells, platelets | CXCL7 (truncation product of CTAP-III) | CXCR2 | Bronchial mucosa | Human | Increased number of CXCL7+ cells/mRNA level in stable severe COPD patients compared to healthy controls | [59] |
| Neutrophils > monocytes | Neutrophils, epithelial cells, macrophages, fibroblasts, airway smooth muscle cells | CXCL8 (IL-8) | CXCR1 CXCR2 CCR2 | Lung fibroblasts | Human | Increased | [58,60,61,62,63,64,65,66,67] |
| Blood | Increased in COPD compared to asthma | ||||||
| Induced sputum | Increased in COPD patients compared to controls (smokers and nonsmokers) and asthma | ||||||
| Lung | Increased in bronchiolar epithelium at the mRNA level | ||||||
| Th1 lymphocytes, Tc1 lymphocytes, B lymphocytes | Macrophages, dendritic cells, bronchial epithelial cells | CXCL9 (MIG) | CXCR3 | Lung section | Human | CXCL9 expressed in and around lung lymphoid follicles; - CXCR3 expressed in lung lymphoid follicles, correlated with GOLD stage, inversely correlated with FEV1 | [68,69,70] |
| Sputum | Human | Increased in the sputum of patients with COPD when compared with nonsmokers (but not smokers without obstruction) | |||||
| Bone marrow | Guinea pig | Decreased at the mRNA level in the bone marrow of CS-exposed guinea pigs compared to controls | |||||
| Th1 lymphocytes, Tc1 lymphocytes, B lymphocytes | Macrophages, dendritic cells, bronchial epithelial cells | CXCL10 (IP-10) | CXCR3 | Lung section | Human | CXCL10 expressed in and around lung lymphoid follicles; also in bronchiolar epithelium, airway smooth muscle cells | [68,69,71,72,73] |
| Serum | Elevated in COPD patients compared to controls | ||||||
| Sputum | Increased in the sputum of patients with COPD when compared with nonsmokers (but not smokers without obstruction) | ||||||
| Th1 lymphocytes, Tc1 lymphocytes, B lymphocytes | Macrophages, dendritic cells, bronchial epithelial cells | CXCL11 (I-TAC) | CXCR3 | Sputum | Human | Increased in the sputum of patients with COPD when compared with nonsmokers (but not smokers without obstruction) | [68] |
| BM-MSCs, EPCs, HPCs, lymphocytes, fibrocytes | Inflammatory cells, epithelial and endothelial cells, perivascular stromal cells, BM-MSCs, | CXCL12 (SDF-1) | CXCR4, CXCR7 | Bone marrow | Human | Reduced in COPD BM-MSCs compared to control subjects (mRNA level) | [74,75,76] |
| Blood | Mouse | Enhanced chemosensitivity to CXCL12 of fibrocytes from exacerbating COPD level of CXCL12 decreased in CS-exposed murine lungs | |||||
| Naïve B cells | Follicular dendritic cells, B cells | CXCL13 | CXCR5 | Serum | Human | Decreased in the serum of BMS-COPD subjects compared to controls | [77] |
| Leucocytes, mononuclear cells | Endothelial, epithelial, and smooth muscle cells; macrophages, dendritic cells, B and T cells, and platelets (transmembrane chemokine) | CXCL16 | CXCR6 | Blood | Human | Percentage of CXCL16 (and CXCR6) expressing platelets is increased in COPD patients compared to controls | [78] |
| Monocytes > T lymphocytes, fibrocytes | Alveolar macrophages, T cells, endothelial and epithelial cells | CCL2 (MCP-1) | CCR2 | Blood | Human | Increased in COPD with prevalent emphysema compared to control subjects | [56,65,66,79,80] |
| Sputum | Human | Increased in COPD compared to nonsmokers and healthy smokers | |||||
| Lung | Human | Increased in bronchiolar epithelium at the mRNA level | |||||
| BALF and lung homogenate | Mouse | Increased in CSE-treated mice compared to control group | |||||
| Macrophages; Th2 lymphocytes | Macrophages, lymphocytes, fibroblasts, epithelial cells | CCL3 (MIP-1alpha) | CCR1, 4, 5 | Induced sputum | Human | Increased in COPD patients compared to controls | [81] |
| Macrophages, neutrophils and dendritic cells; memory T cells; basophils, eosinophils; fibrocytes | Endothelial cells, smooth muscle cells; T lymphocytes, epithelial cells | CCL5 (RANTES) | CCR1, 3, 5 | Bronchial mucosa | Human | Increased number of CCL5+ cells/mRNA level in stable severe COPD patients compared to healthy controls | [59,68] |
| Sputum | Increased in the sputum of patients with COPD when compared with nonsmokers (but not smokers without obstruction) | ||||||
| Eosinophils; Th2 lymphocytes | Epithelial cells, endothelial cells, T lymphocytes, macrophages, eosinophils | CCL11 (Eotaxin-1) | CCR3 | BALF | Human | Increased in BALF of COPD with a bronchodilator response, and correlated with emphysema | [82,83,84,85] |
| Blood | Increased in COPD (particularly rapidly progressive) compared to control subjects, and associated with decreased FEV1% and FEV1/FVC ratio | ||||||
| Lamina propria | Number of eotaxin+ and CCR3+ cells significantly higher in exacerbated COPD compared to healthy subjects | ||||||
| T cells, monocytes;Eosinophils; endothelial cells | Basophils, lung leucocytes, alveolar macrophages, airway smooth muscle cells | CCL15 | CCR1 > CCR3 | Serum | Human | Decreased in the serum of BMS-COPD subjects compared to controls | [77] |
| TH2 lymphocytes | Dendritic cells, activated Langerhans cells, airway epithelial cells | CCL17 (TARC) | CCR4 | Serum | Human | Decreased in the serum of BMS-COPD subjects compared to controls rs9302690 SNP significantly associated with higher CCL17 levels in COPD patients | [77,82] |
| T lymphocytes, immature dendritic cells | Dendritic cells, monocytes, alveolar macrophages | CCL18 (PARC/MIP-4/AMAC-1/DC-CK1/SCYA-18) | CCR8 | Serum | Human | Increased in COPD compared to non-obstructive smokers and never smokers | [86,87] |
| Naïve T lymphocytes, mature dendritic cells | Fibroblasts | CCL19 | CCR7 | Bone marrow | Human | Reduced in COPD BM-MSCs compared to control subjects (mRNA level) | [75] |
| Dendritic cells; neutrophils, lymphocytes | Airway epithelial cells, macrophages | CCL20 (MIP-3alpha) | CCR6 | Total lung and induced sputum | Human | Increased at the mRNA level in total lung and at the protein level in induced sputum, compared to never smokers and smokers without COPD | [88] |
| T lymphocytes, mature dendritic cells | Lymphatic endothelial cells | CCL21 | CCR7 | Bone marrow | Human | Reduced in COPD BM-MSCs compared to control subjects (mRNA level) | [75] |
| Memory T lymphocytes | Epithelial cells? (highly produced by keratinocytes in the skin) | CCL27 | CCR10 | Serum | Human | Decreased in the serum of BMS-COPD subjects compared to controls | [77] |
| T lymphocytes, monocytes | Mature dendritic cells, endothelial cells | CX3CL1 (Fractalkin) | CX3CR1 | Serum | Human | Associated with emphysema in chinese COPD patients | [89] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henrot, P.; Prevel, R.; Berger, P.; Dupin, I. Chemokines in COPD: From Implication to Therapeutic Use. Int. J. Mol. Sci. 2019, 20, 2785. https://doi.org/10.3390/ijms20112785
Henrot P, Prevel R, Berger P, Dupin I. Chemokines in COPD: From Implication to Therapeutic Use. International Journal of Molecular Sciences. 2019; 20(11):2785. https://doi.org/10.3390/ijms20112785
Chicago/Turabian StyleHenrot, Pauline, Renaud Prevel, Patrick Berger, and Isabelle Dupin. 2019. "Chemokines in COPD: From Implication to Therapeutic Use" International Journal of Molecular Sciences 20, no. 11: 2785. https://doi.org/10.3390/ijms20112785
APA StyleHenrot, P., Prevel, R., Berger, P., & Dupin, I. (2019). Chemokines in COPD: From Implication to Therapeutic Use. International Journal of Molecular Sciences, 20(11), 2785. https://doi.org/10.3390/ijms20112785