Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing

 , ,
, ,
Abstract
1. Introduction
2. Results
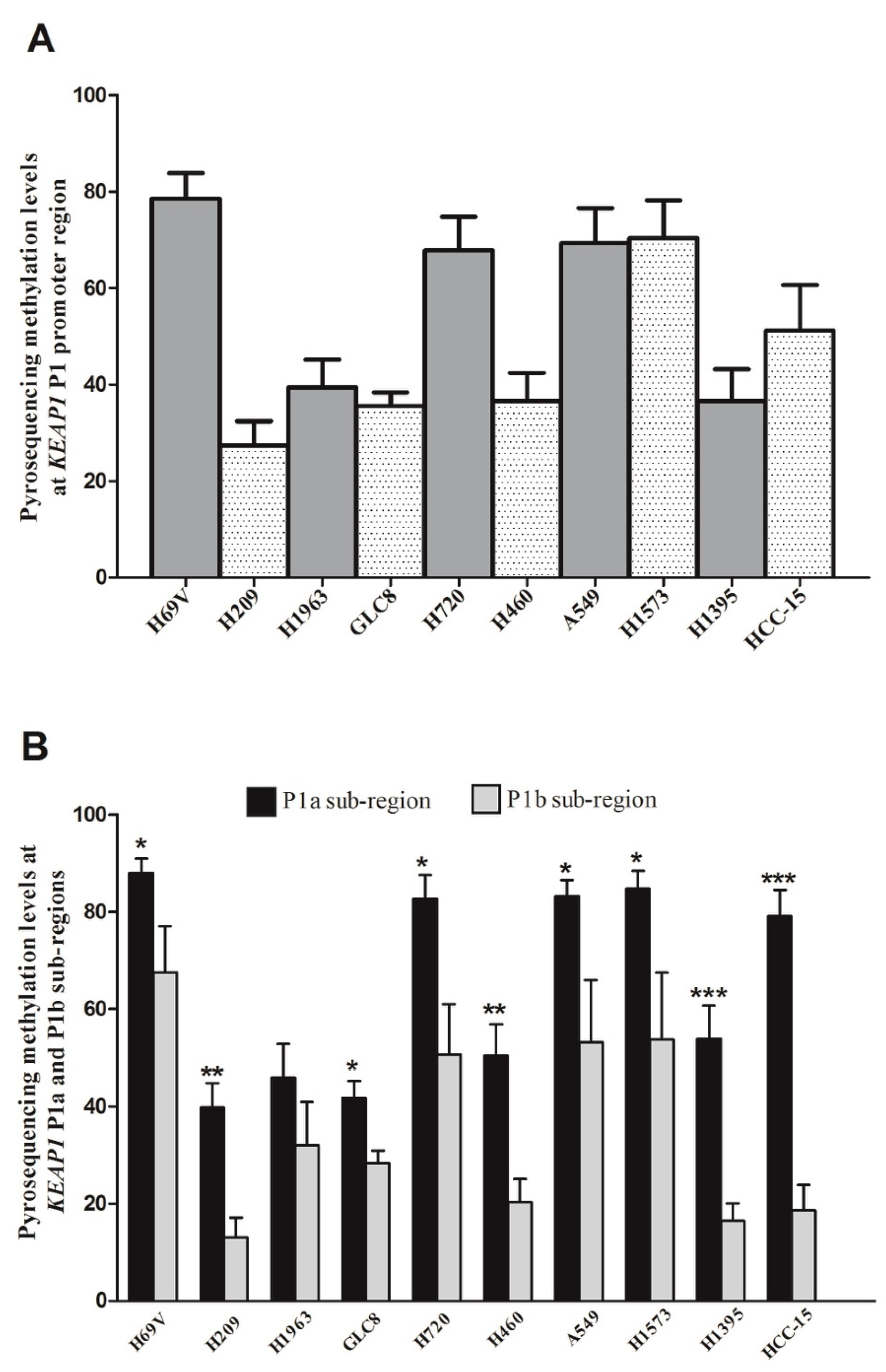
2.1. KEAP1 Promoter Methylation Patterns among Different Lung Cancer Histologies
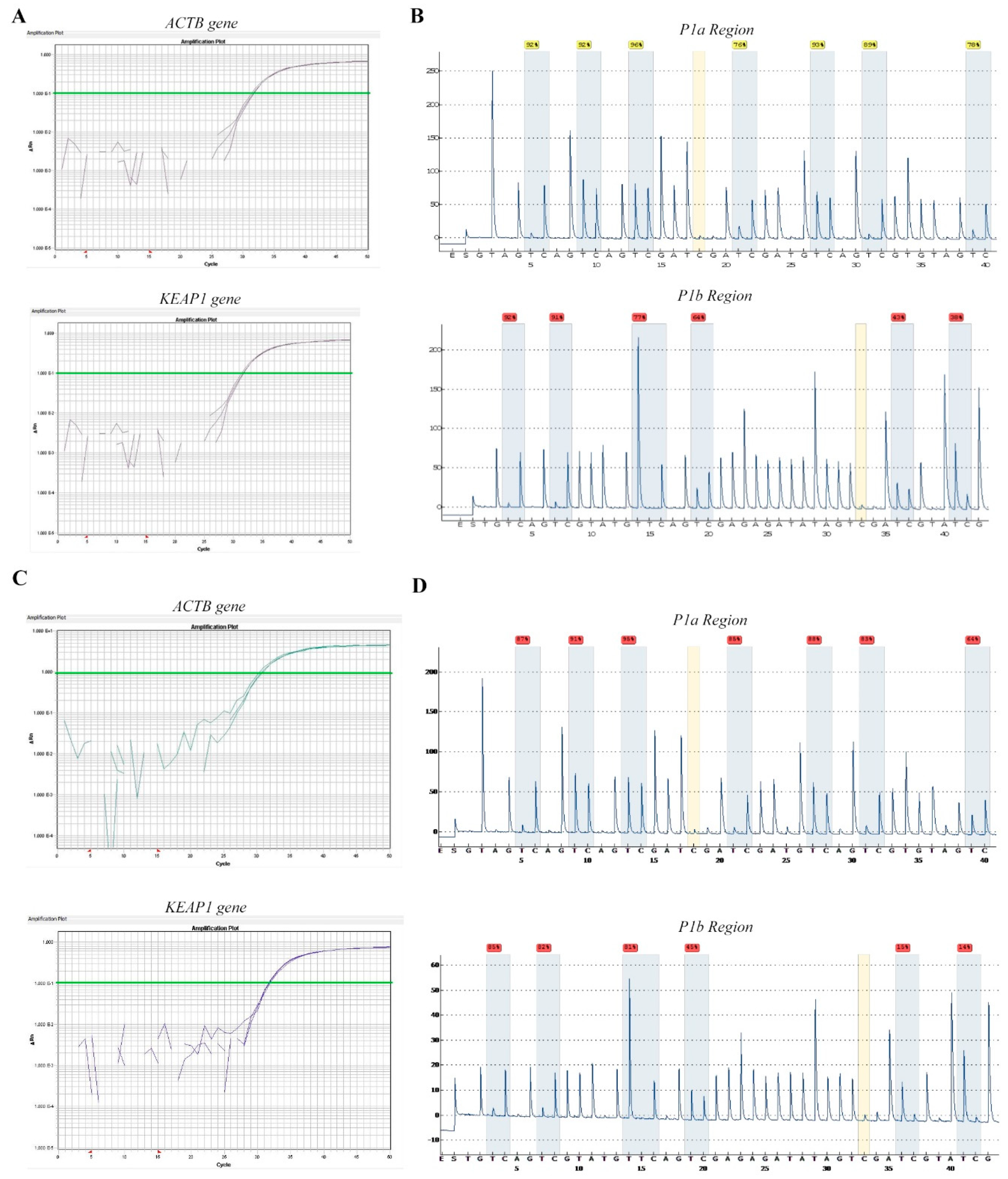
2.2. Technical Evaluation of Pyrosequencing
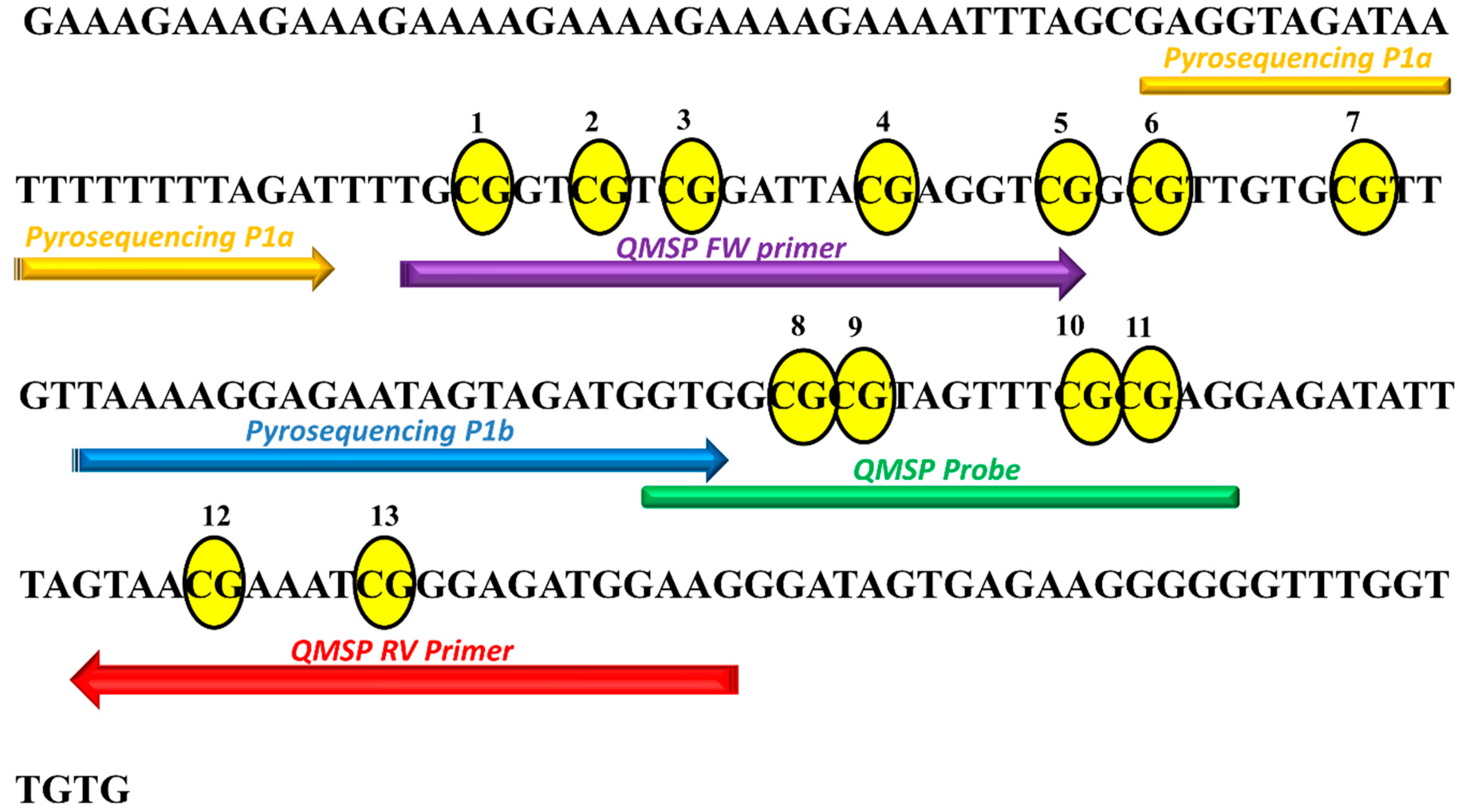
2.3. Pyrosequencing Analysis Reveals Two Distinct P1 Subregions at KEAP1 Promoter
2.4. Somatic Alterations of KEAP1 Detected in Lung Cancer Cell Lines
2.5. The Restoration of KEAP1 Expression Via Its Promoter P1 Region Inversely Correlates with Demethylation by 5-aza-dC
3. Discussion
4. Material and Methods
4.1. Cell Lines
4.2. DNA and RNA Extraction from Cell Lines
4.3. DNA Sodium Bisulfite Conversion and Quantitative Methylation Specific PCR (QMSP)
4.4. Pyrosequencing
4.5. Pyrosequencing Intra and Inter-Assay Analysis
4.6. Mutation Profiling of Cell Lines
4.7. Quantification of KEAP1 Expression by Real-Time Quantitative PCR Analysis
4.8. In vitro 5-Aza-2′-deoxycytidine (5-aza-dC) Treatment
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chio, C., II; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P. Weissman BE, Major MB: NRF2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Sparaneo, A.; Trombetta, D.; Muscarella, L.A. Epigenetic versus Genetic Deregulation of the KEAP1/NRF2 Axis in Solid Tumors: Focus on Methylation and Noncoding RNAs. Oxid. Med. Cell Longev. 2018, 2018, 2492063. [Google Scholar] [CrossRef] [PubMed]
- Sparaneo, A.; Fabrizio, F.P.; Muscarella, L.A. Nrf2 and Notch Signaling in Lung Cancer: Near the Crossroad. Oxid. Med. Cell Longev. 2016, 2016, 7316492. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008, 135, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys Res. Commun. 2007, 362, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Eichenmuller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Haberle, B.; Cairo, S.; Leuschner, I.; von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Pikor, L.A.; Lam, S.; Lam, W.L. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: Association with poor prognosis in head and neck cancer. Head. Neck. 2015, 37, 727–734. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Sasaki, H.; Shitara, M.; Yokota, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. Increased NRF2 gene (NFE2L2) copy number correlates with mutations in lung squamous cell carcinomas. Mol. Med. Rep. 2012, 6, 391–394. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef]
- Ooi, A.; Dykema, K.; Ansari, A.; Petillo, D.; Snider, J.; Kahnoski, R.; Anema, J.; Craig, D.; Carpten, J.; The, B.T.; et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013, 73, 2044–2051. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Costantini, M.; Copetti, M.; la Torre, A.; Sparaneo, A.; Fontana, A.; Poeta, L.; Gallucci, M.; Sentinelli, S.; Graziano, P.; et al. Keap1/Nrf2 pathway in kidney cancer: Frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 2017, 8, 11187–11198. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Barbano, R.; D’Angelo, V.; Copetti, M.; Coco, M.; Balsamo, T.; la Torre, A.; Notarangelo, A.; Troiano, M.; Parisi, S.; et al. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics 2011, 6, 317–325. [Google Scholar] [CrossRef]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef]
- Barbano, R.; Muscarella, L.A.; Pasculli, B.; Valori, V.M.; Fontana, A.; Coco, M.; la Torre, A.; Balsamo, T.; Poeta, M.L.; Marangi, G.F.; et al. Aberrant Keap1 methylation in breast cancer and association with clinicopathological features. Epigenetics 2013, 8, 105–112. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Parrella, P.; D’Alessandro, V.; la Torre, A.; Barbano, R.; Fontana, A.; Tancredi, A.; Guarnieri, V.; Balsamo, T.; Coco, M.; et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 2011, 6, 710–719. [Google Scholar] [CrossRef]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; Konig, K.; Michels, S.; Fischer, R.; et al. Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef]
- Derks, J.L.; Leblay, N.; Thunnissen, E.; van Suylen, R.J.; den Bakker, M.; Groen, H.J.M.; Smit, E.F.; Damhuis, R.; van den Broek, E.C.; Charbrier, A.; et al. Molecular Subtypes of Pulmonary Large-cell Neuroendocrine Carcinoma Predict Chemotherapy Treatment Outcome. Clin. Cancer Res. 2018, 24, 33–42. [Google Scholar] [CrossRef]
- Namani, A.; Matiur Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef]
- Liu, X.; Sun, C.; Liu, B.; Jin, X.; Li, P.; Zheng, X.; Zhao, T.; Li, F.; Li, Q. Genistein mediates the selective radiosensitizing effect in NSCLC A549 cells via inhibiting methylation of the keap1 gene promoter region. Oncotarget 2016, 7, 27267–27279. [Google Scholar] [CrossRef]
- Rekhtman, N.; Pietanza, M.C.; Hellmann, M.D.; Naidoo, J.; Arora, A.; Won, H.; Halpenny, D.F.; Wang, H.; Tian, S.K.; Litvak, A.M.; et al. Next-Generation Sequencing of Pulmonary Large Cell Neuroendocrine Carcinoma Reveals Small Cell Carcinoma-like and Non-Small Cell Carcinoma-like Subsets. Clin. Cancer Res. 2016, 22, 3618–3629. [Google Scholar] [CrossRef]
- Tost, J. Current and Emerging Technologies for the Analysis of the Genome-Wide and Locus-Specific DNA Methylation Patterns. Adv. Exp. Med. Biol. 2016, 945, 343–430. [Google Scholar]
- Guo, D.; Wu, B.; Yan, J.; Li, X.; Sun, H.; Zhou, D. A possible gene silencing mechanism: Hypermethylation of the Keap1 promoter abrogates binding of the transcription factor Sp1 in lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 428, 80–85. [Google Scholar] [CrossRef]
- Lania, L.; Majello, B.; De Luca, P. Transcriptional regulation by the Sp family proteins. Int. J. Biochem. Cell Biol. 1997, 29, 1313–1323. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lung Cancer Cells | Histology | QMSP | CpG1 | CpG2 | CpG3 | CpG4 | CpG5 | CpG6 | CpG7 | CpG8 | CpG9 | CpG10 | CpG11 | CpG12 | CpG13 | Pyrosequencing (Mean % ± SD) * | P1 Region Mean (%) ± SD ** | P1a Region Mean (%) ± SD ** | P1b Region Mean (%) ± SD ** |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H69V | SCLC | 138 | 92 | 92 | 96 | 76 | 93 | 89 | 78 | 92 | 91 | 77 | 64 | 43 | 38 | 78.5 ± 19.3 | 79 ± 20 | 88 ± 8 | 68 ± 23 |
| H209 | SCLC | 4.8 | 55 | 47 | 54 | 23 | 40 | 37 | 22 | 21 | 28 | 15 | 7 | 4 | 3 | 27.4 ± 18.0 | 27 ± 13 | 40 ± 13 | 13 ± 10 |
| H1184 | SCLC | 0 | 14 | 9 | 13 | 4 | 7 | 5 | 6 | 8 | 9 | 8 | 2 | 2 | 2 | 6.9 ± 3.9 | 7 ± 5 | 8 ± 4 | 5 ± 3 |
| N417 | SCLC | 0 | 21 | 19 | 19 | 7 | 11 | 15 | 10 | 12 | 12 | 6 | 3 | 1 | 2 | 10.6 ± 6.7 | 11 ± 6 | 15 ± 5 | 6 ± 5 |
| H2107 | SCLC | 0 | 20 | 24 | 24 | 6 | 18 | 10 | 10 | 7 | 8 | 8 | 3 | 1 | 2 | 10.8 ± 8.0 | 11 ± 5 | 16 ± 7 | 5 ± 3 |
| H1963 | SCLC | 15 | 61 | 57 | 68 | 13 | 46 | 40 | 36 | 56 | 51 | 44 | 28 | 7 | 6 | 39.5 ± 20.6 | 39 ± 32 | 46 ± 19 | 32 ± 22 |
| GLC1 | SCLC | 0 | 21 | 25 | 24 | 5 | 12 | 10 | 9 | 19 | 15 | 8 | 3 | 1 | 2 | 11.8 ± 8.3 | 12 ± 8 | 15 ± 8 | 8 ± 7 |
| GLC2 | SCLC | 0 | 26 | 24 | 21 | 4 | 10 | 13 | 8 | 17 | 13 | 9 | 5 | 2 | 2 | 11.8 ± 8.1 | 12 ± 8 | 15 ± 9 | 8 ± 6 |
| GLC8 | SCLC | 228 | 54 | 46 | 53 | 31 | 38 | 37 | 33 | 34 | 35 | 32 | 26 | 21 | 22 | 35.5 ± 10.4 | 36 ± 28 | 42 ± 9 | 28 ± 6 |
| GLC14 | SCLC | 0 | 22 | 15 | 17 | 5 | 10 | 10 | 8 | 9 | 9 | 5 | 2 | 1 | 2 | 8.8 ± 6.2 | 9 ± 5 | 12 ± 6 | 5 ± 4 |
| H510 | SCLC | 0 | 8 | 4 | 7 | 1 | 3 | 3 | 3 | 2 | 3 | 4 | 1 | 0 | 1 | 3.1 ± 2.3 | 3 ± 2 | 4 ± 2 | 2 ± 1 |
| H2141 | SCLC | 0 | 17 | 12 | 14 | 4 | 7 | 9 | 6 | 6 | 7 | 6 | 2 | 2 | 2 | 7.2 ± 4.7 | 7 ± 4 | 10 ± 5 | 4 ± 2 |
| H720 | AC | 78.8 | 92 | 88 | 96 | 61 | 89 | 84 | 68 | 77 | 74 | 65 | 42 | 15 | 31 | 67.8 ± 25.0 | 68 ± 51 | 83 ± 13 | 51 ± 25 |
| H727 | TC | 0 | 21 | 14 | 21 | 7 | 11 | 11 | 8 | 7 | 7 | 11 | 3 | 2 | 5 | 9.8 ± 6.0 | 10 ± 6 | 13 ± 6 | 6 ± 3 |
| H460 | LCC | 66 | 65 | 64 | 72 | 28 | 50 | 41 | 33 | 23 | 32 | 33 | 9 | 4 | 21 | 36.5 ± 21.2 | 37 ± 20 | 50 ± 17 | 20 ± 12 |
| H2126 | ADC | 0 | 68 | 58 | 65 | 15 | 29 | 27 | 18 | 20 | 17 | 16 | 5 | 3 | 8 | 21.4 ± 14.4 | 27 ± 12 | 40 ± 23 | 12 ± 7 |
| A549 | ADC | 492 | 88 | 88 | 95 | 73 | 87 | 81 | 70 | 87 | 84 | 69 | 43 | 18 | 18 | 26.8 ± 22.4 | 69 ± 53 | 83 ± 9 | 53 ± 31 |
| H1573 | ADC | 123 | 87 | 91 | 95 | 85 | 88 | 83 | 64 | 85 | 82 | 81 | 45 | 15 | 14 | 69.3 ± 26.3 | 70 ± 54 | 85 ± 10 | 54 ± 34 |
| H2228 | ADC | 0 | 43 | 47 | 37 | 13 | 19 | 24 | 16 | 13 | 25 | 12 | 9 | 3 | 6 | 70.4 ± 28.0 | 21 ± 11 | 28 ± 14 | 11 ± 8 |
| H1975 | ADC | 0 | 16 | 10 | 14 | 6 | 10 | 1 | 8 | 4 | 4 | 9 | 2 | 2 | 4 | 20.5 ± 14.1 | 7 ± 4 | 9 ± 5 | 4 ± 3 |
| HCC4006 | ADC | 0 | 40 | 35 | 48 | 11 | 24 | 17 | 15 | 6 | 19 | 23 | 4 | 2 | 20 | 6.9 ± 4.7 | 20 ± 12 | 27 ± 14 | 12 ± 9 |
| H1395 | ADC | 17 | 72 | 74 | 68 | 36 | 56 | 36 | 35 | 24 | 26 | 23 | 11 | 5 | 10 | 20.3 ± 13.9 | 37 ± 17 | 54 ± 18 | 17 ± 9 |
| H1581 | ADC | 30 | 44 | 36 | 38 | 12 | 26 | 31 | 5 | 30 | 27 | 16 | 8 | 2 | 3 | 36.6 ± 23.9 | 21 ± 14 | 27 ± 14 | 14 ± 12 |
| HCC-15 | SqCC | 12 | 85 | 87 | 94 | 54 | 89 | 78 | 67 | 25 | 38 | 24 | 7 | 5 | 13 | 51.2 ± 33.9 | 51 ± 19 | 79 ± 14 | 19 ± 13 |
| H520 | SqCC | 0 | 45 | 34 | 41 | 12 | 25 | 23 | 14 | 14 | 18 | 14 | 5 | 2 | 8 | 19.6 ± 13.4 | 20 ± 10 | 28 ± 13 | 10 ± 6 |
| (A) | ||||||||
| Normal Cell Lines | RN1 Mean ± SD | RN2 Mean ± SD | RN3 Mean ± SD | RN4 Mean ± SD | RN5 Mean ± SD | ALL RPs Mean ± SD | SD | CV% |
| MRC5 | 19.8 ± 12.4 | 16 ± 10.4 | 20.2 ± 12.3 | 22.3 ± 9.6 | 19.7 ± 11.9 | 19.6 ± 1.2 | 1.2 | 6.1 |
| BEAS-2B | 24.8 ± 21.6 | 24.8 ± 18.2 | 23.2 ± 19.5 | 20.3 ± 17.5 | 22.5 ± 20.8 | 23.1 ± 1.7 | 1.7 | 7.4 |
| (B) | ||||||||
| Normal Cell Lines | RP1 Mean ± SD | RP2 Mean ± SD | RP3 Mean ± SD | ALL RPs Mean ± SD | SD | CV% | ||
| MRC5 | 20.2 ± 12.3 | 22.3 ± 9.6 | 19.7 ± 11.9 | 20.7 ± 1.4 | 1.4 | 6.8 | ||
| BEAS-2B | 23.2 ± 19.5 | 20.3 ± 17.5 | 22.5 ± 20.8 | 22 ± 1.7 | 1.7 | 7.7 | ||
| (A) | |||
| CpG | MRC5 (Mean ± SD) | BEAS-2B (Mean ± SD) | CV % |
| 1 | 37 ± 3.7 | 56.4 ± 6.1 | 2.6 |
| 2 | 34.6 ± 4.3 | 32.8 ± 4.7 | 0.6 |
| 3 | 39.2 ± 5.2 | 40.4 ± 3.1 | 2.6 |
| 4 | 17 ± 1.4 | 13.6 ± 2.1 | 2.2 |
| 5 | 22 ± 5.7 | 51.8 ± 5.7 | 0.1 |
| 6 | 23.8 ± 4.0 | 49 ± 2.2 | 2.4 |
| 7 | 17.8 ± 1.6 | 20.6 ± 4.2 | 6.6 |
| 8 | 17.6 ± 5.8 | 8.8 ± 1.1 | 17.7 |
| 9 | 16.4 ± 2.7 | 9.6 ±1.9 | 2.9 |
| 10 | 11.2 ± 2.9 | 7.4 ± 4.4 | 8.2 |
| 11 | 8.8 ± 2.0 | 4.6 ± 1.5 | 4.0 |
| 12 | 4.6 ± 3.0 | 2.6 ± 1.5 | 21.3 |
| 13 | 4.8 ± 2.5 | 3.2 ± 2.7 | 2.4 |
| (B) | |||
| CpG | MRC5 (Mean ± SD) | BEAS-2B (Mean ± SD) | CV % |
| 1 | 37.3 ± 2.5 | 54.3 ± 7.5 | 5.4 |
| 2 | 36.7 ± 1.2 | 29.7 ± 1.2 | 0.0 |
| 3 | 40.0 ± 1.7 | 40.0 ± 4.4 | 3.3 |
| 4 | 17.0 ± 2.0 | 12.3 ± 0.6 | 4.8 |
| 5 | 24.7 ± 1.2 | 49.3 ± 4.7 | 4.8 |
| 6 | 25.3 ± 1.5 | 50.0 ± 2.0 | 0.6 |
| 7 | 17.3 ±2.1 | 18.3 ± 3.8 | 4.8 |
| 8 | 20.7 ± 5.5 | 8.3 ± 1.2 | 15.0 |
| 9 | 17.7 ± 1.5 | 9.0 ± 2.0 | 1.8 |
| 10 | 12.3 ± 2.3 | 6.7 ± 4.0 | 9.1 |
| 11 | 9.3 ± 2.5 | 4.3 ± 2.1 | 3.2 |
| 12 | 5.7 ± 3.8 | 2.0 ± 1.0 | 36.3 |
| 13 | 5.7 ± 3.1 | 2.0 ± 0.0 | 39.8 |
| Cell Line | Histology | Gene | Protein Domain | Nucleotidic Change | Amino Acid Change |
|---|---|---|---|---|---|
| H1184 | SCLC | KEAP1 | KELCH2 | c.1090G > T | p.G364C |
| H460 | LCC | KEAP1 | IVR | c.706G > C | p.D236H |
| H2126 | ADC | KEAP1 | IVR | c.814C > T | p.R272C |
| A549 | ADC | KEAP1 | KELCH1 | c.997G > T | p.G333C |
| H1573 | ADC | KEAP1 | KELCH3 | c.1238G > T | p.L413R |
| H2228 | ADC | KEAP1 | BTB | c.269C > T | p.A90V |
| H1395 | ADC | KEAP1 | KELCH1 | c.1048G > A | p.G350S |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabrizio, F.P.; Sparaneo, A.; Centra, F.; Trombetta, D.; Storlazzi, C.T.; Graziano, P.; Maiello, E.; Fazio, V.M.; Muscarella, L.A. Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing. Int. J. Mol. Sci. 2019, 20, 2697. https://doi.org/10.3390/ijms20112697
Fabrizio FP, Sparaneo A, Centra F, Trombetta D, Storlazzi CT, Graziano P, Maiello E, Fazio VM, Muscarella LA. Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing. International Journal of Molecular Sciences. 2019; 20(11):2697. https://doi.org/10.3390/ijms20112697
Chicago/Turabian StyleFabrizio, Federico Pio, Angelo Sparaneo, Flavia Centra, Domenico Trombetta, Clelia Tiziana Storlazzi, Paolo Graziano, Evaristo Maiello, Vito Michele Fazio, and Lucia Anna Muscarella. 2019. "Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing" International Journal of Molecular Sciences 20, no. 11: 2697. https://doi.org/10.3390/ijms20112697
APA StyleFabrizio, F. P., Sparaneo, A., Centra, F., Trombetta, D., Storlazzi, C. T., Graziano, P., Maiello, E., Fazio, V. M., & Muscarella, L. A. (2019). Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing. International Journal of Molecular Sciences, 20(11), 2697. https://doi.org/10.3390/ijms20112697