Potential Dissociative Glucocorticoid Receptor Activity for Protopanaxadiol and Protopanaxatriol

 and
and 
Abstract
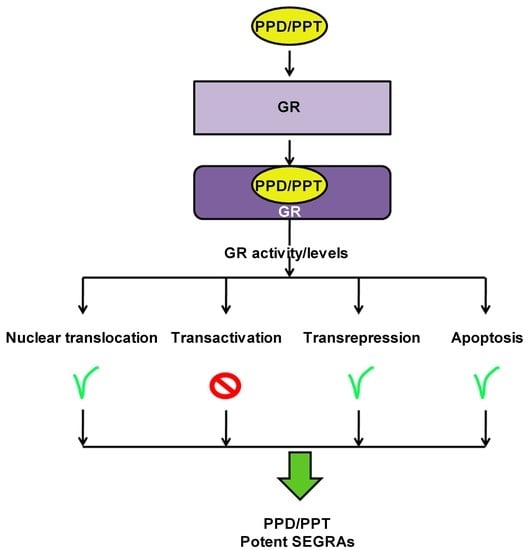
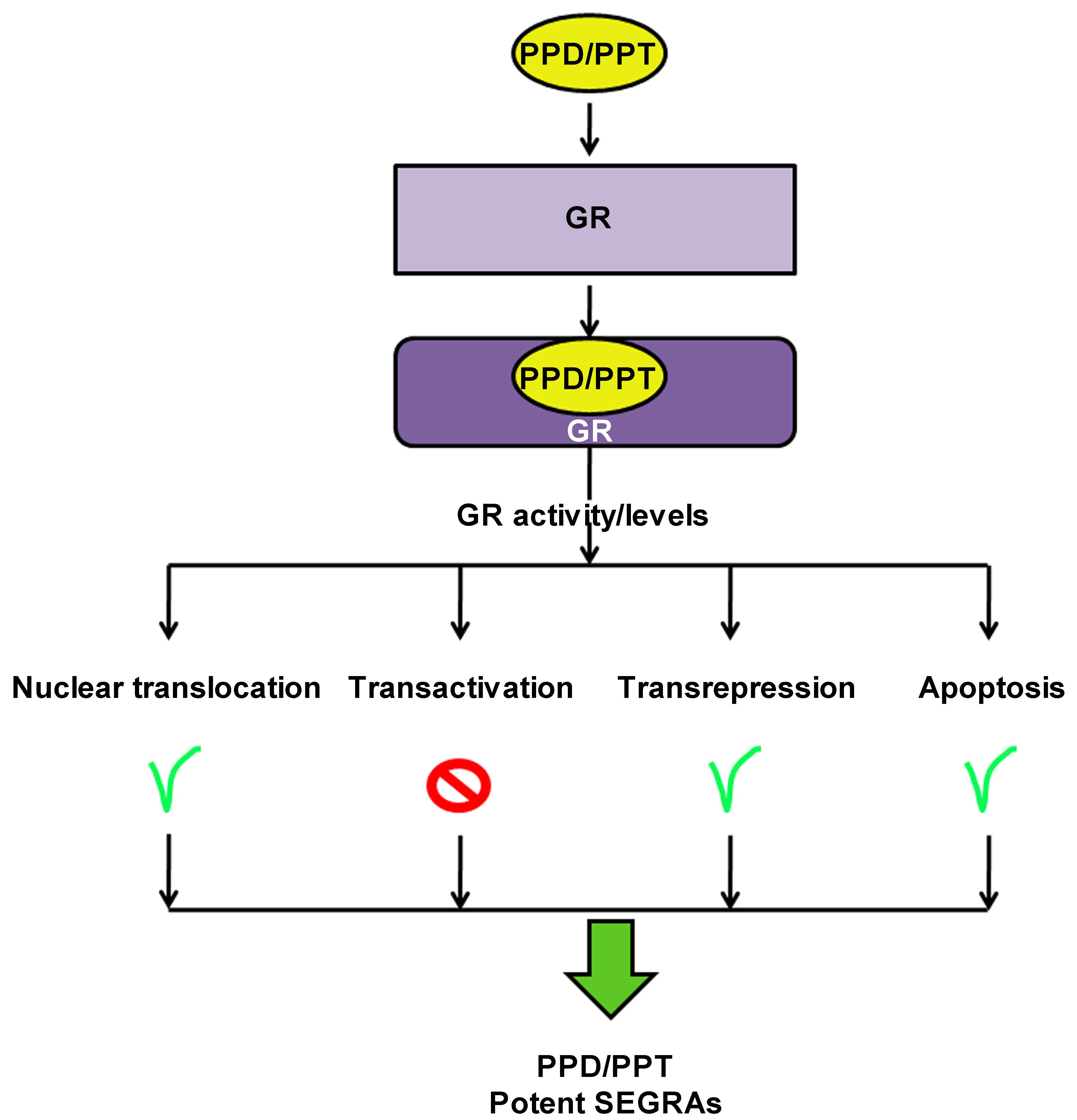
1. Introduction
2. Results
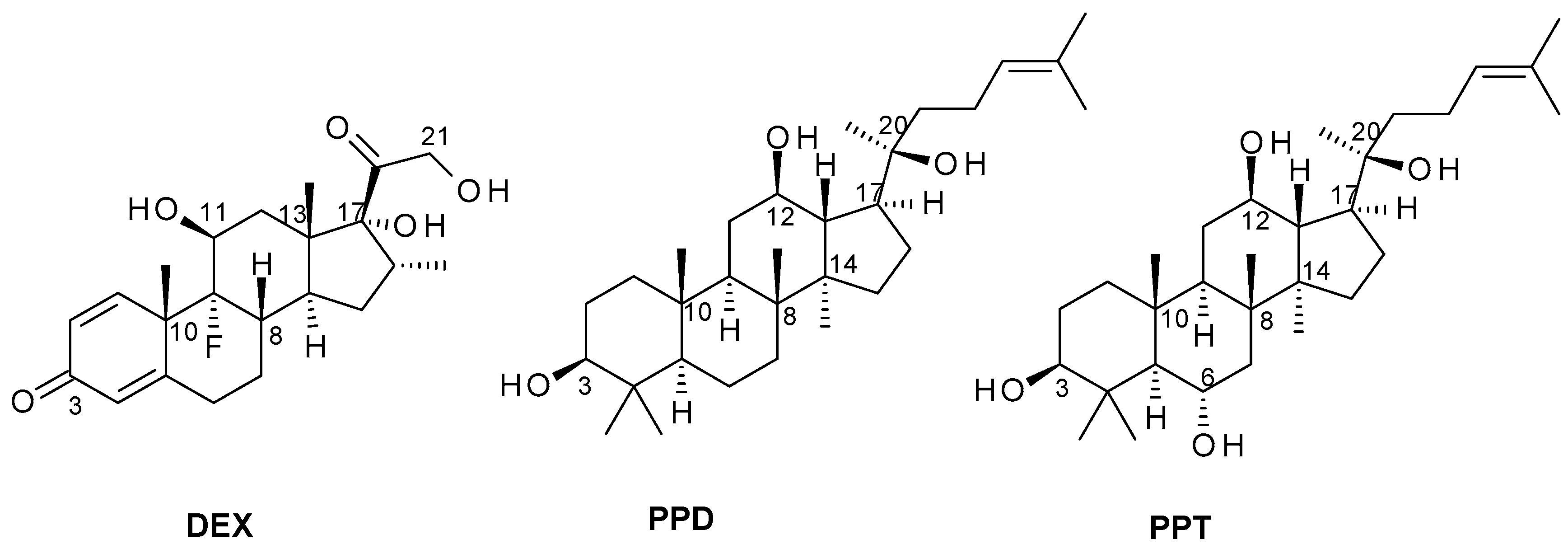
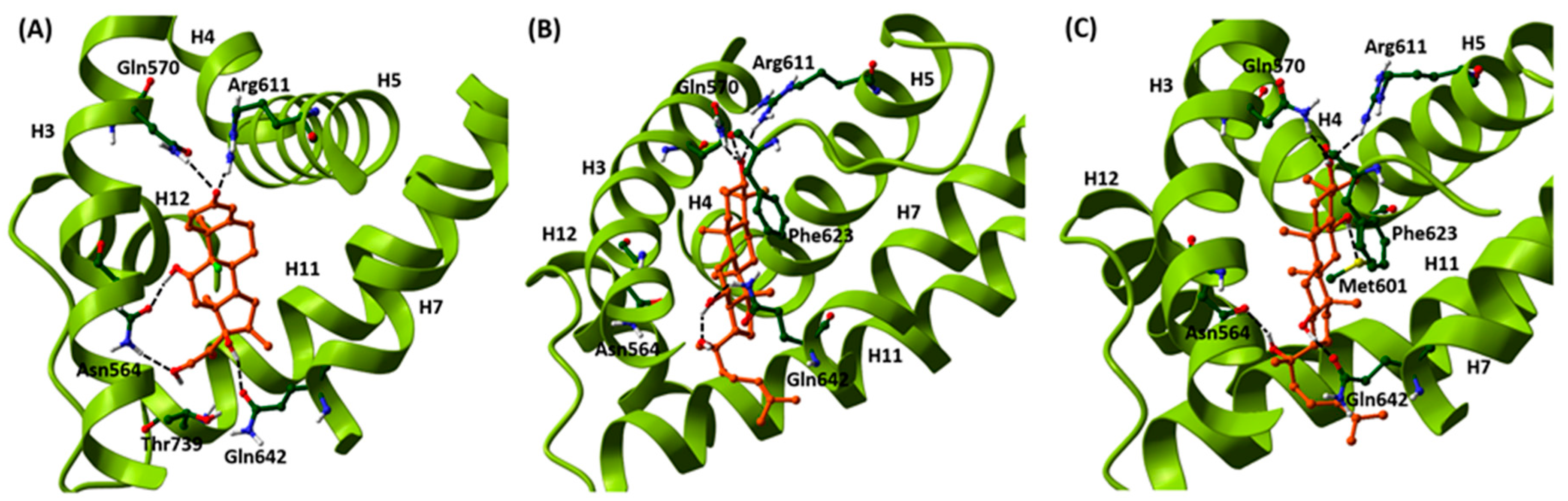
2.1. Induced-Fit Docking Analysis Reveals Potential Binding of PPD and PPT to GR Ligand Binding Domain
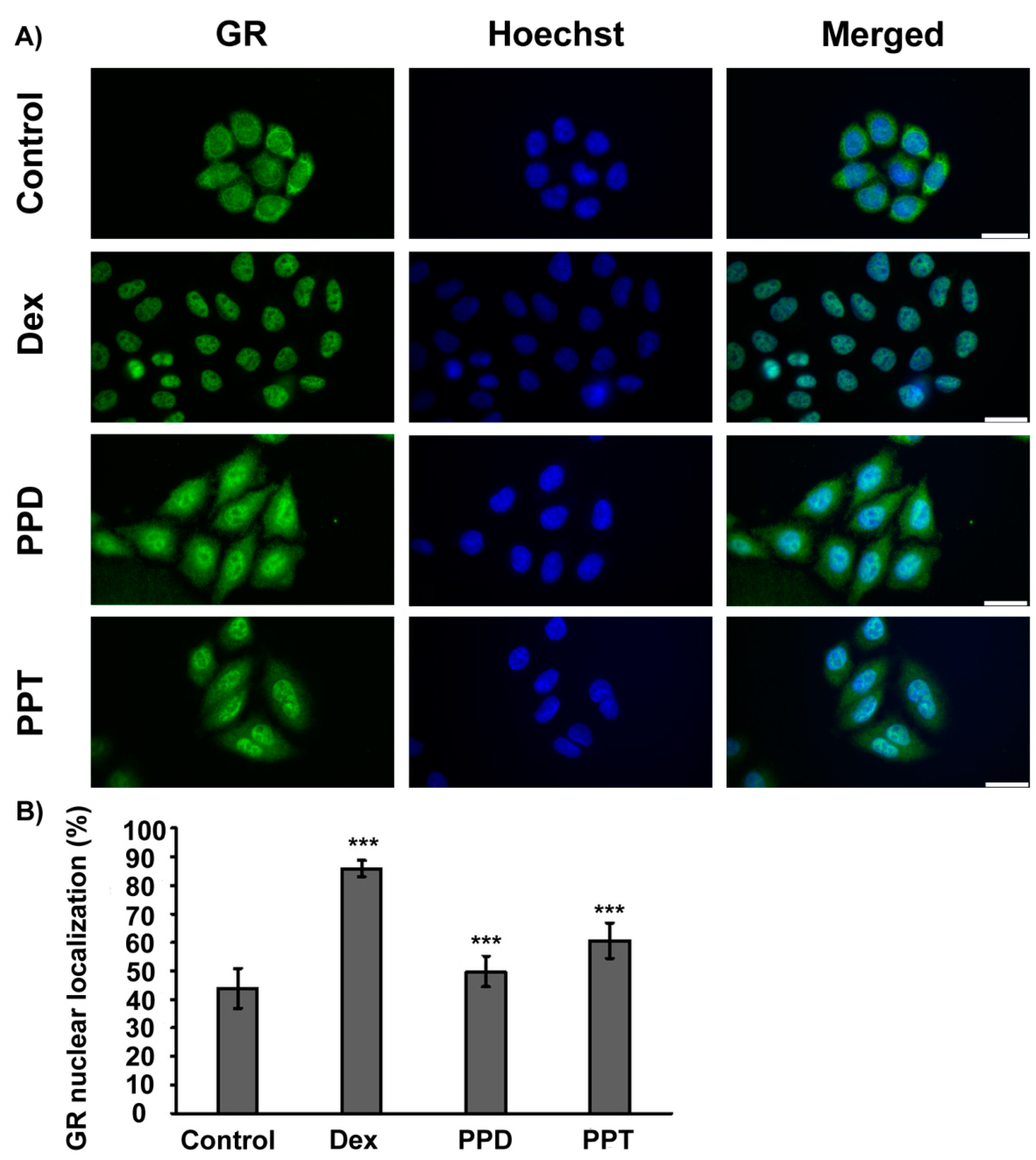
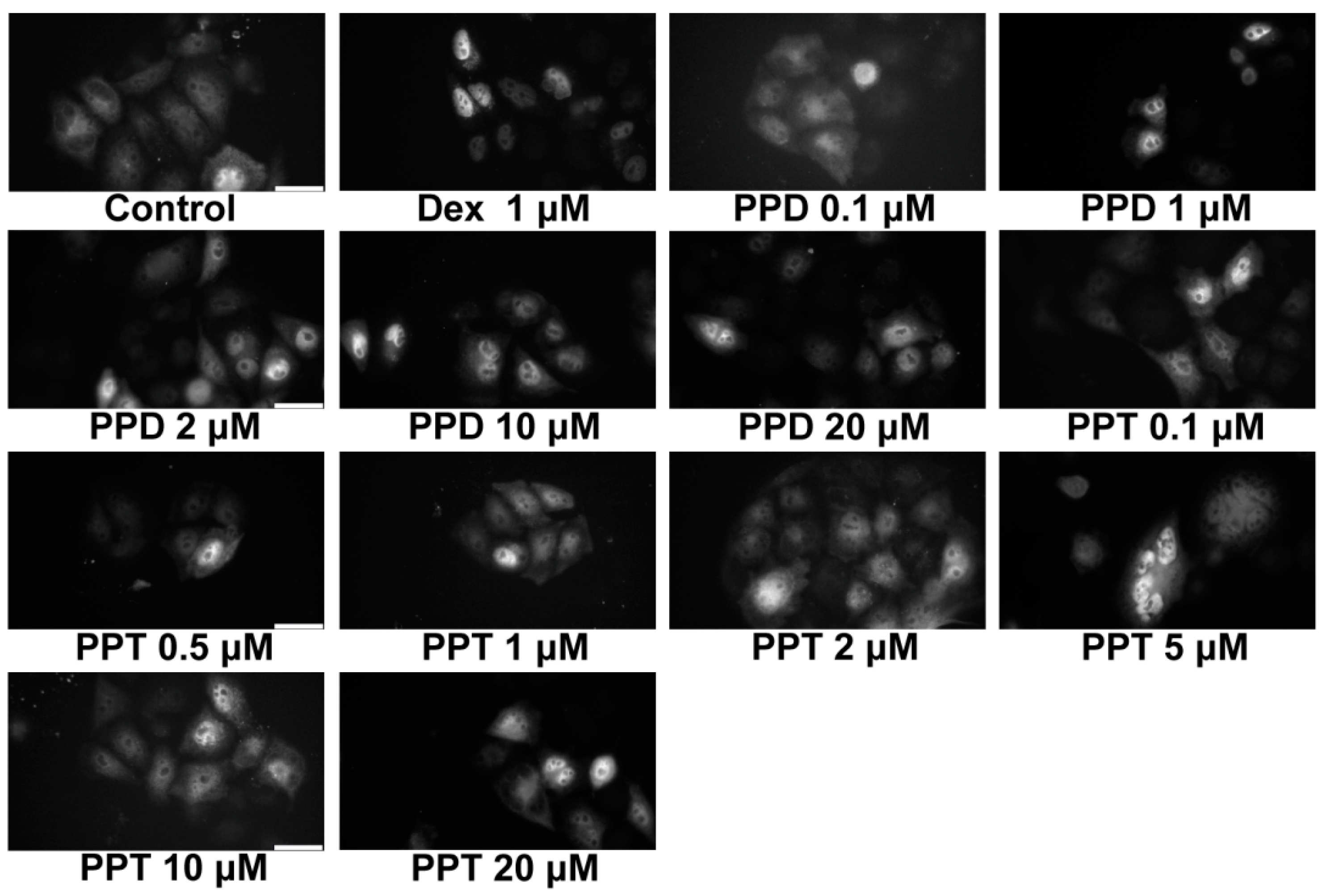
2.2. PPD and PPT Induce GR Nuclear Translocation
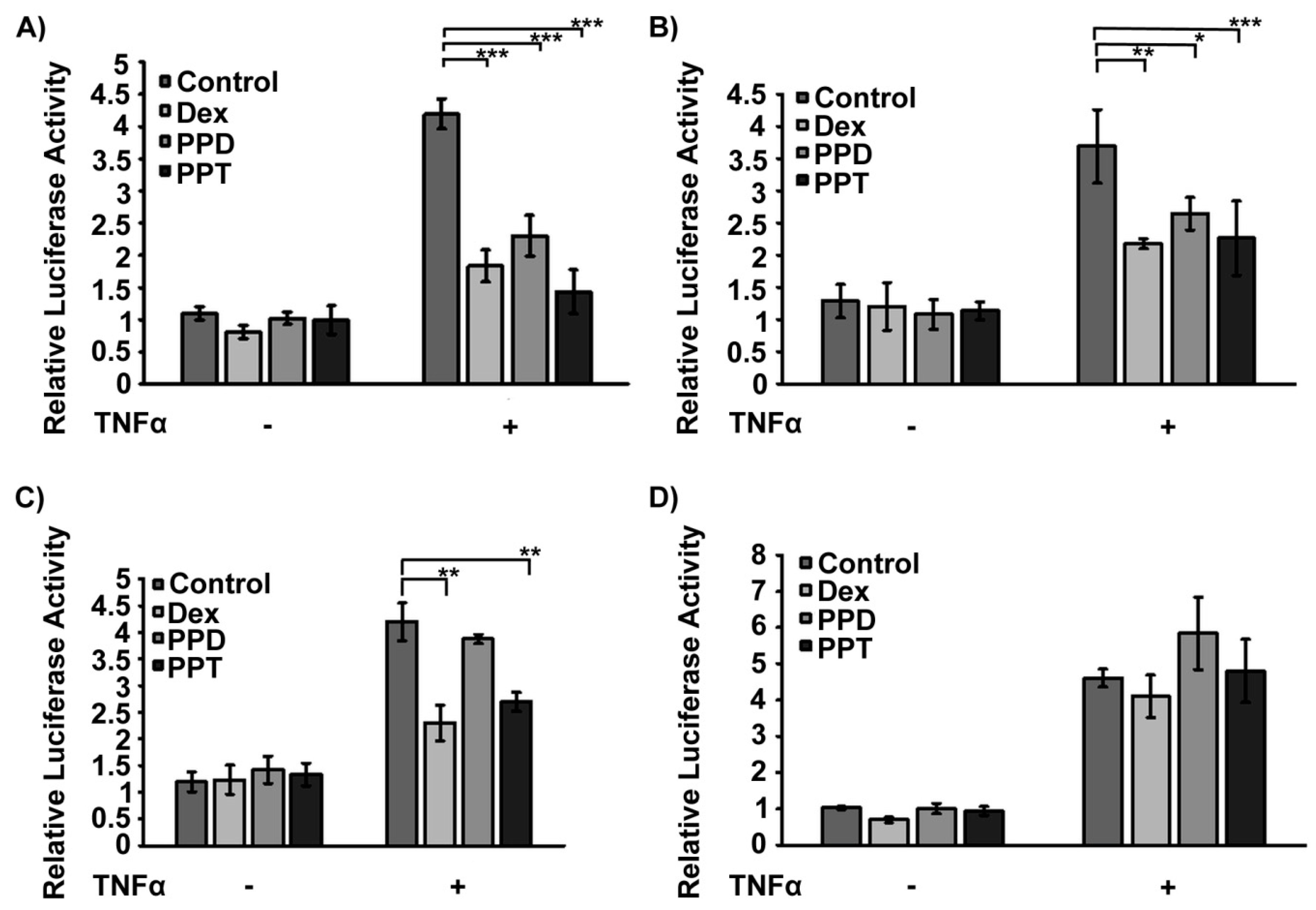
2.3. PPD and PPT Suppress the DEX Induced GR Transactivation
2.4. Transcriptional Inactivation of NF-κΒ by PPD and PPT via Potential Activation of GR Transrepression Activity
2.5. PPD and PPT Induce Mitochondrial-Dependent Apoptosis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Antibodies
4.3. Cell Lines-Cell Culture
4.4. Immunofluorescence Microscopy
4.5. NF-κB and GR Activity
4.6. Electrophoresis and Western Blotting
4.7. Statistical Analysis
4.8. Induced-Fit Docking Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| AMPK | AMP activated protein kinase |
| ANOVA | Analysis of Variance |
| DEX | Dexamethasone |
| GCs | Glucocorticoids |
| GLUT | glucose transporters |
| GR | glucocorticoid receptor |
| GRE | Glucocorticoid Response Element |
| IFD | Induced-Fit Docking Analysis |
| IL | Interleukin |
| JNK | c-Jun terminal NH2-terminal kinase |
| NF-κΒ | Nuclear factor-kappa beta |
| LBD | Ligand Binding Domain |
| MMTV | Mouse Mammary Tumor Virus |
| PEPCK | Phosphoenolopyruvate Carboxykinase |
| PI3K | phosphoinositide-3 kinase |
| PPARgamma | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PPD | Protopanaxadiol |
| PPT | Protopanaxatriol |
| PtdIns(4,5)P2 | Phosphotidyloinositol-4,5 bisphosphate 3-kinase |
| SEGRAs | Selective Glucocorticoid Receptors Activators |
| SEGRMs | Selective Glucocorticoid Receptors Modulators |
| Stat3 | Signal Transducer and Activator of Transcription – 3 |
| STAT5 | signal transducer and activator of transcription 5 |
| TNFα | Tumor Necrosis Factor α |
References
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef]
- Revollo, J.R.; Cidlowski, J.A. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann. N. Y. Acad. Sci. 2009, 1179, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Losel, R.; Wehling, M. Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 2003, 4, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Cellular processing of the glucocorticoid receptor gene and protein: New mechanisms for generating tissue-specific actions of glucocorticoids. J. Biol. Chem. 2011, 286, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, M.; Verhaar, A.P.; van den Brink, G.R.; Hommes, D.W. Glucocorticoid signaling: A nongenomic mechanism for T-cell immunosuppression. Trends Mol. Med. 2007, 13, 158–163. [Google Scholar] [CrossRef]
- Nocentini, G.; Migliorati, G.; Riccardi, C. The Molecular and Cellular Mechanisms Responsible for the Anti-inflammatory and Immunosuppressive Effects of Glucocorticoids. In Systemic Corticosteroids for Inflammatory Disorders in Pediatrics; Cimaz, R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 25–41. [Google Scholar] [CrossRef]
- Sionov, R.V.; Cohen, O.; Kfir, S.; Zilberman, Y.; Yefenof, E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J. Exp. Med. 2006, 203, 189–201. [Google Scholar] [CrossRef]
- Psarra, A.M.; Sekeris, C.E. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: Role of the mitochondrial glucocorticoid receptor. Biochim. Biophys. Acta 2011, 1813, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.; Sekeris, C.E. Nuclear receptors and other nuclear transcription factors in mitochondria: Regulatory molecules in a new environment. Biochim. Biophys. Acta 2008, 1783, 1–11. [Google Scholar] [CrossRef]
- Hunter, R.G.; Seligsohn, M.; Rubin, T.G.; Griffiths, B.B.; Ozdemir, Y.; Pfaff, D.W.; Datson, N.A.; McEwen, B.S. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 9099–9104. [Google Scholar] [CrossRef]
- Baschant, U.; Culemann, S.; Tuckermann, J. Molecular determinants of glucocorticoid actions in inflammatory joint diseases. Mol. Cell. Endocrinol. 2013, 380, 108–118. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef]
- Schmidt, S.; Rainer, J.; Ploner, C.; Presul, E.; Riml, S.; Kofler, R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: Molecular mechanisms and clinical relevance. Cell Death Differ. 2004, 11 (Suppl. 1), S45–S55. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Refojo, D.; Liberman, A.C.; Holsboer, F.; Arzt, E. Transcription factor-mediated molecular mechanisms involved in the functional cross-talk between cytokines and glucocorticoids. Immunol. Cell Biol. 2001, 79, 385–394. [Google Scholar] [CrossRef] [PubMed]
- King, E.M.; Chivers, J.E.; Rider, C.F.; Minnich, A.; Giembycz, M.A.; Newton, R. Glucocorticoid Repression of Inflammatory Gene Expression Shows Differential Responsiveness by Transactivation- and Transrepression-Dependent Mechanisms. PLoS ONE 2013, 8, e53936. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Gilson, H.; Kalista, S.; Thissen, J.P. Mechanisms of muscle atrophy induced by glucocorticoids. Horm. Res. 2009, 72 (Suppl. 1), 36–41. [Google Scholar] [CrossRef] [PubMed]
- Lofberg, E.; Gutierrez, A.; Wernerman, J.; Anderstam, B.; Mitch, W.E.; Price, S.R.; Bergstrom, J.; Alvestrand, A. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Investig. 2002, 32, 345–353. [Google Scholar] [CrossRef]
- Rose, A.J.; Herzig, S. Metabolic control through glucocorticoid hormones: An update. Mol. Cell. Endocrinol. 2013, 380, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef]
- Kleiman, A.; Tuckermann, J.P. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: Lessons from conditional knockout mice. Mol. Cell. Endocrinol. 2007, 275, 98–108. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 2010, 10, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New insights into the anti-inflammatory mechanisms of glucocorticoids: An emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Souffriau, J.; Eggermont, M.; Van Ryckeghem, S.; Van Looveren, K.; Van Wyngene, L.; Van Hamme, E.; Vuylsteke, M.; Beyaert, R.; De Bosscher, K.; Libert, C. A screening assay for Selective Dimerizing Glucocorticoid Receptor Agonists and Modulators (SEDIGRAM) that are effective against acute inflammation. Sci. Rep. 2018, 8, 12894. [Google Scholar] [CrossRef] [PubMed]
- Haridas, V.; Xu, Z.X.; Kitchen, D.; Jiang, A.; Michels, P.; Gutterman, J.U. The anticancer plant triterpenoid, avicin D, regulates glucocorticoid receptor signaling: Implications for cellular metabolism. PLoS ONE 2011, 6, e28037. [Google Scholar] [CrossRef]
- Georgatza, D.; Gorgogietas, V.A.; Kylindri, P.; Charalambous, M.C.; Papadopoulou, K.K.; Hayes, J.M.; Psarra, A.G. The triterpene echinocystic acid and its 3-O-glucoside derivative are revealed as potent and selective glucocorticoid receptor agonists. Int. J. Biochem. Cell Biol. 2016, 79, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.W.; Leung, F.P.; Mak, N.K.; Tombran-Tink, J.; Huang, Y.; Wong, R.N. Protopanaxadiol and protopanaxatriol bind to glucocorticoid and oestrogen receptors in endothelial cells. Br. J. Pharmacol. 2009, 156, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Lau, A.J.; Wang, R.; Chang, T.K.H. Comparative analysis of ginsenosides in human glucocorticoid receptor binding, transactivation, and transrepression. Eur. J. Pharmacol. 2017, 815, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Gao, J.; Wei, F.; Zhao, J.; Wang, D.; Wei, J. Therapeutic Potential of Ginsenosides as an Adjuvant Treatment for Diabetes. Front. Pharmacol. 2018, 9, 423. [Google Scholar] [CrossRef]
- Leung, K.W.; Wong, A.S. Pharmacology of ginsenosides: A literature review. Chin. Med. 2010, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, P.; Subramaniyam, S.; Mathiyalagan, R.; Yang, D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J. Ginseng Res. 2018, 42, 123–132. [Google Scholar] [CrossRef]
- Azam, M.; Powers, J.T.; Einhorn, W.; Huang, W.S.; Shakespeare, W.C.; Zhu, X.; Dalgarno, D.; Clackson, T.; Sawyer, T.K.; Daley, G.Q. AP24163 inhibits the gatekeeper mutant of BCR-ABL and suppresses in vitro resistance. Chem. Biol. Drug Des. 2010, 75, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Chatzileontiadou, D.S.M.; Tsika, A.C.; Diamantopoulou, Z.; Delbe, J.; Badet, J.; Courty, J.; Skamnaki, V.T.; Parmenopoulou, V.; Komiotis, D.; Hayes, J.M.; et al. Evidence for Novel Action at the Cell-Binding Site of Human Angiogenin Revealed by Heteronuclear NMR Spectroscopy, in silico and in vivo Studies. ChemMedChem 2018, 13, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Drakou, C.E.; Malekkou, A.; Hayes, J.M.; Lederer, C.W.; Leonidas, D.D.; Oikonomakos, N.G.; Lamond, A.I.; Santama, N.; Zographos, S.E. hCINAP is an atypical mammalian nuclear adenylate kinase with an ATPase motif: Structural and functional studies. Proteins 2012, 80, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Vashishtha, S.C.; Erve, J.C. Characterization of glutathione conjugates of duloxetine by mass spectrometry and evaluation of in silico approaches to rationalize the site of conjugation for thiophene containing drugs. Chem. Res. Toxicol. 2010, 23, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Jang, S.; Wang, S.M.; Chen, C.H.; Li, F.Y. Investigation on critical structural motifs of ligands for triggering glucocorticoid receptor nuclear migration through molecular docking simulations. J. Biomol. Struct. Dyn. 2016, 34, 1214–1231. [Google Scholar] [CrossRef]
- Psarra, A.M.; Hermann, S.; Panayotou, G.; Spyrou, G. Interaction of mitochondrial thioredoxin with glucocorticoid receptor and NF-kappaB modulates glucocorticoid receptor and NF-kappaB signalling in HEK-293 cells. Biochem. J. 2009, 422, 521–531. [Google Scholar] [CrossRef]
- Scott, D.K.; Stromstedt, P.E.; Wang, J.C.; Granner, D.K. Further characterization of the glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. The role of the glucocorticoid receptor-binding sites. Mol. Endocrinol. 1998, 12, 482–491. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protocols 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, R.H.; Wang, M.; Xu, G.B.; Liao, S.G. Prodrugs of triterpenoids and their derivatives. Eur. J. Med. Chem. 2017, 131, 222–236. [Google Scholar] [CrossRef]
- Park, J.; Bui, P.T.C.; Song, H.; Kim, S.K.; Rhee, D.K.; Kim, E.Y.; Rhyu, M.R.; Lee, M.S.; Lee, Y.J. Ginseng on Nuclear Hormone Receptors. Am. J. Chin. Med. 2017, 45, 1147–1156. [Google Scholar] [CrossRef]
- Leung, K.W.; Cheng, Y.K.; Mak, N.K.; Chan, K.K.; Fan, T.P.; Wong, R.N. Signaling pathway of ginsenoside-Rg1 leading to nitric oxide production in endothelial cells. FEBS Lett. 2006, 580, 3211–3216. [Google Scholar] [CrossRef]
- Leung, K.W.; Leung, F.P.; Huang, Y.; Mak, N.K.; Wong, R.N. Non-genomic effects of ginsenoside-Re in endothelial cells via glucocorticoid receptor. FEBS Lett. 2007, 581, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Chung, E.; Lee, K.Y.; Lee, Y.H.; Huh, B.; Lee, S.K. Ginsenoside-Rg1, one of the major active molecules from Panax ginseng, is a functional ligand of glucocorticoid receptor. Mol. Cell. Endocrinol. 1997, 133, 135–140. [Google Scholar] [CrossRef]
- Wu, J.; Pan, Z.; Wang, Z.; Zhu, W.; Shen, Y.; Cui, R.; Lin, J.; Yu, H.; Wang, Q.; Qian, J.; et al. Ginsenoside Rg1 protection against beta-amyloid peptide-induced neuronal apoptosis via estrogen receptor alpha and glucocorticoid receptor-dependent anti-protein nitration pathway. Neuropharmacology 2012, 63, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Ko, S.R.; Cho, B.G.; Shin, D.M.; Yuk, J.M.; Li, S.; Kim, J.M.; Evans, R.M.; Jung, J.S.; Song, D.K.; et al. The ginsenoside metabolite compound K, a novel agonist of glucocorticoid receptor, induces tolerance to endotoxin-induced lethal shock. J. Cell. Mol. Med. 2008, 12, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Lee, H.Y.; Chung, H.Y.; Kim, S.I.; Lee, S.K.; Park, B.C.; Kim, K.W. In vitro induction of differentiation by ginsenoides in F9 teratocarcinoma cells. Eur. J. Cancer 1996, 32A, 1420–1428. [Google Scholar] [CrossRef]
- Lee, Y.N.; Lee, H.Y.; Lee, Y.M.; Chung, H.Y.; Kim, S.I.; Lee, S.K.; Park, B.C.; Kim, K.W. Involvement of glucocorticoid receptor in the induction of differentiation by ginsenosides in F9 teratocarcinoma cells. J. Steroid Biochem. Mol. Biol. 1998, 67, 105–111. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jin, Y.R.; Lim, W.C.; Ji, S.M.; Cho, J.Y.; Ban, J.J.; Lee, S.K. Ginsenoside Rc and Re stimulate c-fos expression in MCF-7 human breast carcinoma cells. Arch. Pharm. Res. 2003, 26, 53–57. [Google Scholar] [CrossRef]
- Lee, Y.; Jin, Y.; Lim, W.; Ji, S.; Choi, S.; Jang, S.; Lee, S. A ginsenoside-Rh1, a component of ginseng saponin, activates estrogen receptor in human breast carcinoma MCF-7 cells. J. Steroid Biochem. Mol. Biol. 2003, 84, 463–468. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jin, Y.R.; Lim, W.C.; Park, W.K.; Cho, J.Y.; Jang, S.; Lee, S.K. Ginsenoside-Rb1 acts as a weak phytoestrogen in MCF-7 human breast cancer cells. Arch. Pharm. Res. 2003, 26, 58–63. [Google Scholar] [CrossRef]
- Niu, C.S.; Yeh, C.H.; Yeh, M.F.; Cheng, J.T. Increase of adipogenesis by ginsenoside (Rh2) in 3T3-L1 cell via an activation of glucocorticoid receptor. Horm. Metab. Res. 2009, 41, 271–276. [Google Scholar] [CrossRef]
- Wang, Q.; Blackford, J.A., Jr.; Song, L.N.; Huang, Y.; Cho, S.; Simons, S.S., Jr. Equilibrium interactions of corepressors and coactivators with agonist and antagonist complexes of glucocorticoid receptors. Mol. Endocrinol. 2004, 18, 1376–1395. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Haas, M.; Wang, D.Q.; May, A.; Lo, C.C.; Obici, S.; Tso, P.; Woods, S.C.; Liu, M. Ginsenoside Rb1 increases insulin sensitivity by activating AMP-activated protein kinase in male rats. Physiol. Rep. 2015, 3, e12543. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Liu, J.; Zhou, J.; Lu, W.; Zhou, H.Y.; Long, L.H.; Hu, Z.L.; Ni, L.; Wang, Y.; Chen, J.G.; et al. AMPK Mediates Glucocorticoids Stress-Induced Downregulation of the Glucocorticoid Receptor in Cultured Rat Prefrontal Cortical Astrocytes. PLoS ONE 2016, 11, e0159513. [Google Scholar] [CrossRef] [PubMed]
- Clarisse, D.; Van Wesemael, K.; Tavernier, J.; Offner, F.; Beck, I.M.; De Bosscher, K. Effect of combining glucocorticoids with Compound A on glucocorticoid receptor responsiveness in lymphoid malignancies. PLoS ONE 2018, 13, e0197000. [Google Scholar] [CrossRef]
- Cidlowski, N.B.; Cidlowski, J.A. Regulation of Glucocorticoid Receptors by Glucocorticoids in Cultured HeLa S3 Cells. Endocrinology 1981, 109, 1975–1982. [Google Scholar] [CrossRef]
- Shimojo, M.; Hiroi, N.; Yakushiji, F.; Ueshiba, H.; Yamaguchi, N.; Miyachi, Y. Differences in Down-Regulation of Glucocorticoid Receptor mRNA by Cortisol Prednisolone and Dexamethasone in HeLa Cells. Endocr. J. 1995, 42, 629–636. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Ligand-induced repression of the glucocorticoid receptor gene is mediated by an NCoR1 repression complex formed by long-range chromatin interactions with intragenic glucocorticoid response elements. Mol. Cell. Biol. 2013, 33, 1711–1722. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Jeong, H.G. Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol. Appl. Pharmacol. 2010, 242, 18–28. [Google Scholar] [CrossRef]
- Shang, W.; Yang, Y.; Zhou, L.; Jiang, B.; Jin, H.; Chen, M. Ginsenoside Rb1 stimulates glucose uptake through insulin-like signaling pathway in 3T3-L1 adipocytes. J. Endocrinol. 2008, 198, 561–569. [Google Scholar] [CrossRef]
- Chen, W.; Wang, J.; Luo, Y.; Wang, T.; Li, X.; Li, A.; Li, J.; Liu, K.; Liu, B. Ginsenoside Rb1 and compound K improve insulin signaling and inhibit ER stress-associated NLRP3 inflammasome activation in adipose tissue. J. Ginseng Res. 2016, 40, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Lee, O.H.; Kim, K.J.; Lee, B.Y. Ginsenoside Rg1 promotes glucose uptake through activated AMPK pathway in insulin-resistant muscle cells. Phytother. Res. 2012, 26, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Yuan, H.D.; Chung, S.H. Ginsenoside Rg1 suppresses hepatic glucose production via AMP-activated protein kinase in HepG2 cells. Biol. Pharm. Bull. 2010, 33, 325–328. [Google Scholar] [CrossRef]
- Prima, V.; Depoix, C.; Masselot, B.; Formstecher, P.; Lefebvre, P. Alteration of the glucocorticoid receptor subcellular localization by non steroidal compounds. J. Steroid Biochem. Mol. Biol. 2000, 72, 1–12. [Google Scholar] [CrossRef]
- Scheinman, R.I.; Gualberto, A.; Jewell, C.M.; Cidlowski, J.A.; Baldwin, A.S., Jr. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol. Cell. Biol. 1995, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Liden, J.; Delaunay, F.; Rafter, I.; Gustafsson, J.; Okret, S. A new function for the C-terminal zinc finger of the glucocorticoid receptor. Repression of RelA transactivation. J. Biol. Chem. 1997, 272, 21467–21472. [Google Scholar] [CrossRef]
- Beck, I.M.; Van Crombruggen, K.; Holtappels, G.; Daubeuf, F.; Frossard, N.; Bachert, C.; De Bosscher, K. Differential cytokine profiles upon comparing selective versus classic glucocorticoid receptor modulation in human peripheral blood mononuclear cells and inferior turbinate tissue. PLoS ONE 2015, 10, e0123068. [Google Scholar] [CrossRef]
- Wu, Y.; Yu, Y.; Szabo, A.; Han, M.; Huang, X.F. Central inflammation and leptin resistance are attenuated by ginsenoside Rb1 treatment in obese mice fed a high-fat diet. PLoS ONE 2014, 9, e92618. [Google Scholar] [CrossRef]
- Wang, N.; Wan, J.B.; Chan, S.W.; Deng, Y.H.; Yu, N.; Zhang, Q.W.; Wang, Y.T.; Lee, S.M. Comparative study on saponin fractions from Panax notoginseng inhibiting inflammation-induced endothelial adhesion molecule expression and monocyte adhesion. Chin. Med. 2011, 6, 37. [Google Scholar] [CrossRef]
- Jang, S.; Lim, Y.; Valacchi, G.; Sorn, S.; Park, H.; Park, N.Y.; Lee, M. Preventive effects of protopanaxadiol and protopanaxatriol ginsenosides on liver inflammation and apoptosis in hyperlipidemic apoE KO mice. Genes Nutr. 2012, 7, 319–329. [Google Scholar] [CrossRef]
- De Bosscher, K.; Beck, I.M.; Dejager, L.; Bougarne, N.; Gaigneaux, A.; Chateauvieux, S.; Ratman, D.; Bracke, M.; Tavernier, J.; Vanden Berghe, W.; et al. Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-kappaB and AP-1. Cell. Mol. Life Sci. 2014, 71, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.G.; Zhu, L.J.; Chen, C.; Wu, H.Y.; Luo, C.X.; Chang, L.; Zhu, D.Y. Hippocampal neuronal nitric oxide synthase mediates the stress-related depressive behaviors of glucocorticoids by downregulating glucocorticoid receptor. J. Neurosci. 2011, 31, 7579–7590. [Google Scholar] [CrossRef] [PubMed]
- Gruver-Yates, A.L.; Cidlowski, J.A. Tissue-specific actions of glucocorticoids on apoptosis: A double-edged sword. Cells 2013, 2, 202–223. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Psarra, A.M.; Solakidi, S.; Trougakos, I.P.; Margaritis, L.H.; Spyrou, G.; Sekeris, C.E. Glucocorticoid receptor isoforms in human hepatocarcinoma HepG2 and SaOS-2 osteosarcoma cells: Presence of glucocorticoid receptor alpha in mitochondria and of glucocorticoid receptor beta in nucleoli. Int. J. Biochem. Cell Biol. 2005, 37, 2544–2558. [Google Scholar] [CrossRef]
- Schrodinger, LLC. Modeling Software Suite; Schrodinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Suino-Powell, K.; Xu, Y.; Zhang, C.; Tao, Y.G.; Tolbert, W.D.; Simons, S.S., Jr.; Xu, H.E. Doubling the size of the glucocorticoid receptor ligand binding pocket by deacylcortivazol. Mol. Cell. Biol. 2008, 28, 1915–1923. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | GlideScore | IFDScore |
|---|---|---|
| DEX a | −15.59 | −551.7 |
| PPD b | −14.93 | −557.7 |
| PPT c | −15.23 | −560.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karra, A.G.; Konstantinou, M.; Tzortziou, M.; Tsialtas, I.; Kalousi, F.D.; Garagounis, C.; Hayes, J.M.; Psarra, A.-M.G. Potential Dissociative Glucocorticoid Receptor Activity for Protopanaxadiol and Protopanaxatriol. Int. J. Mol. Sci. 2019, 20, 94. https://doi.org/10.3390/ijms20010094
Karra AG, Konstantinou M, Tzortziou M, Tsialtas I, Kalousi FD, Garagounis C, Hayes JM, Psarra A-MG. Potential Dissociative Glucocorticoid Receptor Activity for Protopanaxadiol and Protopanaxatriol. International Journal of Molecular Sciences. 2019; 20(1):94. https://doi.org/10.3390/ijms20010094
Chicago/Turabian StyleKarra, Aikaterini G., Maria Konstantinou, Maria Tzortziou, Ioannis Tsialtas, Foteini D. Kalousi, Constantine Garagounis, Joseph M. Hayes, and Anna-Maria G. Psarra. 2019. "Potential Dissociative Glucocorticoid Receptor Activity for Protopanaxadiol and Protopanaxatriol" International Journal of Molecular Sciences 20, no. 1: 94. https://doi.org/10.3390/ijms20010094
APA StyleKarra, A. G., Konstantinou, M., Tzortziou, M., Tsialtas, I., Kalousi, F. D., Garagounis, C., Hayes, J. M., & Psarra, A.-M. G. (2019). Potential Dissociative Glucocorticoid Receptor Activity for Protopanaxadiol and Protopanaxatriol. International Journal of Molecular Sciences, 20(1), 94. https://doi.org/10.3390/ijms20010094