How Glucocorticoids Affect the Neutrophil Life



Abstract
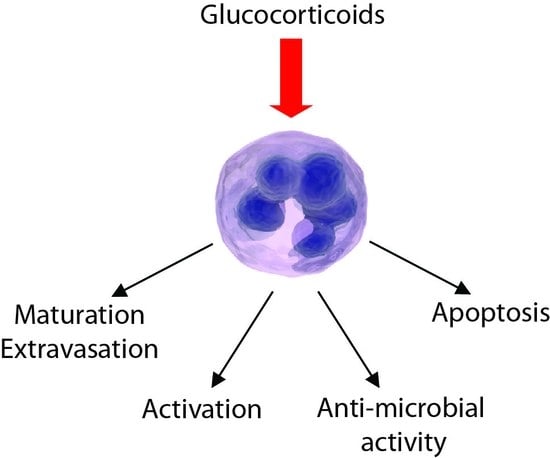
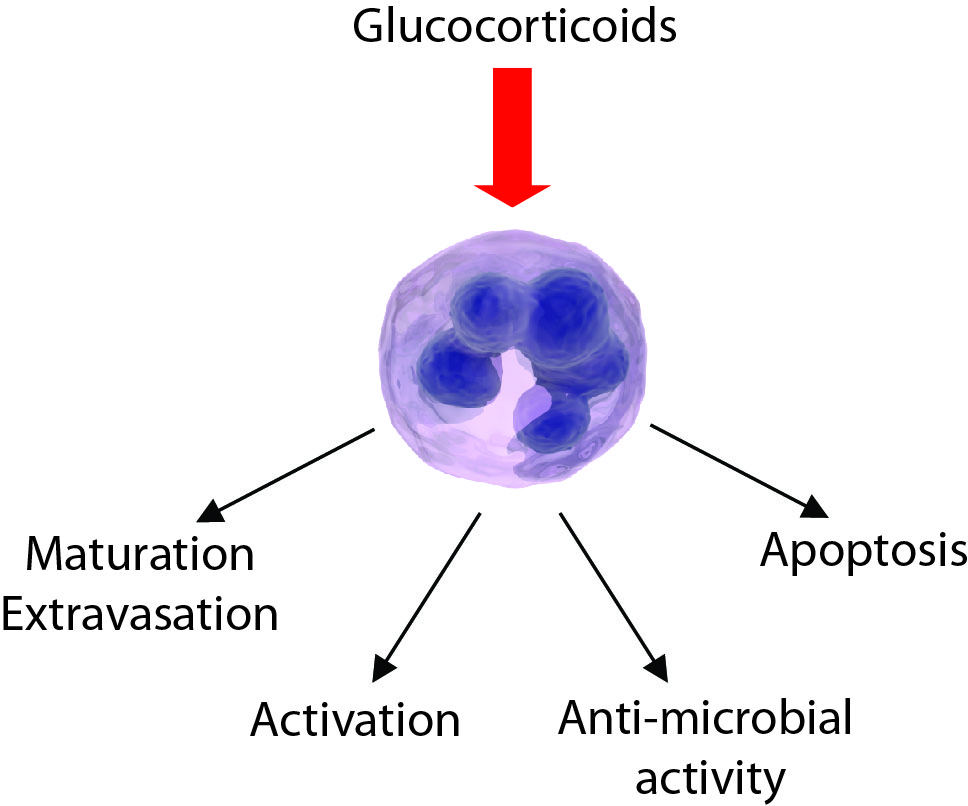{kind=link}
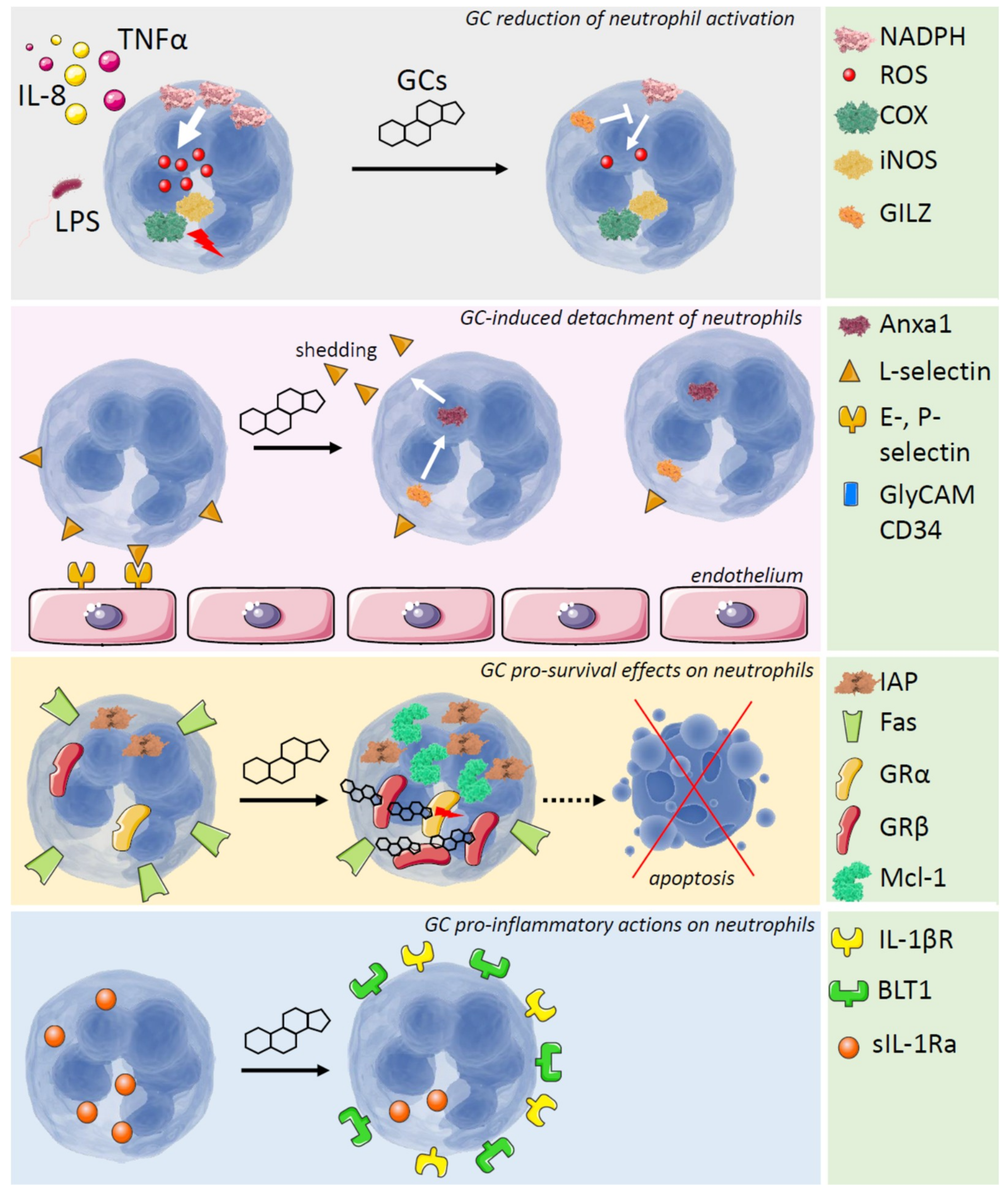{kind=link}
1. Introduction
2. Role of GCs in the Control of Neutrophil Maturation and Extravasation
3. Anti-Inflammatory Effects of GCs on Neutrophils
4. Pro-Inflammatory Effects of GCs on Neutrophils
5. GCs and Neutrophil Apoptosis
6. Neutrophils, Disease and GC Treatment
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Pick, R.; Brechtefeld, D.; Walzog, B. Intraluminal crawling versus interstitial neutrophil migration during inflammation. Mol. Immunol. 2013, 55, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Arase, H. Regulation of immune responses by neutrophils. Ann. N. Y. Acad. Sci. 2014, 1319, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Migliorati, G.; Bruscoli, S.; Riccardi, C. Defining the role of glucocorticoids in inflammation. Clin. Sci. 2018, 132, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Bereshchenko, O.; Bruscoli, S.; Riccardi, C. Glucocorticoids, Sex Hormones, and Immunity. Front. Immunol. 2018, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- Solano, M.E.; Holmes, M.C.; Mittelstadt, P.R.; Chapman, K.E.; Tolosa, E. Antenatal endogenous and exogenous glucocorticoids and their impact on immune ontogeny and long-term immunity. Semin. Immunopathol. 2016, 38, 739–763. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, D.M.; Lotufo, C.M.; Borelli, P.; Ferreira, Z.S.; Markus, R.P.; Farsky, S.H. Endogenous glucocorticoids control neutrophil mobilization from bone marrow to blood and tissues in non-inflammatory conditions. Br. J. Pharmacol. 2007, 152, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, D.M.; Lotufo, C.M.; Borelli, P.; Tavassi, A.M.; Pereira, A.L.; Markus, R.P.; Farsky, S.H. Adrenal deficiency alters mechanisms of neutrophil mobilization. Mol. Cell. Endocrinol. 2006, 249, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Lund-Johansen, F.; Terstappen, L.W. Differential surface expression of cell adhesion molecules during granulocyte maturation. J. Leukoc. Biol. 1993, 54, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268. [Google Scholar] [CrossRef]
- Ivetic, A. A head-to-tail view of L-selectin and its impact on neutrophil behaviour. Cell Tissue Res. 2018, 371, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Schultz, J.B.; Knauf, P.A.; King, M.R. Mechanical shedding of L-selectin from the neutrophil surface during rolling on sialyl Lewis x under flow. J. Biol. Chem. 2007, 282, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- Strausbaugh, H.J.; Rosen, S.D. A potential role for annexin 1 as a physiologic mediator of glucocorticoid-induced L-selectin shedding from myeloid cells. J. Immunol. 2001, 166, 6294–6300. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Ronchetti, S.; Pericolini, E.; Gabrielli, E.; Cari, L.; Gentili, M.; Roselletti, E.; Migliorati, G.; Vecchiarelli, A.; Riccardi, C. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. Fed. Am. Soc. Exp. Biol. J. 2017, 31, 3054–3065. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Migliorati, G.; Riccardi, C. GILZ as a Mediator of the Anti-Inflammatory Effects of Glucocorticoids. Front. Endocrinol. 2015, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- Petri, B.; Sanz, M.J. Neutrophil chemotaxis. Cell Tissue Res. 2018, 371, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Pinho, V.; Russo, R.C.; Amaral, F.A.; de Sousa, L.P.; Barsante, M.M.; de Souza, D.G.; Alves-Filho, J.C.; Cara, D.C.; Hayflick, J.S.; Rommel, C.; et al. Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. J. Immunol. 2007, 179, 7891–7898. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kreisel, D.; Goldstein, D.R. Processes of sterile inflammation. J. Immunol. 2013, 191, 2857–2863. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. Fed. Am. Soc. Exp. Biol. J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Nonresolving macrophage-mediated inflammation in malignancy. Fed. Am. Soc. Exp. Biol. J. 2018, 285, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.; Bickel, C.A.; Bochner, B.S.; Schleimer, R.P. The effects of the potent glucocorticoid budesonide on adhesion of eosinophils to human vascular endothelial cells and on endothelial expression of adhesion molecules. J. Pharmacol. Exp. Ther. 1993, 267, 245–249. [Google Scholar] [PubMed]
- Burton, J.L.; Kehrli, M.E., Jr.; Kapil, S.; Horst, R.L. Regulation of L-selectin and CD18 on bovine neutrophils by glucocorticoids: Effects of cortisol and dexamethasone. J. Leukoc. Biol. 1995, 57, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Filep, J.G.; Delalandre, A.; Payette, Y.; Foldes-Filep, E. Glucocorticoid receptor regulates expression of L-selectin and CD11/CD18 on human neutrophils. Circulation 1997, 96, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Ignacchiti, M.D.; Sesti-Costa, R.; Marchi, L.F.; Chedraoui-Silva, S.; Mantovani, B. Effect of academic psychological stress in post-graduate students: The modulatory role of cortisol on superoxide release by neutrophils. Stress 2011, 14, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Ghanim, H.; Mohanty, P.; Hamouda, W.; Al-Haddad, W. Acute suppressive effect of hydrocortisone on p47 subunit of nicotinamide adenine dinucleotide phosphate oxidase. Metabolism 2001, 50, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mohanty, P.; Hamouda, W.; Aljada, A.; Kumbkarni, Y.; Garg, R. Effect of dexamethasone on reactive oxygen species generation by leukocytes and plasma interleukin-10 concentrations: A pharmacodynamic study. Clinical Pharmacol. Ther. 1999, 66, 58–65. [Google Scholar] [CrossRef]
- Satoh, S.; Oishi, K.; Iwagaki, A.; Senba, M.; Akaike, T.; Akiyama, M.; Mukaida, N.; Atsushima, K.M.; Nagatake, T. Dexamethasone impairs pulmonary defence against Pseudomonas aeruginosa through suppressing iNOS gene expression and peroxynitrite production in mice. Clin. Exp. Immunol. 2001, 126, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Maloney, C.G.; Kutchera, W.A.; Albertine, K.H.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Inflammatory agonists induce cyclooxygenase type 2 expression by human neutrophils. J. Immunol. 1998, 160, 1402–1410. [Google Scholar] [PubMed]
- Llewellyn-Jones, C.G.; Hill, S.L.; Stockley, R.A. Effect of fluticasone propionate on neutrophil chemotaxis, superoxide generation, and extracellular proteolytic activity in vitro. Thorax 1994, 49, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Petroni, K.C.; Shen, L.; Guyre, P.M. Modulation of human polymorphonuclear leukocyte IgG Fc receptors and Fc receptor-mediated functions by IFN-gamma and glucocorticoids. J. Immunol. 1988, 140, 3467–3472. [Google Scholar] [PubMed]
- Franco, E.L.; Trottier, H. A new window into the natural history of human papillomavirus infection: A view from the ALTS (Atypical Squamous Cells of Undetermined Significance/Low-Grade Squamous Intraepithelial Lesions Triage Study) trial. J. Infect. Dis. 2007, 195, 1560–1562. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, G.; Lavoie-Lamoureux, A.; Beauchamp, G.; Lavoie, J.P. Neutrophils are not less sensitive than other blood leukocytes to the genomic effects of glucocorticoids. PLoS ONE 2012, 7, e44606. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; de Oliveira, M.C.; Verlengia, R.; Curi, R.; Pithon-Curi, T.C. Effect of dexamethasone on neutrophil metabolism. Cell Biochem. Funct. 2003, 21, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Ronchetti, S.; Gabrielli, E.; Pericolini, E.; Gentili, M.; Roselletti, E.; Vecchiarelli, A.; Riccardi, C. GILZ restrains neutrophil activation by inhibiting the MAPK pathway. J. Leukoc. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schleimer, R.P. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc. Am. Thorac. Soc. 2004, 1, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.H.; Peterson, R.H.; Moore, M.A. Cytokines and dexamethasone modulation of IL-1 receptors on human neutrophils in vitro. J. Immunol. 1993, 150 Pt 1, 3515–3524. [Google Scholar]
- Liles, W.C.; Dale, D.C.; Klebanoff, S.J. Glucocorticoids inhibit apoptosis of human neutrophils. Blood 1995, 86, 3181–3188. [Google Scholar] [PubMed]
- Obinata, H.; Yokomizo, T.; Shimizu, T.; Izumi, T. Glucocorticoids up-regulate leukotriene B4 receptor-1 expression during neutrophilic differentiation of HL-60 cells. Biochem. Biophys. Res. Commun. 2003, 309, 114–119. [Google Scholar] [CrossRef]
- Langereis, J.D.; Oudijk, E.J.; Schweizer, R.C.; Lammers, J.W.; Koenderman, L.; Ulfman, L.H. Steroids induce a disequilibrium of secreted interleukin-1 receptor antagonist and interleukin-1beta synthesis by human neutrophils. Eur. Respir. J. 2011, 37, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Cox, G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 1995, 154, 4719–4725. [Google Scholar] [PubMed]
- Nittoh, T.; Fujimori, H.; Kozumi, Y.; Ishihara, K.; Mue, S.; Ohuchi, K. Effects of glucocorticoids on apoptosis of infiltrated eosinophils and neutrophils in rats. Eur. J. Pharmacol. 1998, 354, 73–81. [Google Scholar] [CrossRef]
- Saffar, A.S.; Dragon, S.; Ezzati, P.; Shan, L.; Gounni, A.S. Phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase regulate induction of Mcl-1 and survival in glucocorticoid-treated human neutrophils. J. Allergy Clin. Immunol. 2008, 121, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Espinasse, M.A.; Pepin, A.; Virault-Rocroy, P.; Szely, N.; Chollet-Martin, S.; Pallardy, M.; Biola-Vidamment, A. Glucocorticoid-Induced Leucine Zipper Is Expressed in Human Neutrophils and Promotes Apoptosis through Mcl-1 Down-Regulation. J. Innate Immun. 2016, 8, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Marwick, J.A.; Dorward, D.A.; Lucas, C.D.; Jones, K.O.; Sheldrake, T.A.; Fox, S.; Ward, C.; Murray, J.; Brittan, M.; Hirani, N.; et al. Oxygen levels determine the ability of glucocorticoids to influence neutrophil survival in inflammatory environments. J. Leukoc. Biol. 2013, 94, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Madsen-Bouterse, S.A.; Rosa, G.J.; Burton, J.L. Glucocorticoid modulation of Bcl-2 family members A1 and Bak during delayed spontaneous apoptosis of bovine blood neutrophils. Endocrinology 2006, 147, 3826–3834. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Vucic, D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014, 545, 35–65. [Google Scholar] [PubMed]
- Chang, L.C.; Madsen, S.A.; Toelboell, T.; Weber, P.S.; Burton, J.L. Effects of glucocorticoids on Fas gene expression in bovine blood neutrophils. J. Endocrinol. 2004, 183, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.; Kisich, K.; Hauk, P.J.; Vottero, A.; Chrousos, G.P.; Klemm, D.J.; Leung, D.Y. High constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J. Exp. Med. 2001, 193, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Takeda, Y.; Nakada, T.; Sendo, F. Inhibition by dexamethasone of human neutrophil apoptosis in vitro. Nat. Immun. 1995, 14, 198–208. [Google Scholar] [PubMed]
- Zhang, X.; Moilanen, E.; Kankaanranta, H. Beclomethasone, budesonide and fluticasone propionate inhibit human neutrophil apoptosis. Eur. J. Pharmacol. 2001, 431, 365–371. [Google Scholar] [CrossRef]
- Macdowell, A.L.; Peters, S.P. Neutrophils in asthma. Curr. Allergy Asthma Rep. 2007, 7, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jatakanon, A.; Uasuf, C.; Maziak, W.; Lim, S.; Chung, K.F.; Barnes, P.J. Neutrophilic inflammation in severe persistent asthma. Am. J. Respir. Crit. Care Med. 1999, 160 Pt 1, 1532–1539. [Google Scholar] [CrossRef]
- Ray, A.; Kolls, J.K. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol. 2017, 38, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Green, B.J.; Wiriyachaiporn, S.; Grainge, C.; Rogers, G.B.; Kehagia, V.; Lau, L.; Carroll, M.P.; Bruce, K.D.; Howarth, P.H. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS ONE 2014, 9, e100645. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Higham, A.; Cadden, P.; Southworth, T.; Rossall, M.; Kolsum, U.; Lea, S.; Knowles, R.; Singh, D. Leukotriene B4 levels in sputum from asthma patients. ERJ Open Res. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lindo, T.; Jackson, N.; Meng-Choong, L.; Reynolds, P.; Hill, A.; Haswell, M.; Jackson, S.; Kilfeather, S. Reversal of human neutrophil survival by leukotriene B(4) receptor blockade and 5-lipoxygenase and 5-lipoxygenase activating protein inhibitors. Am. J. Respir. Crit. Care Med. 1999, 160, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, P.; Wu, X.; Chen, Y.; Feng, Y.; Yang, Q.; Xu, Y.; Zhao, J.; Xie, J. Impaired anti-inflammatory action of glucocorticoid in neutrophil from patients with steroid-resistant asthma. Respir. Res. 2016, 17, 153. [Google Scholar] [CrossRef] [PubMed]
- Velthove, K.J.; Leufkens, H.G.; Souverein, P.C.; Schweizer, R.C.; Bracke, M.; van Solinge, W.W. Effects of glucocorticoids on the neutrophil count: A cohort study among hospitalized patients. Pulm. Pharmacol. Ther. 2010, 23, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Oudijk, E.J.; Nijhuis, E.H.; Zwank, M.D.; van de Graaf, E.A.; Mager, H.J.; Coffer, P.J.; Lammers, J.W.; Koenderman, L. Systemic inflammation in COPD visualised by gene profiling in peripheral blood neutrophils. Thorax 2005, 60, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agusti, A.G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.A.; Clarke, M.S.; Sim, E.H.; Fong, K.M. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2012, 7, CD002991. [Google Scholar] [CrossRef] [PubMed]
- Roche, N.; Marthan, R.; Berger, P.; Chambellan, A.; Chanez, P.; Aguilaniu, B.; Brillet, P.Y.; Burgel, P.R.; Chaouat, A.; Devillier, P.; et al. Beyond corticosteroids: Future prospects in the management of inflammation in COPD. Eur. Respir. Rev. 2011, 20, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Mechanisms and resistance in glucocorticoid control of inflammation. J. Steroid Biochem. Mol. Biol. 2010, 120, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.; Gaffey, K.; Kane, B.; Malia-Milanes, B.; Shah, R.; Bentley, A.; Ray, D.; Singh, D. Reduced glucocorticoid receptor expression and function in airway neutrophils. Int. Immunopharmacol. 2012, 12, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Lluch, J.; Almudever, P.; Freire, J.; Xiaozhong, Q.; Cortijo, J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2014, 134, 314–322. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Yan, W.; Zhao, Y.; Yu, K. Enhanced neutrophil autophagy and increased concentrations of IL-6, IL-8, IL-10 and MCP-1 in rheumatoid arthritis. Int. Immunopharmacol. 2018, 65, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Youssef, P.P.; Cormack, J.; Evill, C.A.; Peter, D.T.; Roberts-Thomson, P.J.; Ahern, M.J.; Smith, M.D. Neutrophil trafficking into inflamed joints in patients with rheumatoid arthritis, and the effects of methylprednisolone. Arthritis Rheum. 1996, 39, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, K.R.; Korotkova, M.; Catrina, A.I.; Backman, L.; af Klint, E.; Claesson, H.E.; Radmark, O.; Jakobsson, P.J. Expression of 5-lipoxygenase and 15-lipoxygenase in rheumatoid arthritis synovium and effects of intraarticular glucocorticoids. Arthritis Res. Ther. 2009, 11, R83. [Google Scholar] [CrossRef] [PubMed]
- Wittkowski, H.; Foell, D.; af Klint, E.; De Rycke, L.; De Keyser, F.; Frosch, M.; Ulfgren, A.K.; Roth, J. Effects of intra-articular corticosteroids and anti-TNF therapy on neutrophil activation in rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Torsteinsdottir, I.; Arvidson, N.G.; Hallgren, R.; Hakansson, L. Enhanced expression of integrins and CD66b on peripheral blood neutrophils and eosinophils in patients with rheumatoid arthritis, and the effect of glucocorticoids. Scand. J. Immunol. 1999, 50, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Lampinen, M.; Sangfelt, P.; Taha, Y.; Carlson, M. Accumulation, activation, and survival of neutrophils in ulcerative colitis: Regulation by locally produced factors in the colon and impact of steroid treatment. Int. J. Colorectal Dis. 2008, 23, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Camacho, K.; Ottum, P.A.; Franco-Munoz, D.; De la Fuente, M.; Torres-Riquelme, A.; Diaz-Jimenez, D.; Olivares-Morales, M.; Astudillo, G.; Quera, R.; Hermoso, M.A. Glucocorticosteroid therapy in inflammatory bowel diseases: From clinical practice to molecular biology. World J. Gastroenterol. 2017, 23, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, M.; Funakoshi, S.; Sakuraba, A.; Takagi, H.; Inoue, N.; Ogata, H.; Iwao, Y.; Ishi, H.; Hibi, T. Granulocytapheresis is useful as an alternative therapy in patients with steroid-refractory or -dependent ulcerative colitis. Inflamm. Bowel Dis. 2004, 10, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Saniabadi, A.R.; Tanaka, T.; Ohmori, T.; Sawada, K.; Yamamoto, T.; Hanai, H. Treating inflammatory bowel disease by adsorptive leucocytapheresis: A desire to treat without drugs. World J. Gastroenterol. 2014, 20, 9699–9715. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, S.; Takeda, H.; Kawata, S.; Yamakawa, M. The relationship between the expression of the glucocorticoid receptor in biopsied colonic mucosa and the glucocorticoid responsiveness of ulcerative colitis patients. Clin. Immunol. 2009, 133, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Sprung, C.L.; Annane, D.; Keh, D.; Moreno, R.; Singer, M.; Freivogel, K.; Weiss, Y.G.; Benbenishty, J.; Kalenka, A.; Forst, H.; et al. Hydrocortisone therapy for patients with septic shock. N. Engl. J. Med. 2008, 358, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, E.L.; Koper, J.W.; Joosten, K.; de Jong, F.H.; Hazelzet, J.A.; Lamberts, S.W.; Hokken-Koelega, A.C. Glucocorticoid receptor mRNA levels are selectively decreased in neutrophils of children with sepsis. Intensive Care Med. 2009, 35, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, M.; Lindholm, C.; Strinnholm, M.; Hedenstierna, G.; Rylander, C. Impairment of neutrophilic glucocorticoid receptor function in patients treated with steroids for septic shock. Intensive Care Med. Exp. 2015, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Parlato, M.; Souza-Fonseca-Guimaraes, F.; Philippart, F.; Misset, B.; Captain Study, G.; Adib-Conquy, M.; Cavaillon, J.M. CD24-triggered caspase-dependent apoptosis via mitochondrial membrane depolarization and reactive oxygen species production of human neutrophils is impaired in sepsis. J. Immunol. 2014, 192, 2449–2459. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on Neutrophil Function in Severe Inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronchetti, S.; Ricci, E.; Migliorati, G.; Gentili, M.; Riccardi, C. How Glucocorticoids Affect the Neutrophil Life. Int. J. Mol. Sci. 2018, 19, 4090. https://doi.org/10.3390/ijms19124090
Ronchetti S, Ricci E, Migliorati G, Gentili M, Riccardi C. How Glucocorticoids Affect the Neutrophil Life. International Journal of Molecular Sciences. 2018; 19(12):4090. https://doi.org/10.3390/ijms19124090
Chicago/Turabian StyleRonchetti, Simona, Erika Ricci, Graziella Migliorati, Marco Gentili, and Carlo Riccardi. 2018. "How Glucocorticoids Affect the Neutrophil Life" International Journal of Molecular Sciences 19, no. 12: 4090. https://doi.org/10.3390/ijms19124090
APA StyleRonchetti, S., Ricci, E., Migliorati, G., Gentili, M., & Riccardi, C. (2018). How Glucocorticoids Affect the Neutrophil Life. International Journal of Molecular Sciences, 19(12), 4090. https://doi.org/10.3390/ijms19124090