Multiple Aspects of PIP2 Involvement in C. elegans Gametogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PIP2 Is Present in C. elegans Prophase I Nuclei and Embryos
2.2. Depletion of PIP2 Decreases the Brood Size and Increases Male Incidence
2.3. PIP2 Depletion Causes Aneuploidy and Altered Chromosome Structure in Oocytes
2.4. DNA Transcription Increases Upon PIP2 Depletion in Germ Cells Nuclei
2.5. PIP2 Depletion Increases Germ Cells Apoptosis
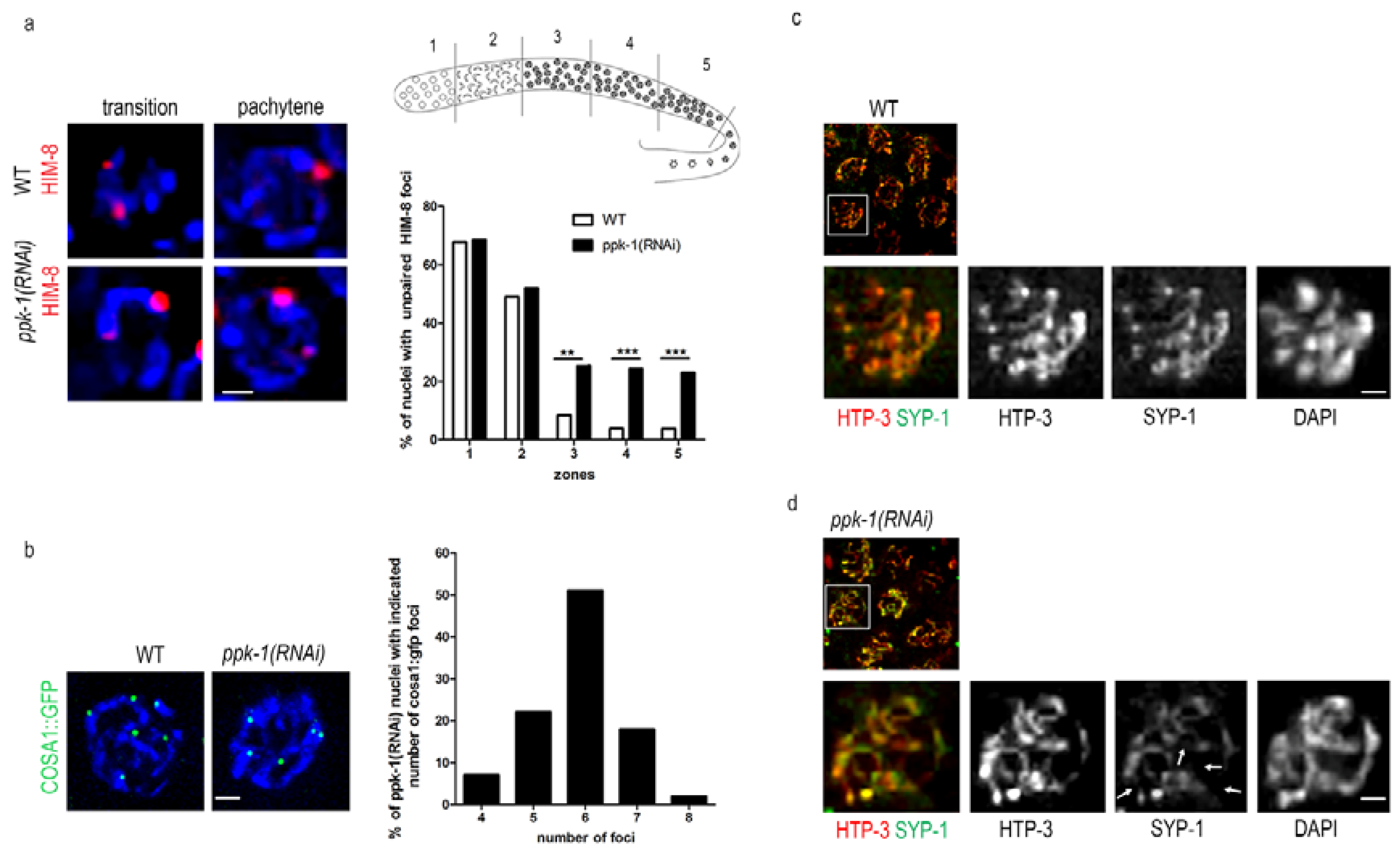
2.6. Impaired Chromosome Pairing and Synapsis Defect in ppk-1(RNAi) Worms
2.7. PIP2 Interacts with Proteins Involved in Uubiquitin–Proteasome Pathway
3. Discussion
4. Materials and Methods
4.1. Strains and Culture Conditions
4.2. RNA Interference
4.3. Antibodies
4.4. Indirect Immunofluorescence, 5-FU Treatment, and Actinomycin D Inhibition
4.5. Mass-Spectrometry
4.6. Immunoprecipitation and Dot Blot
4.7. Characterization of Brood Sizes and Embryonic Lethality
4.8. Quantitation of COSA-1::GFP, HIM-8::mCherry and RAD-51 Foci
4.9. Detection of DAPI Stained Bodies in Oocyte Nuclei
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacQueen, A.V.; Villeneuve, A.M. Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev. 2001, 15, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, B.R. Chromosome segregation mechanisms. Genetics 1974, 78, 205–213. [Google Scholar] [PubMed]
- Ostergren, G. The mechanism of co-orientation in bivalents and multivalents. Hereditas 1951, 37, 85–156. [Google Scholar] [CrossRef]
- Bell, O.; Tiwari, V.K.; Thoma, N.H.; Schubeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011, 12, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, Y.; Wong, K.; Ehlers, P.; Kohara, Y.; Jones, S.J.; Marra, M.A.; Holt, R.A.; Moerman, D.G.; Hansen, D. Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genom. 2009, 10, 213. [Google Scholar] [CrossRef] [PubMed]
- Capitani, S.; Mazzotti, G.; Jovine, R.; Papa, S.; Maraldi, N.M.; Manzoli, F.A. Effect of phosphatidylcholine vesicles on the activity of DNA polymerase-alpha. Mol. Cell. Biochem. 1979, 27, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Capitani, S.; Caramelli, E.; Felaco, M.; Miscia, S.; Manzoli, F.A. Effect of phospholipid vesicles on endogenous RNA polymerase activity of isolated rat liver nuclei. Physiol. Chem. Phys. 1981, 13, 153–158. [Google Scholar] [PubMed]
- Manzoli, F.A.; Capitani, S.; Mazzotti, G.; Barnabei, O.; Maraldi, N.M. Role of chromatin phospholipids on template availability and ultrastructure of isolated nuclei. Adv. Enzym. Regul. 1982, 20, 247–262. [Google Scholar] [CrossRef]
- Maraldi, N.M.; Capitani, S.; Caramelli, E.; Cocco, L.; Barnabei, O.; Manzoli, F.A. Conformational changes of nuclear chromatin related to phospholipid induced modifications of the template availability. Adv. Enzym. Regul. 1984, 22, 447–464. [Google Scholar] [CrossRef]
- Cocco, L.; Gilmour, R.S.; Maraldi, N.M.; Martelli, A.M.; Papa, S.; Manzoli, F.A. Increase of globin RNA synthesis induced by phosphatidylserine liposomes in isolated erythroleukemic cell nuclei. Morphological and functional features. Biol. Cell 1985, 54, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Capitani, S.; Cocco, L.; Maraldi, N.M.; Papa, S.; Manzoli, F.A. Effect of phospholipids on transcription and ribonucleoprotein processing in isolated nuclei. Adv. Enzym. Regul. 1986, 25, 425–438. [Google Scholar] [CrossRef]
- Kuvichkin, V.V. DNA-lipid interactions in vitro and in vivo. Bioelectrochemistry 2002, 58, 3–12. [Google Scholar] [CrossRef]
- Mazzotti, G.; Zini, N.; Rizzi, E.; Rizzoli, R.; Galanzi, A.; Ognibene, A.; Santi, S.; Matteucci, A.; Martelli, A.M.; Maraldi, N.M. Immunocytochemical detection of phosphatidylinositol 4,5-bisphosphate localization sites within the nucleus. J. Histochem. Cytochem. 1995, 43, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Boronenkov, I.V.; Loijens, J.C.; Umeda, M.; Anderson, R.A. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell 1998, 9, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Osborne, S.L.; Thomas, C.L.; Gschmeissner, S.; Schiavo, G. Nuclear PtdIns(4,5)P2 assembles in a mitotically regulated particle involved in pre-mRNA splicing. J. Cell Sci. 2001, 114, 2501–2511. [Google Scholar] [PubMed]
- Yildirim, S.; Castano, E.; Sobol, M.; Philimonenko, V.V.; Dzijak, R.; Venit, T.; Hozák, P. Involvement of phosphatidylinositol 4,5-bisphosphate in RNA polymerase I transcription. J. Cell Sci. 2013, 126, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Ulicna, L.; Kalendova, A.; Kalasova, I.; Vacik, T.; Hozák, P. PIP2 epigenetically represses rRNA genes transcription interacting with PHF8. Biochim. Biophys. Acta 2018, 1863, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Sobol, M.; Krausová, A.; Yildirim, S.; Kalasová, I.; Fáberová, V.; Vrkoslav, V.; Philimonenko, V.; Marášek, P.; Pastorek, L.; Čapek, M.; et al. Nuclear phosphatidylinositol 4,5-bisphosphate islets contribute to efficient RNA polymerase II-dependent transcription. J. Cell Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Fukami, K.; Watanabe, Y.; Ozaki, C.; Takenawa, T. Phosphatidylinositol 4,5-bisphosphate reverses the inhibition of RNA transcription caused by histone H1. Eur. J. Biochem. 1998, 251, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Toska, E.; Campbell, H.A.; Shandilya, J.; Goodfellow, S.J.; Shore, P.; Medler, K.F.; Roberts, S.G.E. Repression of Transcription by WT1-BASP1 Requires the Myristoylation of BASP1 and the PIP2-Dependent Recruitment of Histone Deacetylase. Cell Rep. 2012, 2, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Loijens, J.C.; Anderson, R.A. Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J. Biol. Chem. 1996, 271, 32937–32943. [Google Scholar] [CrossRef] [PubMed]
- Ogg, S.R.; Ruvkun, G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol. Cell 1998, 2, 887–893. [Google Scholar] [CrossRef]
- Blondeau, F.; Laporte, J.; Bodin, S.; Superti-Furga, G.; Payrastre, B.; Mandel, J.L. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum. Mol. Genet. 2000, 22, 2223–2229. [Google Scholar] [CrossRef]
- Klopfenstein, D.R.; Tomishige, M.; Stuurman, N.; Vale, R.D. Role of phosphatidylinositol (4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell 2002, 109, 347–358. [Google Scholar] [CrossRef]
- Nicot, A.S.; Fares, H.; Payrastre, B.; Chisholm, A.D.; Labouesse, M.; Laporte, J. The phosphoinositide kinase PIKfyve/Fab1p regulates terminal lysosome maturation in Caenorhabditis elegans. Mol. Biol. Cell 2006, 17, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Kim, E.; L’hernault, S.W.; Barr, M.M. The CIL-1 PI 5-phosphatase localizes TRP Polycystins to cilia and activates sperm in C. elegans. Curr. Biol. 2009, 19, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Mukhopadhyay, A.; Narasimhan, S.D.; Tesz, G.; Czech, M.P.; Tissenbaum, H.A. A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell 2009, 136, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Klaavuniemi, T.; Ono, S. Distinct roles of four gelsolin-like domains of Caenorhabditis elegans gelsolin-like protein-1 in actin filament severing, barbed end capping, and phosphoinositide binding. Biochemistry 2010, 49, 4349–4360. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Shen, Q.; Mahoney, T.R.; Neukomm, L.J.; Wang, Y.; Zhou, Z. Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol. 2012, 10, e1001245. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wang, K.; Zou, W.; Miao, R.; Huang, Y.; Wang, H.; Wang, X. PtdIns(4,5)P2 and PtdIns3P coordinate to regulate phagosomal sealing for apoptotic cell clearance. J. Cell Biol. 2015, 210, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, D.; Bastiani, M.; Chessa, T.A.M.; Joshi, D.; Hauth, L.; Cooke, F.T.; Divecha, N.; Kim, S. Overexpression of PPK-1, the C. elegans Type 1 PIP kinase, inhibits growth cone collapse in the developing nervous system and causes axonal degeneration in adults. Dev. Biol. 2008, 313, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Guo, H.; Wycuff, D.L.; Lee, M. Role of phosphatidylinositol-4-phosphate 5′ kinase (ppk-1) in ovulation of Caenorhabditis elegans. Exp. Cell Res. 2007, 313, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Sobol, M.; Yildirim, S.; Philimonenko, V.V.; Marasek, P.; Castano, E.; Hozak, P. UBF complexes with phosphatidylinositol 4,5-bisphosphate in nucleolar organizer regions regardless of ongoing RNA polymerase I activity. Nucleus 2013, 4, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Mellman, D.L.; Gonzales, M.L.; Song, C.; Barlow, C.A.; Wang, P.; Kendziorski, C.; Anderson, R.A. A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 2008, 451, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Kumsta, C.; Hansen, M.C. Elegans rrf-1 mutations maintain RNAi efficiency in the soma in addition to the germline. PLoS ONE 2012, 7, e35428. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Machacek, T.; Mamnum, Y.M.; Penkner, A.; Gloggnitzer, J.; Wegrostek, C.; Konrat, R.; Jantsch, M.F.; Loidl, J.; Jantsch, V. Mutations in Caenorhabditis elegans him-19 show meiotic defects that worsen with age. Mol. Biol. Cell 2010, 21, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Cortes, D.B.; McNally, K.L.; Mains, P.E.; McNally, F.J. The asymmetry of female meiosis reduces the frequency of inheritance of unpaired chromosomes. eLife 2015, 4, e06056. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hartwieg, E.; Horvitz, H.R. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 2001, 104, 43–56. [Google Scholar] [CrossRef]
- Colaiacovo, M.P.; MacQueen, A.J.; Martinez-Perez, E.; McDonald, K.; Adamo, A.; La Volpe, A.; Villeneuve, A.M. Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev. Cell 2003, 5, 463–474. [Google Scholar] [CrossRef]
- Kim, H.M.; Colaiacovo, M.P. ZTF-8 interacts with the 9-1-1 complex and is required for DNA damage response and double-strand break repair in the C. elegans germline. PLoS Genet. 2014, 10, e1004723. [Google Scholar] [CrossRef] [PubMed]
- Goodyer, W.; Kaitna, S.; Couteau, F.; Ward, J.D.; Boulton, S.J.; Zetka, M. HTP-3 links DSB formation with homolog pairing and crossing over during C. elegans meiosis. Dev. Cell 2008, 14, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, R.; Zawadzki, K.A.; Nabeshima, K.; Drake, M.; Arur, S.; Villeneuve, A.M. COSA-1 Reveals Separable Licensing and Reinforcement Steps and Efficient Homeostasis Governing Meiotic Crossovers. Cell 2012, 149, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Merlet, J.; Burger, J.; Tavernier, N.; Richaudeau, B.; Gomes, J.E.; Pintard, L. The CRL2LRR-1 ubiquitin ligase regulates cell cycle progression during C. elegans development. Development 2010, 137, 3857–3866. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.; Merlet, J.; Tavernier, N.; Richaudeau, B.; Arnold, A.; Ciosk, R.; Bowerman, B.; Pintard, L. CRL2(LRR-1) E3-ligase regulates proliferation and progression through meiosis in the Caenorhabditis elegans germline. PLoS Genet. 2013, 9, e1003375. [Google Scholar] [CrossRef] [PubMed]
- Panbianco, C.; Weinkove, D.; Zanin, E.; Jones, D.; Divecha, N.; Gotta, M.; Ahringer, J. A casein kinase 1 and PAR proteins regulate asymmetry of a PIP(2) synthesis enzyme for asymmetric spindle positioning. Dev. Cell 2008, 15, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Sanyal, S.; Ghosh, A.; Bhar, K.; Das, C.; Siddhanta, A. Phosphatidylinositol-4-phosphate 5-Kinase 1α Modulates Ribosomal RNA Gene Silencing through Its Interaction with Histone H3 Lysine 9 Trimethylation and Heterochromatin Protein HP1-α. J. Biol. Chem. 2015, 290, 20893–20903. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, J.; Horovitz, H.R.; Brenner, S. Nondisjunction mutants of the nematode Caenorhabditis elegans. Genetics 1979, 91, 67–94. [Google Scholar] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Starr, D.A. A nuclear-envelope bridge positions nuclei and moves chromosomes. J. Cell Biol. 2009, 122, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Penkner, A.; Tang, L.; Novatchkova, M.; Ladurner, M.; Fridkin, A.; Gruenbaum, Y.; Schweizer, D.; Loidl, J.; Jantsch, V. The nuclear envelope protein Matefin/SUN-1 is required for homologous pairing in C. elegans meiosis. Dev. Cell 2007, 12, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Jungmichel, S.; Sylvestersen, K.B.; Choudhary, C.; Nguyen, S.; Mann, M.; Nielsen, M.L. Specificity and commonality of the phosphoinositide-binding proteome analyzed by quantitative mass spectrometry. Cell Rep. 2014, 6, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Timmons, L.; Fire, A. Specific interference by ingested dsRNA. Nature 1998, 395, 854. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Perez, E.; Villeneuve, A.M. HTP-1-dependent constraints coordinate homolog pairing and synapsis and promote chiasma formation during C. elegans meiosis. Genes Dev. 2005, 19, 2727–2743. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.A.; Aballay, A. Regulation of DAF-16-mediated Innate Immunity in Caenorhabditis elegans. J. Biol. Chem. 2009, 284, 35580–35587. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase Transfer Surfactant-Aided Trypsin Digestion for Membrane Proteome Analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell. Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Harper, N.; Rillo, R.; Jover-Gil, S.; Assaf, Z.J.; Bhalla, N.; Dernburg, A.F. Pairing centers recruit a Polo-like kinase to orchestrate meiotic chromosome dynamics in C. elegans. Dev. Cell 2011, 21, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, A. A cis-acting locus that promotes crossing over between X chromosomes in Caenorhabditis elegans. Genetics 1994, 136, 887–902. [Google Scholar] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulicna, L.; Rohozkova, J.; Hozak, P. Multiple Aspects of PIP2 Involvement in C. elegans Gametogenesis. Int. J. Mol. Sci. 2018, 19, 2679. https://doi.org/10.3390/ijms19092679
Ulicna L, Rohozkova J, Hozak P. Multiple Aspects of PIP2 Involvement in C. elegans Gametogenesis. International Journal of Molecular Sciences. 2018; 19(9):2679. https://doi.org/10.3390/ijms19092679
Chicago/Turabian StyleUlicna, Livia, Jana Rohozkova, and Pavel Hozak. 2018. "Multiple Aspects of PIP2 Involvement in C. elegans Gametogenesis" International Journal of Molecular Sciences 19, no. 9: 2679. https://doi.org/10.3390/ijms19092679
APA StyleUlicna, L., Rohozkova, J., & Hozak, P. (2018). Multiple Aspects of PIP2 Involvement in C. elegans Gametogenesis. International Journal of Molecular Sciences, 19(9), 2679. https://doi.org/10.3390/ijms19092679