Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3





Abstract
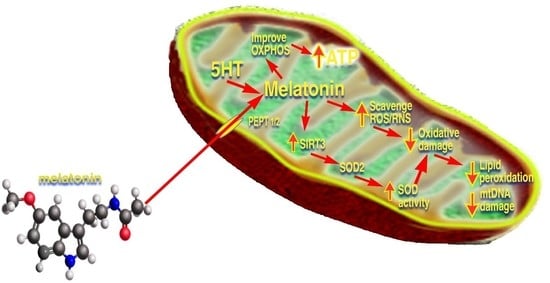
1. Introduction
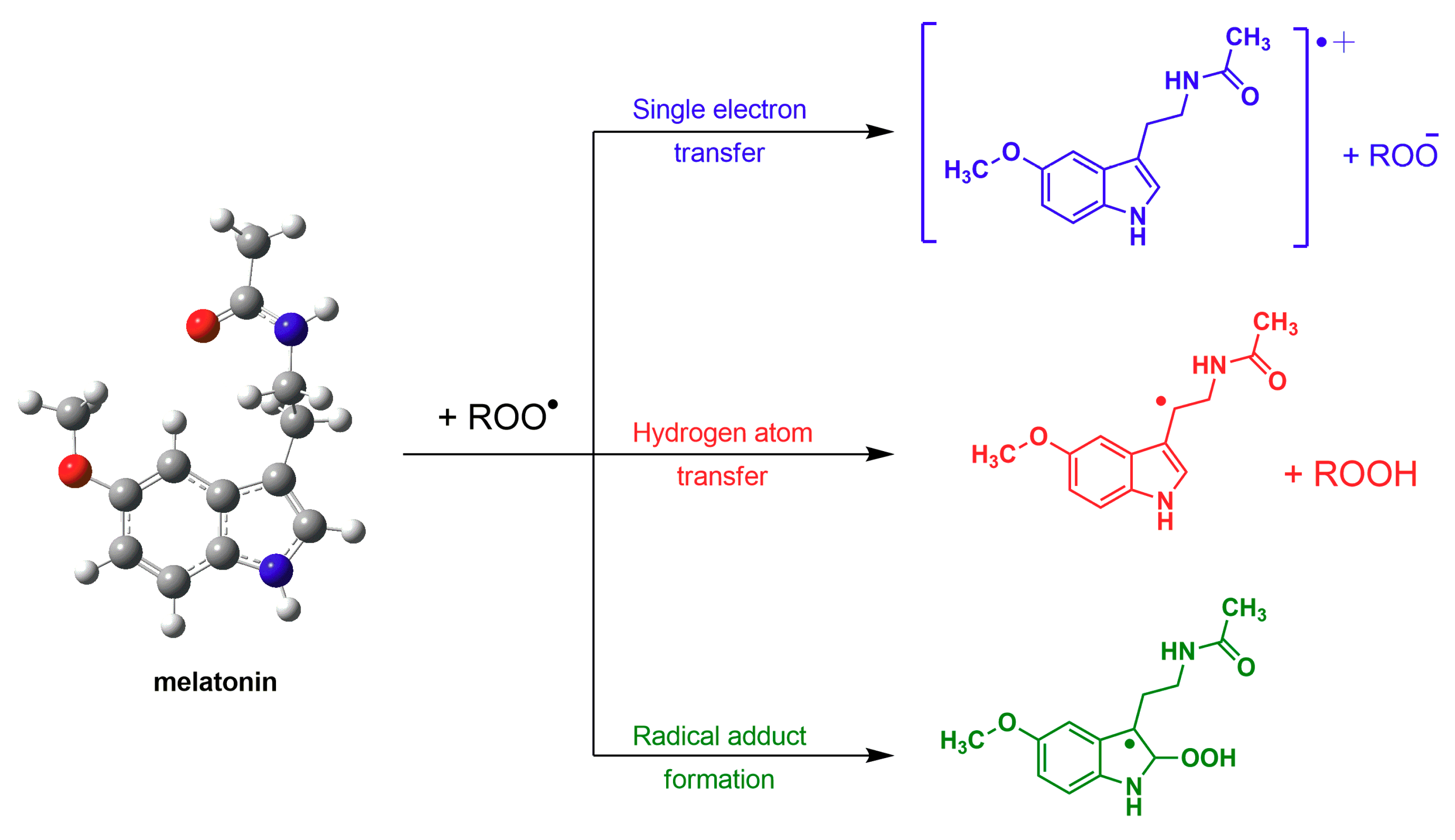
2. Melatonin: Harnessing Free Radicals
3. Intrinsic Sources of Melatonin
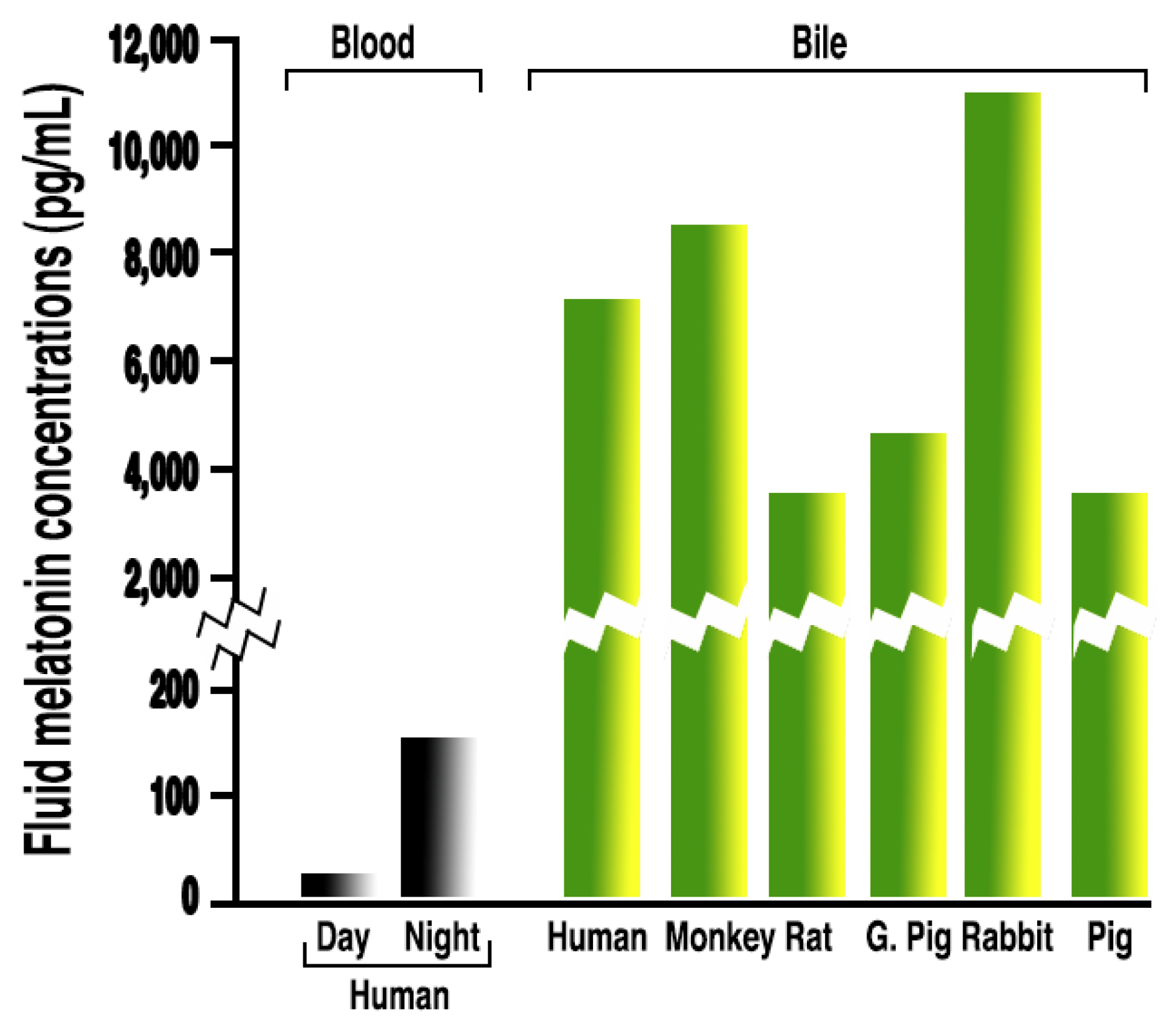
3.1. Melatonin in Bodily Fluids
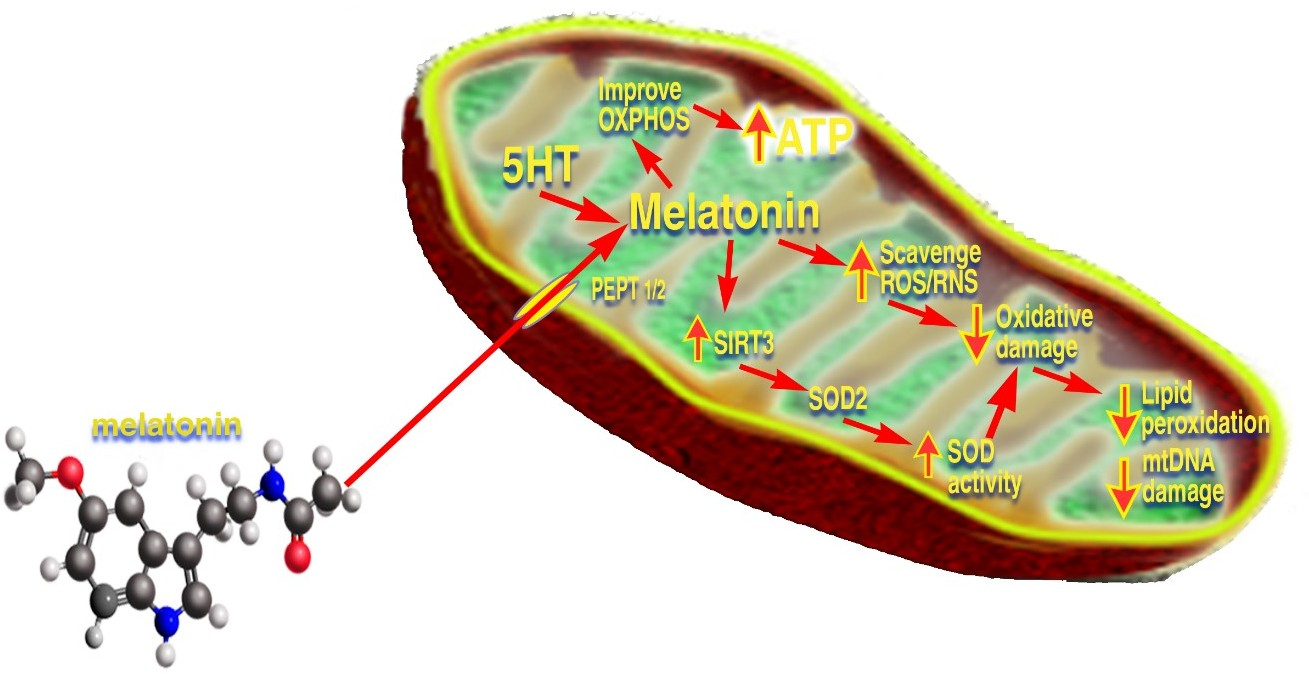
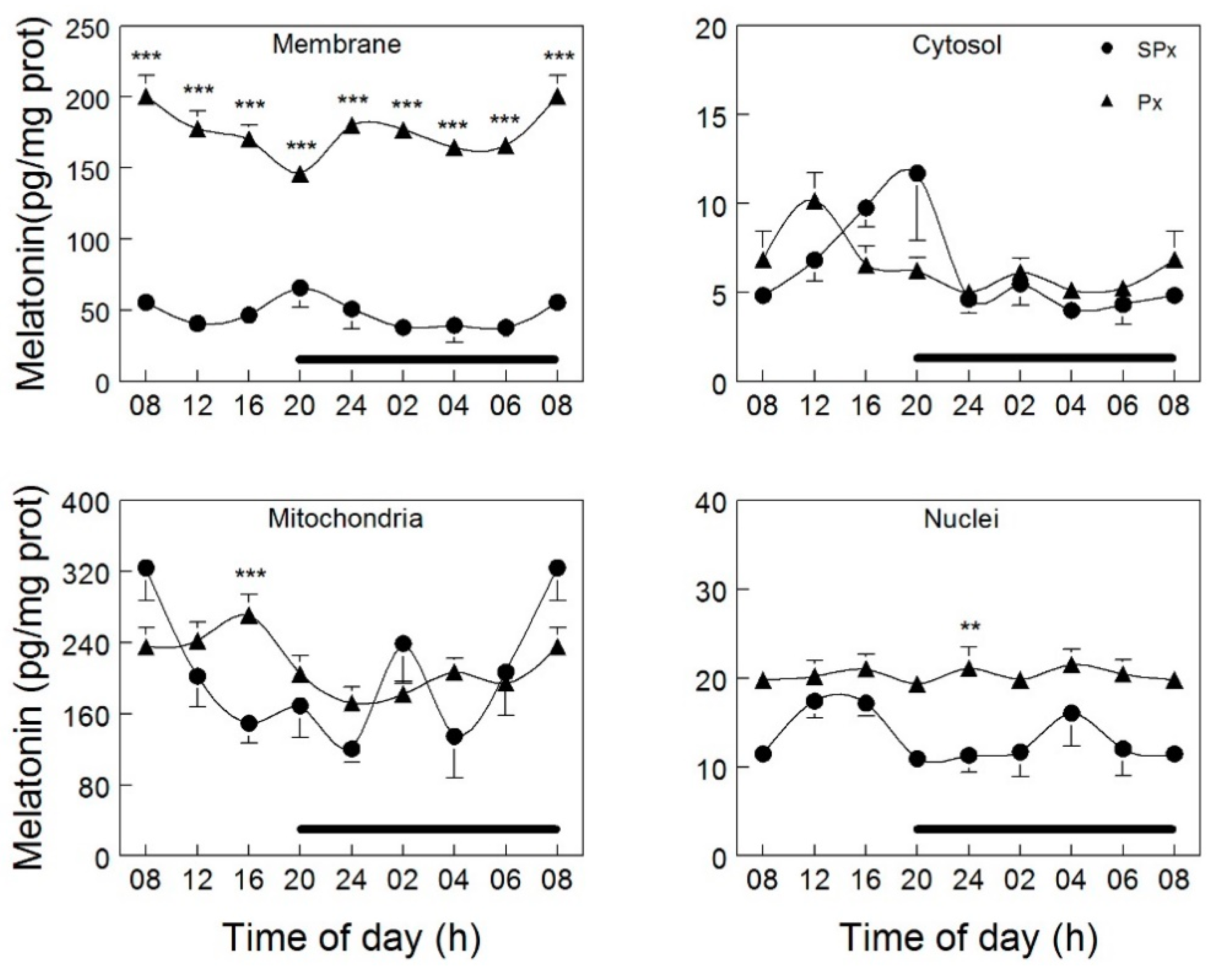
3.2. Mitochondrial Melatonin
3.3. Mitochondrial Synthesis of Melatonin
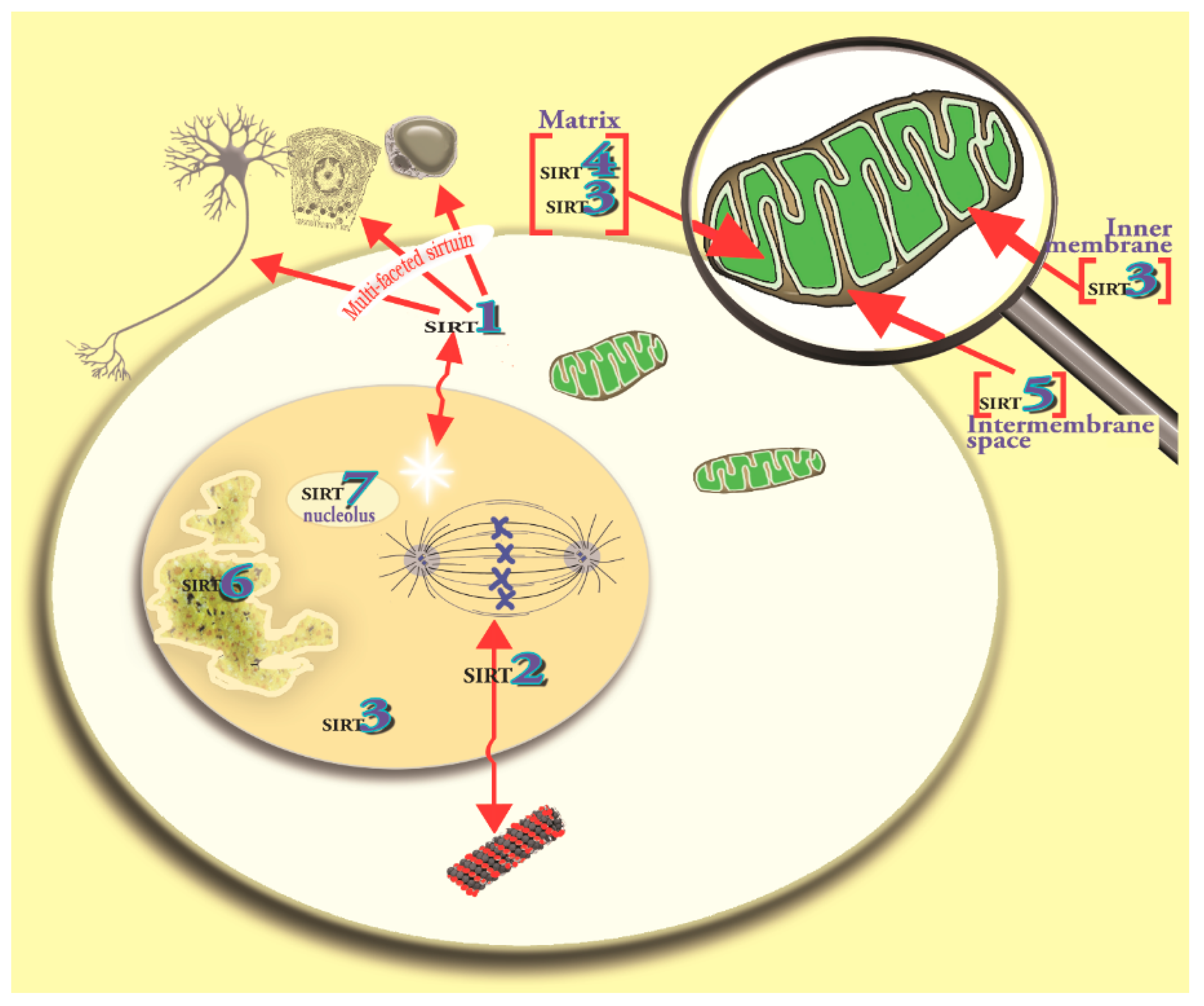
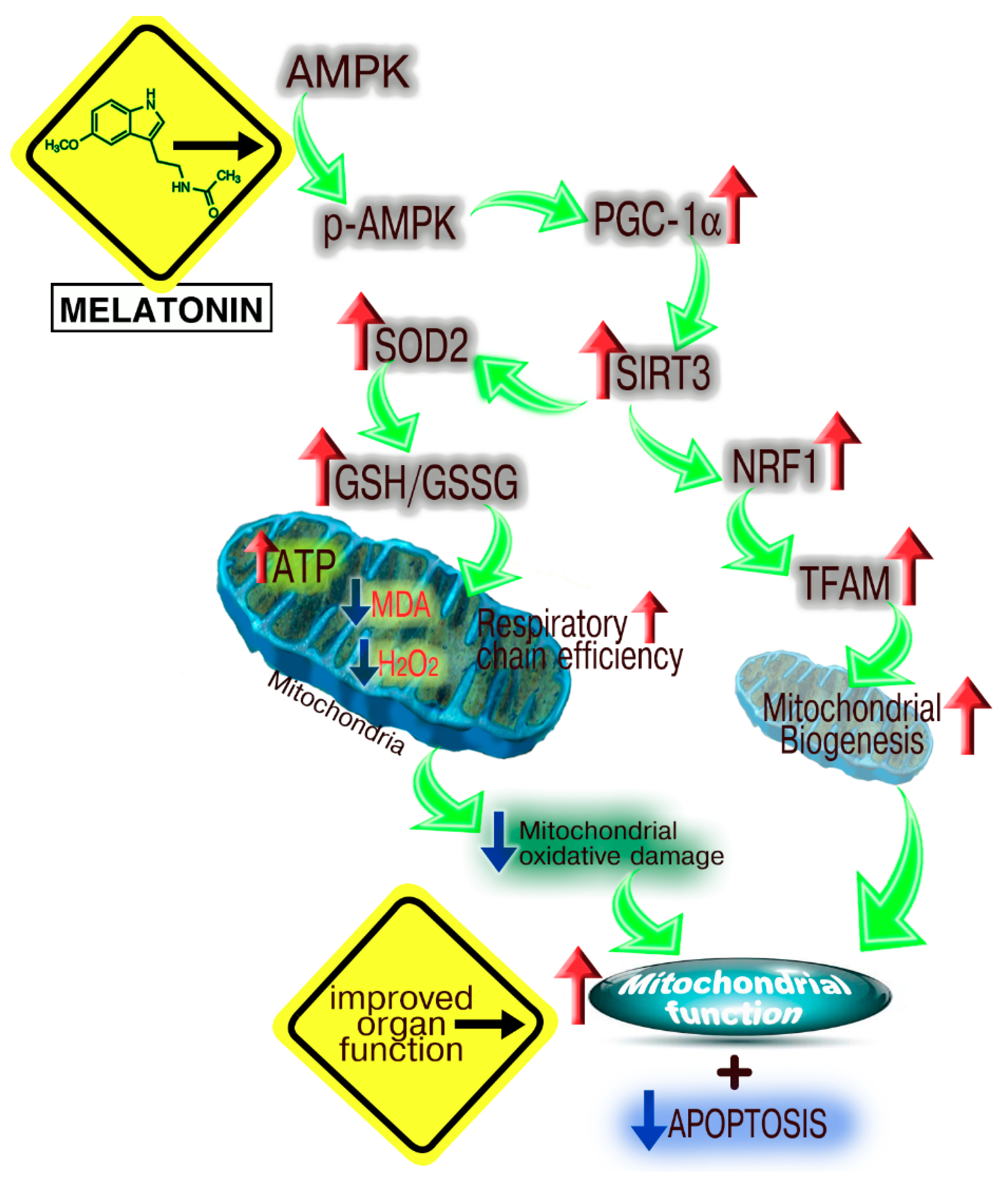
4. The Melatonin/SIRT3 Interactions
4.1. Reproductive Organ Protection
4.2. Cardiovascular Protection
4.3. Hepatic Protection
5. Concluding Remarks
Conflicts of Interest
References
- Mayo, J.C.; Sainz, R.M.; Gonzalez-Menendez, P.; Cepas, V.; Tan, D.X.; Reiter, R.J. Melatonin and sirtuins: A “not-so unexpected” relationship. J. Pineal Res. 2017, 62, e12391. [Google Scholar] [CrossRef] [PubMed]
- Otto, G.M.; Brar, G.A. Seq-ing answers: Uncovering the unexpected in global gene regulation. Curr. Genet. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Chaubai, A.; Pile, L.A. Same agent, different messages: Insight into transcriptional regulation by SIN3 isoforms. Epigenet. Chromatin 2018, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Godini, R.; Lafta, H.Y.; Fallahi, H. Epigenetics modifications in the embryonic and induced pluripotent stem cells. Gene Expr. Patterns 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, A.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Gene regulation by melatonin linked to epigenetic phenomena. Gene 2012, 503, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. The key role of epigenetics in human disease prevention and mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Nadal, S.; Raj, R.; Mohammed, S.; Davis, B.G. Synthetic post-translational modification of histones. Curr. Opin. Chem. Biol. 2018, 45, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Klar, A.J.; Fogel, S.; MacLeod, K. MAR1-a regulator of the HMa and HMalpha loci in Saccharomyces cerevisiae. Genetics 1979, 93, 37–50. [Google Scholar] [PubMed]
- Klar, A.J.; Kakar, S.N.; Ivy, J.M.; Hicks, J.B.; Livi, G.P.; Miglio, L.M. SUM1, an apparent positive regulator of the cryptic mating-type in Saccharomyces cerevisiae. Genetics 1985, 111, 745–758. [Google Scholar] [PubMed]
- Rine, J.; Herskowitz, I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 1987, 116, 9–22. [Google Scholar] [PubMed]
- Rusche, L.N.; Kirchmaier, A.L.; Rine, J. Ordered nucleation and spreading of silenced chromatin in Saccharomyces cerevisiae. Mol. Biol. Cell 2002, 13, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Ajami, M.; Pazoki-Toroudi, H.; Amani, H.; Nabavi, S.F.; Braidy, N.; Vacca, R.A.; Atanasov, A.G.; Mocan, A.; Nabavi, S.M. Therapeutic role of sirtuins in neurodegeneration disease and their modulation by polyphenols. Neurosci. Biobehav. Rev. 2017, 73, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Dang, W. The controversial world of sirtuins. Drug Discov. Today Technol. 2014, 12, e9–e17. [Google Scholar] [CrossRef] [PubMed]
- Watroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szurkiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Reiter, R.J.; Ahmad, N. Sirtuins, melatonin, and circadian rhythms: Building a bridge between aging and cancer. J. Pineal Res. 2010, 48, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.L.C.; Ramasamy, T.S. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res. Rev. 2018, 43, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Masri, S. Sirtuin-dependent clock control: New advances in metabolism, aging and cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, R.H. Associations among metabolism, circadian rhythms and age-associated diseases. Aging Dis. 2017, 8, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The mitochondrial acylome emerges: Proteomics, regulation of sirtuins and metabolic and disease implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Steegborn, C. Function and regulation of the mitochondrial sirtuin isoform Sirt5 in mammalia. Biochim. Biophys. Acta 2010, 1804, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Xing, H.; Zang, T.; Ruan, X.; Wo, L.; He, M. Sirtuins in mitochondrial stress: Indispensable helpers behind the scenes. Ageing Res. Rev. 2018, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslovsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Noqueiras, R.; Habegger, K.M.; Chaudhary, M.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Hausenloy, D.J. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Ther. 2012, 136, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central organelles for melatonin’s antioxidant and anti-aging actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A mitochondrial-targeting molecule involving mitochondrial proteins and energetics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Mostoslavky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of caloric restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devries, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; Smith, L.M.; et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Reiter, R.J. Melatonin: A potent hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Tan, D.X.; Poeggeler, B.; Reiter, R.J.; Chen, L.D.; Chen, S.; Manchester, L.C.; Barlow-Walden, L.R. The pineal hormone melatonin inhibits DNA-adduct formation induced by the chemical carcinogen safrole in vivo. Neurosci. Lett. 1993, 157, 131–134. [Google Scholar] [CrossRef]
- Trush, M.A.; Kensler, T.W. An overview of the relationship between oxidative stress and chemical carcinogenesis. Free Radic. Biol. Med. 1991, 10, 201–209. [Google Scholar] [CrossRef]
- Harman, D. The free radical theory of aging. Antioxid. Redox Signal. 2003, 5, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.; Rosen, G.M.; Rauckmann, E.J. Spin trapping of superoxide and hydroxyl radical: Practical aspects. J. Am. Chem. Soc. 1980, 102, 4994–4999. [Google Scholar] [CrossRef]
- Floyd, R.A. Role of oxygen free radicals in carcinogenesis and brain ischemia. FASEB J. 1990, 4, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L. Pharmacology and function of melatonin receptors. FASEB J. 1988, 2, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Stankov, B.; Reiter, R.J. Melatonin receptors: Current status, facts, and hypotheses. Life Sci. 1990, 46, 971–982. [Google Scholar] [CrossRef]
- Poeggeler, B.; Saarela, S.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C.; Barlow-Walden, L.R. Melatonin—A highly potent endogenous radical scavenger and electron donor: New aspects of the oxidation chemistry of this indole assessed in vitro. Ann. N. Y. Acad. Sci. 1994, 738, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Reiter, R.J.; Orhii, P.B.; Hara, M.; Poeggeler, B. Inhibitory effect of melatonin on cataract formation in newborn rats: Evidence for an antioxidative role for melatonin. J. Pineal Res. 1994, 17, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Vijayalaxmi; Reiter, R.J.; Meltz, M.L. Melatonin protects human blood lymphocytes from radiation-induced chromosome damage. Mutat. Res. 1995, 346, 23–31. [Google Scholar] [CrossRef]
- Melchiorri, D.; Reiter, R.J.; Attia, A.M.; Hara, M.; Burgos, A.; Nistico, G. Potent protective effect of melatonin on in vivo paraquat-induced oxidative damage in rats. Life Sci. 1995, 56, 83–89. [Google Scholar] [CrossRef]
- Sewerynek, E.; Abe, M.; Reiter, R.J.; Barlow-Walden, L.R.; Chen, L.D.; McCabe, T.J.; Roman, L.J.; Diaz-Lopez, B. Melatonin administration prevents lipopolysaccharide-induced oxidative damage in phenobarbital-treated rats. J. Cell. Biochem. 1995, 58, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Sewerynek, E.; Melchiorri, D.; Chen, L.D.; Reiter, R.J. Melatonin reduces both basal and bacterial lipopolysaccharide-induced lipid peroxidation in vitro. Free Radic. Biol. Med. 1995, 19, 903–909. [Google Scholar] [CrossRef]
- Melchiorri, D.; Reiter, R.J.; Sewerynek, E.; Chen, L.D.; Nistico, G. Melatonin reduces kainate-induced lipid peroxidation in homogenates of different brain regions. FASEB J. 1995, 9, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Sewerynek, E.; Reiter, R.J.; Melchiorri, D.; Ortiz, G.G.; Lewinski, A. Oxidative damage in the liver induced by ischemia-reperfusion: Protection by melatonin. Hepato-Gastroenterology 1996, 43, 898–905. [Google Scholar]
- Pieri, C.; Marra, M.; Moroni, F.; Recchioni, R.; Marcheselli, F. Melatonin: A peroxyl radical scavenger more effective than vitamin E. Life Sci. 1994, 55, 271–276. [Google Scholar] [CrossRef]
- Pieri, C.; Moroni, F.; Marra, M.; Marcheselli, F.; Recchioni, R. Melatonin is an efficient antioxidant. Arch. Gerontol. Geriatr. 1995, 20, 159–165. [Google Scholar] [CrossRef]
- Scaiano, J.C. Exploratory laser flash photolysis study of the free radical reactions and magnetic field effects in melatonin chemistry. J. Pineal Res. 1995, 19, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, Z.; Reszka, K.J.; Chignell, C.F. Reaction of melatonin and related indoles with hydroxyl radicals: EPR and spin trapping investigation. Free Radic. Biol. Med. 1997, 23, 367–372. [Google Scholar] [CrossRef]
- Stasica, P.; Ulanski, P.; Rosiak, J.M. Melatonin as a hydroxyl radical scavenger. J. Pineal Res. 1998, 25, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Stasica, P.; Paneth, P.; Rosiak, J.M. Hydroxyl radical reaction with melatonin molecule: A computational study. J. Pineal Res. 2000, 29, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Stasica, P.; Ulanski, P.; Roriak, J.M. Reactions of melatonin with radicals in deoxygenated aqueous solutions. J. Radioanal. Nucl. Chem. 1998, 232, 107–113. [Google Scholar] [CrossRef]
- Livrea, M.A.; Tesoriere, L.; D’Arpa, D.; Morreale, M. Reaction of melatonin with lipoperoxyl radicals in phospholipid bilayers. Free Radic. Biol. Med. 1997, 23, 706–711. [Google Scholar] [CrossRef]
- Zang, L.Y.; Cosma, G.; Gardner, H.; Vallyathan, V. Scavenging of reactive oxygen species by melatonin. Biochim. Biophys. Acta 1998, 1425, 469–477. [Google Scholar] [CrossRef]
- Turjanski, A.G.; Rosenstein, R.E.; Estrin, D.A. Reactions of melatonin and related indoles with free radicals: A computational study. J. Med. Chem. 1998, 41, 3684–3689. [Google Scholar] [CrossRef] [PubMed]
- Brömme, H.J.; Mörke, W.; Peschke, E.; Ebelt, H.; Peschke, D. Scavenging effect of melatonin on hydroxyl radicals generated by alloxan. J. Pineal Res. 2000, 29, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Zavodnik, I.B.; Domanski, A.V.; Lapshina, E.A.; Bryszewska, M.; Reiter, R.J. Melatonin directly scavenges free radicals generated in red blood cells and a cell-free system: Chemiluminescence measurements and theoretical calculations. Life Sci. 2006, 79, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Brömme, H.J.; Ebelt, H.; Peschke, D.; Peschke, E. Alloxan acts as a prooxiant only under reducing conditions: Influence of melatonin. Cell. Mol. Life Sci. 1999, 55, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Mahal, H.S.; Sharma, H.S.; Mukherjie, T. Antioxidant properties of melatonin: A pulse radiolysis study. Free Radic. Biol. Med. 1999, 26, 557–565. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Galano, A. On the direct scavenging activity of melatonin towards hydroxyl and a series of peroxyl radicals. Phys. Chem. Chem. Phys. 2011, 13, 7178–7188. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin and related compounds: Chemical insights into their protective effects against oxidative stress. Curr. Org. Chem. 2017, 21, 2077–2095. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pshenichnyuk, S.A.; Modelli, A.; Jones, D.; Lazneva, E.F.; Komolov, A.S. Low-energy electron interaction with melatonin and related compounds. J. Phys. Chem. B. 2017, 121, 3965–3974. [Google Scholar] [CrossRef] [PubMed]
- Daniels, W.M.O.; Reiter, R.J.; Melchiorri, D.; Seweryneck, E.; Pablos, M.I.; Ortiz, G.G. Melatonin counteracts lipid peroxidation induced by carbon tetrachloride but does not restore glucose-6 phosphatase activity. J. Pineal Res. 1995, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pablos, M.I.; Reiter, R.J.; Chuang, J.I.; Ortiz, G.G.; Guerrero, J.M.; Sewerynek, E.; Agapito, M.T.; Melchiorri, D.; Lawrence, R.; Deneke, S.M. Acutely administered melatonin reduces oxidative damage in lung and brain induced by hyperbaric oxygen. J. Appl. Physiol. 1997, 83, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Reiter, R.J.; Rouvier-Garay, M.V.; El-Sokkary, G.H.; Tan, D.X. 2-Nitropropane-induced lipid peroxidation and DNA damage: Antitoxic effects of melatonin. Toxicology 1998, 19, 117–123. [Google Scholar]
- Qi, W.; Tan, D.X.; Reiter, R.J.; Kim, S.J.; Manchester, L.C.; Cabrera, J.; Sainz, R.M.; Mayo, J.C. Melatonin reduces lipid peroxidation and tissue edema in cerulein-induced acute pancreatitis in rats. Dig. Dis. Sci. 1999, 44, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, J.; Reiter, R.J.; Tan, D.X.; Qi, W.; Sainz, R.M.; Mayo, J.C.; Garcia, J.J.; Kim, S.J.; El-Sokkary, G. Melatonin reduces oxidative neurotoxicity due to quinolinic acid: In vitro and in vivo findings. Neuropharmacology 2000, 39, 507–514. [Google Scholar] [CrossRef]
- Karbownik, M.; Tan, D.X.; Reiter, R.J. Melatonin reduces the oxidation of nuclear DNA and membrane lipids induced by the carcinogen delta-aminolevulinic acid. Int. J. Cancer 2000, 88, 7–11. [Google Scholar] [CrossRef]
- Fulia, F.; Gitto, E.; Cuzzocrea, S.; Reiter, R.J.; Dugo, L.; Gitto, P.; Barberi, S.; Cordaro, S.; Barberi, I. Increased levels of malondialdehyde and nitrite/nitrate in blood of asphyxiated newborns: Reduction by melatonin. J. Pineal Res. 2001, 31, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Meki, A.R.; Hussein, A.A. Melatonin reduces oxidative stress induced by ochratoxin A in rat liver and kidney. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2001, 130, 305–313. [Google Scholar] [CrossRef]
- Fischer, T.W.; Scholz, G.; Knoll, B.; Hipler, U.C.; Elsner, P. Melatonin suppresses reactive oxygen species induced by UV irradiation in leukocytes. J. Pineal Res. 2004, 37, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Gitto, E.; Reiter, R.J.; Amodio, A.; Romeo, C.; Cuzzocrea, E.; Sabatino, G.; Buonocore, G.; Cordaro, V.; Trimarchi, G.; Barberi, I. Early indicators of chronic lung disease in preterm infants with respiratory distress syndrome and their inhibition by melatonin. J. Pineal Res. 2004, 30, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Mohanakumar, K.P. Melatonin protects against oxidative stress caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the mouse nigrostriatum. J. Pineal Res. 2004, 36, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Vairetti, M.; Ferrigno, A.; Bertone, R.; Rizzo, V.; Richelmi, P.; Berte, F.; Reiter, R.J.; Freitas, I. Exogenous melatonin enhances bile flow and ATP levels after cold storage and reperfusion in rat liver: Implications for liver transplantation. J. Pineal Res. 2005, 38, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, W.; Xu, J.; Min, F.; Wang, X.; Cao, S.; Tan, D.X.; Qi, W.; Reiter, R.J. Inhibitory effect of melatonin on diquat-induced lipid peroxidation in vivo assessed by the measurement of F2-isoprostanes. J. Pineal Res. 2008, 40, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Ozacmak, V.H.; Barut, F.; Ozacmak, H.S. Melatonin provides neuroprotection by reducing oxidative stress and HSP70 expression during chronic cerebral hypoperfusion in ovariectomized rats. J. Pineal Res. 2009, 47, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Konturek, S.J.; Celinski, K.; Slomka, M.; Cichoz-Lach, H.; Bielanski, W.; Reiter, R.J. Role of melatonin on mucosal gastroprotection against aspirin-induced gastric lesions in humans. J. Pineal Res. 2010, 48, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Laothong, U.; Pinlaor, P.; Hiraku, Y.; Boonsiri, P.; Prakobwong, S.; Khoontawad, J.; Pinlaor, S. Protective effects of melatonin against Opisthorchis viverrini-induced oxidative and nitrosative damage and liver injury in hamsters. J. Pineal Res. 2010, 49, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Nopparat, C.; Porter, J.E.; Ebadi, M.; Govitrapong, P. The mechanisms for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J. Pineal Res. 2010, 49, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Govender, J.; Loos, B.; Marais, E.; Engelbrecht, A.M. Mitochondrial catastrophe during doxorubicin-induced cardiotoxicity: A review of the protective role of melatonin. J. Pineal Res. 2014, 57, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Abdel Moneim, A.E.; Ortiz, F.; Leonardo-Mendonca, R.C.; Vergano-Villodres, R.; Guerrero-Martinez, D.A.; Lopez, L.C.; Acuna-Castroviejo, D.; Escames, G. Protective effects of melatonin against oxidative damage induced by Egyptian cobra (Naja haje) venom in rats. Acta Trop. 2015, 143, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Borges-Lda, S.; Dermargos, A.; da Silva Junior, E.P.; Weimann, E.; Lambertucci, R.H.; Hatanaks, E. Melatonin decreases muscular oxidative stress and inflammation induced by strenuous exercise and stimulates growth factor synthesis. J. Pineal Res. 2015, 58, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, F.; Acuna-Castroviejo, D.; Doerrier, C.; Dayoub, J.C.; Lopez, L.C.; Venegas, C.; Garcia, J.A.; Lopez, A.; Volt, H.; Luna-Sanchez, M.; et al. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced mucositis. J. Pineal Res. 2015, 58, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Dwaich, K.H.; Al-Amran, F.G.; Al-Sheibani, B.I.; Al-Aubaidy, H.A. Melatonin effects on myocardial ischemia-reperfusion injury: Impact on the outcome in patients undergoing coronary artery bypass grafting surgery. Int. J. Cardiol. 2016, 221, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pi, H.; Zhang, L.; Zhang, N.; Li, Y.; Zhang, H.; Tang, J.; Li, H.; Feng, M.; Deng, P.; et al. Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J. Pineal Res. 2016, 60, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Asghari, M.H.; Abdollahi, M.; de Oliveira, M.R.; Nabavi, S.M. A review of the protective role of melatonin during phosphine-induced cardiotoxicity: Focus on mitochondrial dysfunction, oxidative stress and apoptosis. J. Pharm. Pharmacol. 2017, 69, 236–243. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Marseglia, L.; Reiter, R.J.; Buonocore, G.; Gitto, E. Melatonin and neonatal sepsis: A promising antioxidant adjuvant agent. Am. J. Perinatol. 2017, 34, 1382–1388. [Google Scholar] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; de la Torre-Hernandez, J.M.; Consuegra-Sanchez, L.; Piccolo, R.; Gonzalez-Gonzalez, J.; Garcia-Camarero, T.; Del Mar Garcia-Sainz, M.; Aldea-Perona, A.; Reiter, R.J.; et al. Usefulness of early treatment with melatonin to reduce infarct size in patients with ST-segment elevation myocardial infarction receiving percutaneous coronary intervention (from the melatonin adjunct in the acute myocardial infarction treated with angioplasty trial). Am. J. Cardiol. 2017, 120, 522–526. [Google Scholar] [PubMed]
- Feng, D.; Wang, B.; Wang, L.; Abraham, N.; Tao, K.; Huang, L.; Shi, W.; Dong, Y.; Qu, Y. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signaling. J. Pineal Res. 2017, 62, e12395. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Han, Y.S.; Lee, J.H.; Kim, S.M.; Lee, S.H. Melatonin rescues mesenchymal stem cells from senescence induced by the uremic toxin p-cresol via inhibiting mTOR-dependent autophagy. Biomol. Ther. (Seoul) 2018, 26, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, Z.; Liu, W.; Wang, J.; He, X.; Huang, H.; Zhang, J.; Yang, Z. Melatonin protects against arsenic trioxide-induced liver injury by the upregulation of Nrf2 expression through the activation of Pl3K/AKT pathway. Oncotarget 2017, 8, 3773–3780. [Google Scholar] [PubMed]
- Espino, J.; Rodriguez, A.B.; Pariente, J.A. Melatonin and oxidative stress in the diabetic state: Clinical implications and potential therapeutic applications. Curr. Med. Chem. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhang, P.; Guo, J.; Zhu, Z.; Li, X.; Xu, D.; Zeng, W. Melatonin protects mouse spermatogonial stem cells against hexavalent chromium-induced apoptosis and epigenetic histone modifications. Toxicol. Appl. Pharmacol. 2018, 340, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Wei, C.H.; Wang, W.D.; Wang, J.S.; Zhang, J.; Wang, J.J. Melatonin attenuates lung ischemia-reperfusion injury via inhibition of oxidative stress and inflammation. Interact. Cardiovasc. Thorac. Surg. 2018, 26, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B.; Reiter, R.J.; Hardeland, R.; Tan, D.X.; Barlow-Walden, L.R. Melatonin and structurally-related, endogenous indoles act as potent electron donors and radical scavengers in vitro. Redox Rep. 1996, 2, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Diduk, R.; Galano, A.; Tan, D.X.; Reiter, R.J. The key role of the sequential proton loss electron transfer mechanism on the free radical scavenging activity of some melatonin-related compounds. Theor. Chem. Acc. 2016, 139, 1–5. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin: A versatile protector against oxidative DNA damage. Molecule 2018, 23, 530. [Google Scholar] [CrossRef] [PubMed]
- Abuja, P.M.; Leibmann, P.; Hayn, M.; Schauenstein, K.; Esterbauer, H. Antioxidant role of melatonin in lipid peroxidation of human LDL. FEBS Lett. 1997, 413, 289–293. [Google Scholar] [CrossRef]
- Antunes, F.; Barclay, L.R.; Ingold, K.U.; King, M.; Norris, J.Q.; Scaiano, J.C.; Xi, F. On the antioxidant activity of melatonin. Free Radic. Biol. Med. 1999, 26, 117–128. [Google Scholar] [CrossRef]
- Marshall, K.C.; Reiter, R.J.; Poeggeler, B.; Aruoma, O.I.; Halliwell, B. Evaluation of the antioxidant activity of melatonin in vitro. Free Radic. Biol. Med. 1996, 21, 307–315. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: The resurrection of an almost forgotten class of biogenic amines. J. Pineal Res. 2009, 47, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Reiter, R.J.; Poeggeler, B.; Tan, D.X. The significance of the metabolism of the neurohormone melatonin: Antioxidative protection and formation of bioactive substances. Neurosci. Biobehav. Rev. 1993, 17, 347–357. [Google Scholar] [CrossRef]
- Tsia, P.L.; Hu, M.K. Free radical scavenging and antioxidative activity of melatonin derivatives. J. Pharm. Pharmacol. 2003, 55, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Burkhardt, S.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Shohami, E.; Huo, Y.S.; Hardeland, R.; Reiter, R.J. N1-acetyl-N2-formyl-5-methoxykynuramine, a biogenic amine and melatonin metabolite, functions as a potent antioxidant. FASEB J. 2001, 15, 2294–2296. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, R.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin directly scavenges hydrogen peroxide: A potentially new metabolic pathway of melatonin biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 2013, 54, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Limson, J.; Nyokong, T.; Daya, S. The interaction of melatonin and its precursors with aluminum, cadmium, copper, iron, lead, and zinc: An absorptive voltammetric study. J. Pineal Res. 1998, 24, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Medina, M.D.; Tan, D.X.; Reiter, R.J. Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: A physicochemical study. J. Pineal Res. 2015, 58, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Barlow-Walden, L.R.; Reiter, R.J.; Abe, M.; Pablos, M.I.; Menendez-Pelaez, A.; Chen, L.D.; Poeggeler, B. Melatonin stimulates brain glutathione peroxidase activity. Neurochem. Int. 1995, 26, 497–502. [Google Scholar] [CrossRef]
- Pablos, M.I.; Agapito, M.T.; Gutierrez, R.; Recio, J.M.; Reiter, R.J.; Barlow-Walden, L.R.; Acuna-Castroviejo, D.; Menendez-Pelaez, A. Melatonin stimulates the activity of the detoxifying enzyme glutathione peroxidase in several tissues of chicks. J. Pineal Res. 1995, 19, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Pablos, M.I.; Reiter, R.J.; Ortiz, G.G.; Guerrero, J.M.; Agapito, M.T.; Chuang, J.J.; Sewerynek, E. Rhythms of glutathione peroxidase and glutathione reductase in brain of chick and their inhibition by light. Neurochem. Int. 1998, 32, 69–75. [Google Scholar] [CrossRef]
- Antolin, I.; Rodriquez, C.; Sainz, R.M.; Mayo, J.C.; Uria, H.; Kotler, M.L.; Rodriquez-Colunga, M.J.; Tolivia, D.; Menendez-Pelaez, A. Neurohormone melatonin prevents cell damage: Effect on gene expression for antioxidant enzymes. FASEB J. 1996, 10, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Kotler, M.L.; Rodriguez, C.; Sainz, R.M.; Antolin, I.; Menendez-Pelaez, A. Melatonin increases gene expression for antioxidant enzymes in rat brain cortex. J. Pineal Res. 1998, 24, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Pablos, M.I.; Guerrero, J.M.; Ortiz, G.G.; Agapito, M.T.; Reiter, R.J. Both melatonin and a putative nuclear melatonin receptor agonist CGP 52608 stimulate glutathione peroxidase and glutathione reductase activities in mouse brain in vivo. Neuroendocrinol. Lett. 1997, 18, 49–58. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress: A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Rodriquez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Manchester, L.C. The universal nature, unequal distribution and antioxidant functions of melatonin and its derivatives. Mini Rev. Med. Chem. 2013, 13, 373–384. [Google Scholar] [PubMed]
- Gitto, E.; Tan, D.X.; Reiter, R.J.; Karbownik, M.; Manchester, L.C.; Cuzzocrea, S.; Fulia, F.; Barberi, I. Individual and synergistic antioxidative actions of melatonin: Studies with vitamin E, vitamin C, glutathione and desferrioxamine (desferoxamine) in rat liver homogenates. J. Pharm. Pharmacol. 2001, 53, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Milczarek, R.; Hallmann, A.; Sokolowska, E.; Kaletha, K.; Klimek, J. Melatonin enhances antioxidant action of α-tocopherol and ascorbate against NADPH- and iron-dependent lipid peroxidation in human placental mitochondria. J. Pineal Res. 2010, 49, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.A. Quinone reductase 2 as a promising target of melatonin therapeutic actions. Expert Opin. Ther. Targets 2016, 20, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Antioxidant protection by melatonin: Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 2005, 27, 119–130. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and the electron transport chain. Cell. Mol. Life Sci. 2017, 74, 3883–3896. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- Axelrod, J.; Wurtman, R.J.; Snyder, S.H. Control of hydroxyindole O-methyltransferase activity in the rat pineal gland by environmental lighting. J. Biol. Chem. 1965, 240, 949–954. [Google Scholar] [PubMed]
- Champney, T.H.; Holtorf, A.P.; Steger, R.W.; Reiter, R.J. Concurrent determination of enzymatic activities and substrate concentrations in the melatonin synthetic pathway within the same rat pineal gland. J. Neurosci. Res. 1984, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Rosner, J.M. Ocular distribution of hydroxyindole O-methyltransferase (HIOMT) in the duck (Anas platyrhynchos). Gen. Comp. Endocrinol. 1972, 18, 407–409. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Wurtman, R.J. Hydroxyindole-O-methyltransferases in rat pineal, retina and Harderian gland. Endocrinology 1972, 91, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Tricoire, H.; Moller, M.; Chermineau, P.; Malpaux, B. Origin of cerebrospinal fluid melatonin and possible function in the integration of photoperiod. Reprod. Suppl. 2003, 61, 311–321. [Google Scholar] [PubMed]
- Reiter, R.J.; Tan, D.X.; Kim, S.J.; Cruz, M.H. Delivery of pineal melatonin to the brain and SCN: Role of canaliculi, cerebrospinal fluid, tanycytes and Virchow-Robin perivascular spaces. Brain Struct. Funct. 2014, 219, 1873–1887. [Google Scholar] [CrossRef] [PubMed]
- Legros, C.; Chesneau, D.; Boutin, J.A.; Barc, L.; Malpaux, B. Melatonin from cerebrospinal fluid but not from blood reaches sheep cerebral tissues under physiological conditions. J. Neuroendocrinol. 2014, 26, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Goldman, H.; Wurtman, R.J. Flow of blood to the pineal body of the rat. Nature 1964, 203, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Reiter, R.J. Ultrastructural observations at pineal gland capillaries in four rodent species. Am. J. Anat. 1975, 143, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A. Localization, physiological significance and possible clinical implications of gastrointestinal melatonin. Biol. Signals Recept. 2001, 10, 350–366. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.J.; Ozaki, Y.; Shakal, D.; Wurtman, R.J. Melatonin excretion of man and rats: Effect of time of day, sleep, Pinealectomy and food consumption. Int. J. Biometeorol. 1975, 19, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Tetsno, M.; Perlow, M.J.; Mishkin, M.; Markey, S.P. Light exposure reduces and pinealectomy virtually stops urinary excretion of 6-hydroxymelatonin by rhesus monkeys. Endocrinology 1982, 110, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Waldhauser, F.; Waldhauser, M.; Lieberman, H.R.; Deng, M.H.; Lynch, H.J.; Wurtman, R.J. Bioavailability of oral melatonin in humans. Neuroendocrinology 1984, 39, 307–314. [Google Scholar] [CrossRef] [PubMed]
- English, J.; Bojkowski, L.J.; Poulton, A.L.; Symons, A.M.; Arendt, J. Metabolism pharmacokinetics of melatonin in the ewe. J. Pineal Res. 1987, 4, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Qi, W.; Hanes, M.A.; Farley, N.J. High physiological levels of melatonin in the bile of mammals. Life Sci. 1999, 65, 2523–2529. [Google Scholar] [CrossRef]
- Tamura, J.; Nakamura, Y.; Korkmaz, A.; Manchester, L.C.; Tan, D.X.; Sugino, N.; Reiter, R.J. Melatonin and the ovary: Physiological and pathophysiological implications. Fertil. Steril. 2009, 92, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Coelho, L.A.; Peres, R.; Amaral, F.G.; Reiter, R.J.; Cipolla-Neto, J. Daily differential expression of melatonin-related genes and clock genes in rat cumulus-oocyte complex: Changes after pinealectomy. J. Pineal Res. 2015, 58, 490–499. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, J.; Zhang, Z.; Yang, M.; Li, Y.; Tian, X.; Ma, T.; Tao, J.; Zhu, K.; Song, Y.; et al. Mitochondria synthesize melatonin to ameliorate its function and improve mice oocyte’s quality under in vitro conditions. Int. J. Mol. Sci. 2016, 17, 939. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, G.M.; Pelham, R.W.; Pang, S.F.; Loughlin, L.L.; Wilson, K.M.; Sandock, L.; Vaughn, M.K.; Koslow, S.H.; Reiter, R.J. Nocturnal elevation of plasma melatonin and urinary 5-hydroxyindoleacetic acid in young men: Attempts at modification by brief changes in environmental lighting and sleep and by autonomic drugs. J. Clin. Endocrinol. Metab. 1976, 42, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Stehle, J.H.; Saade, A.; Rawashdeh, O.; Ackermann, K.; Jilg, A.; Sebesteny, T.; Maronde, E. A survey of molecular details in the human pineal gland in the light of phylogeny, structure function and chronobiological diseases. J. Pineal Res. 2011, 51, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Houdek, P.; Novakova, M.; Polidarova, L.; Sladek, M.; Sumova, A. Melatonin is a redundant entraining signal in the rat circadian system. Horm. Behav. 2016, 83, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pevet, P. Melatonin receptors as therapeutic targets in the suprachiasmatic nucleus. Expert Opin. Ther. Targets 2016, 20, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Korf, H.W.; Wicht, H. Synchronizing effects of melatonin on diurnal and circadian rhythms. Gen. Comp. Endocrinol. 2018, 258, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tamura, H.; Tau, D.X.; Xu, X.Y. Melatonin and the circadian system: Contributions to successful female reproduction. Fertil. Steril. 2014, 102, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Zerman, M.; Herichova, I. Melatonin and clock genes expression in the cardiovascular system. Front. Biosci. 2013, 5, 743–753. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Korkmaz, A.; Rosales-Corral, S.A. Melatonin and stable circadian rhythms optimize maternal, placental and fetal physiology. Hum. Reprod. Update 2014, 20, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin and circadian oscillators in aging—A dynamic approach to the multiply connected players. Interdiscip. Top. Gerontol. 2015, 40, 128–140. [Google Scholar] [PubMed]
- Faraut, B.; Bayon, V.; Leger, D. Neuroendocrine, immune and oxidative stress in shift workers. Sleep Med. Dev. 2013, 17, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, M.A.; Nihal, M.; Wood, G.S.; Ahmad, N. Skin, reactive oxygen species, and circadian clocks. Antioxid. Redox Signal. 2014, 20, 2982–2996. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Alatorre-Jimenez, M.; Lopez, C. Circadian dysregulation and melatonin rhythm suppression in the context of aging. In Circadian Rhythms and Their Impact on Aging; Jazwinski, S.M., Belancio, V.P., Hill, S.M., Eds.; Springer: Chambersburg, PA, USA, 2017; pp. 1–25. [Google Scholar]
- Troini, M.E.; Reiter, R.J.; Tannenbaum, M.G.; Puig-Domingo, M.; Guerrero, M.M.; Menendez-Pelaez, A. Neither the pituitary gland nor the sympathetic nervous system is responsible for eliciting the large drop in elevated rat pineal melatonin levels due to swimming. J. Neural Transm. 1988, 74, 149–160. [Google Scholar] [CrossRef]
- Wu, W.; Chen, Y.C.; Reiter, R.J. Day-night differences in the response of the pineal gland to swimming stress. Proc. Soc. Exp. Biol. Med. 1988, 187, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.M.; Llesuy, S.F.; Pasquini, J.M.; Boveris, A. Brain chemiluminescence and oxidative stress in hyperthyroid rats. Biochem. J. 1989, 263, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Sjodin, B.; Hellsten Westing, Y.; Apple, F.S. Biochemical mechanisms for oxygen free radical formation during exercise. Sport Med. 1990, 10, 236–254. [Google Scholar] [CrossRef]
- Jou, M.J.; Peng, T.I.; Hsu, L.F.; Jou, S.B.; Reiter, R.J.; Yang, C.M.; Chiao, C.C.; Lin, Y.F.; Chen, C.C. Visualization of melatonin’s multiple mitochondrial levels of protection against mitochondrial Ca(2+)-mediated permeability transition and beyond in rat brain astrocytes. J. Pineal Res. 2010, 48, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Jou, M.J.; Peng, T.I.; Reiter, R.J.; Jou, S.B.; Wu, H.Y.; Wen, S.T. Visualization of the antioxidant effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J. Pineal Res. 2004, 37, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Jou, M.J.; Peng, T.I.; Yu, P.Z.; Jou, S.B.; Reiter, R.J.; Chen, J.Y.; Wu, H.Y.; Chen, C.C.; Hsu, L.F. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J. Pineal Res. 2007, 43, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Martin, M.; Macias, M.; Escames, G.; Leon, J.; Khaldy, H.; Reiter, R.J. Melatonin, mitochondria, and cellular bioenergetics. J. Pineal Res. 2001, 30, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Rodriguez, M.I.; Lopez, L.C. Melatonin role in the mitochondrial function. Front. Biosci. 2007, 12, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Symbiotic theory of the origin of eukaryotic organelles: Criteria for proof. Symp. Soc. Exp. Biol. 1975, 29, 21–38. [Google Scholar]
- Archibald, J.M. Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Poeggeler, B.; Alvares, F.L.; Ogden, G.B.; Reiter, R.J. Melatonin immunoreactivity in the photosynthetic prokaryote Rhodospirillum rubrum: Implications for an ancient antioxidant system. Cell. Mol. Biol. Res. 1995, 41, 391–395. [Google Scholar] [PubMed]
- Tilden, A.R.; Becker, M.A.; Amma, L.L.; Arciniega, J.; McGaw, A.K. Melatonin production in an aerobic photosynthetic bacterium: An evolutionarily early association with darkness. J. Pineal Res. 1997, 22, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Konopelski, P.; Ufnal, M. Indoles—Gut bacteria metabolites of tryptophan with pharmacotherapeutic potential. Curr. Drug Metab. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Garcia, J.A.; Escames, G.; Ortiz, F.; Lopez, A.; Doerrier, C.; Garcia-Corzo, L.; Lopez, L.C.; Reiter, R.J.; Acuna-Castroviejo, D. Extrapineal melatonin: Analysis of its subcellular distribution and daily fluctuations. J. Pineal Res. 2012, 52, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Hevia, D.; Gonzalez-Menendez, P.; Quiros-Gonzalez, I.; Miar, A.; Rodriguez-Garcia, A.; Tan, D.X.; Reiter, R.J.; Mayo, J.C.; Sainz, R.M. Melatonin uptake through glucose transporters: A new target for melatonin inhibition of cancer. J. Pineal Res. 2015, 58, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Sun, P.; Yan, C.; Ke, M.; Jiang, X.; Xiong, L.; Ren, W.; Hirata, K.; Yamamoto, M.; Fan, S.; et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Wang, C.; Yu, Z.; Peng, Y.; Wang, S.; Feng, S.; Zhang, S.; Tian, X.; Sun, C.; Liu, K.; et al. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: An implication of the therapeutic potential. J. Pineal Res. 2017, 62, e12390. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, N.A.; Balogh, I.; Somogyi, E.; Satonyi, P. Cytochemical investigation of acetyl-serotonin-transferase activity in the pineal gland. Cell. Mol. Biol. Incl. Cyto-Enzymol. 1979, 25, 259–262. [Google Scholar] [PubMed]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome C release. Proc. Nat. Acad. Sci. USA 2017, 115, E1944. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Macias, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuna-Castroviejo, D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Aranda, A.; Fernandez-Vazquez, G.; Mohammad A-Serrano, M.; Reiter, R.J.; Agil, A. Melatonin improves mitochondrial function in inguinal white adipose tissue of Zucker diabetic fatty rats. J. Pineal Res. 2014, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Lopez, L.C.; Escames, G.; Lopez, A.; Garcia, J.A.; Reiter, R.J. Melatonin-mitochondria interplay in health and disease. Curr. Top. Med. Chem. 2011, 11, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Qin, Q.; Qian, Z.H.; Shi, M.; Deng, Q.C.; Zhu, W.P.; Zhang, H.; Tao, X.M.; Liu, Y. Protective effects of melatonin on ischemia-reperfusion induced myocardial damage and hemodynamic recovery in rats. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3681–3686. [Google Scholar] [PubMed]
- Miller, E.; Morel, A.; Saso, L.; Saluk, J. Melatonin redox activity. Its potential clinical applications in neurodegenerative disorders. Curr. Top. Med. Chem. 2015, 15, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Skobowiat, C.; Brozyna, A.A.; Janyetovic, Z.; Jeayeng, S.; Oak, A.S.W.; Kim, T.K.; Panich, U.; Reiter, R.J.; Slominski, A.T. Melatonin and its derivatives counteract the ultraviolet B radiation-induced damage in human and porcine skin ex vivo. J. Pineal Res. 2018, e312501. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, K.; Fraczyk, T.; Maliniska, D.; Drozak, J.; Bryla, J. Melatonin attenuates diabetes-induced oxidative stress in rabbits. J. Pineal Res. 2006, 40, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Urata, Y.; Honma, S.; Goto, S.; Todoroki, S.; Iida, T.; Cho, S.; Honma, K.; Kondo, T. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic. Biol. Med. 1999, 27, 838–847. [Google Scholar] [CrossRef]
- Esteban-Zubero, E.; Garcia-Gil, F.A.; Lopez-Pingarron, L.; Alatorre-Jimenez, M.A.; Inigo-Gil, P.; Tan, D.X.; Garcia, J.J.; Reiter, R.J. Potential benefits of melatonin in organ transplantation: A review. J. Endocrinol. 2016, 229, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Cristofanon, S.; Patemoster, L.; D’Alessio, M.; DeNicola, M.; Cerella, C.; Dicato, M.; Diederich, M.; Ghibelli, L. Melatonin antagonizes the intrinsic pathway of apoptosis via mitochondrial targeting of Bcl-2. J. Pineal Res. 2008, 44, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Paredes, S.D.; Forman, K.A.; Garcia, C.; Vara, E.; Escames, G.; Tresguerres, J.A. Protective actions of melatonin and growth hormone on the aged cardiovascular system. Horm. Mol. Biol. Clin. Investig. 2014, 18, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Ordonez, R.; Reiter, R.J.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal Res. 2015, 59, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xin, Z.; Di, W.; Yan, X.; Li, X.; Reiter, R.J.; Yang, Y. Melatonin and mitochondrial function during ischemia/reperfusion injury. Cell. Mol. Life Sci. 2017, 74, 3989–3998. [Google Scholar] [CrossRef] [PubMed]
- Tatone, C.; Di Emidio, G.; Barbonetti, A.; Carta, G.; Luciano, A.M.; Falone, S.; Amicarelli, F. Sirtuins in gamete biology and reproductive physiology: Emerging roles and therapeutic potential in female and male infertility. Hum. Reprod. Update 2018, 24, 267–289. [Google Scholar] [CrossRef] [PubMed]
- Watroba, M.; Szukiewicz, D. The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 2016, 61, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathophysiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Bin, P.; Wang, T.; Ren, W.; Zhong, J.; Liang, J.; Hu, C.A.; Zeng, Z.; Yin, Y. DNA methylation and the potential role of methyl-containing nutrients in cardiovascular diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1670815. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Taleb-Belkadi, O.; Chaib, H.; Zemour, L.; Fatah, A.; Chafi, B.; Mekki, K. Lipid prolife, inflammation, and oxidative status in peri- and postmenopausal women. Gynecol. Endocrinol. 2016, 32, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Pertynska-Marczewska, M.; Diamanti-Kandarakis, E. Aging ovary and the role for advanced glycation end products. Menopause 2017, 24, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Takasaki, A.; Miwa, I.; Taniguchi, K.; Maekawa, R.; Asada, H.; Taketani, T.; Matsuoka, A.; Yamagata, Y.; Shimamura, K.; et al. Oxidative stress impairs oocyte quality and melatonin protects oocytes from free radical damage and improves fertilization rate. J. Pineal Res. 2008, 44, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Budani, M.C.; Carletti, E.; Tiboni, G.M. Cigarette smoke is associated with altered expression of antioxidant enzymes in granulosa cells from women undergoing in vitro fertilization. Zygote 2017, 25, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Ripperdan, R.; Ortiz, L.; Luderer, U. Very low doses of heavy oxygen ion radiation induce premature ovarian failure. Reproduction 2017, 154, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gao, Y.Y.; Chen, L.; Nei, Z.W.; Cheng, W.; Liu, X.; Schatten, H.; Zhang, X.; Miao, Y.L. Melatonin prevents postovulatory oocyte aging and promotes subsequent embryonic development in the pig. Aging 2017, 9, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Zhao, S.; Sun, Y.; Jiang, X.; Hao, H.; Du, W.; Zhu, H. Protective effects of melatonin on the in vitro developmental competence of bovine oocytes. Anim. Sci. J. 2018, 89, 648–660. [Google Scholar] [CrossRef] [PubMed]
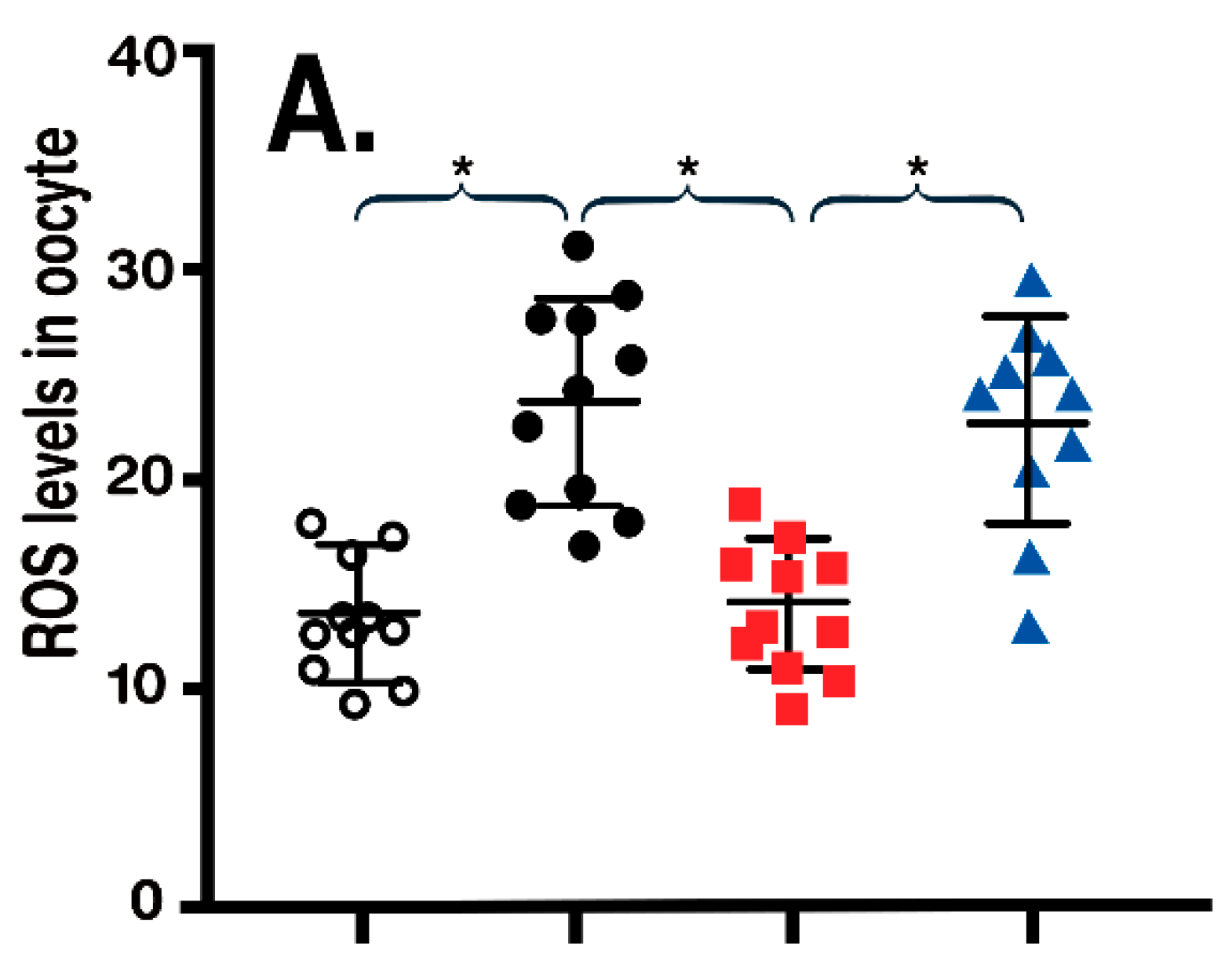
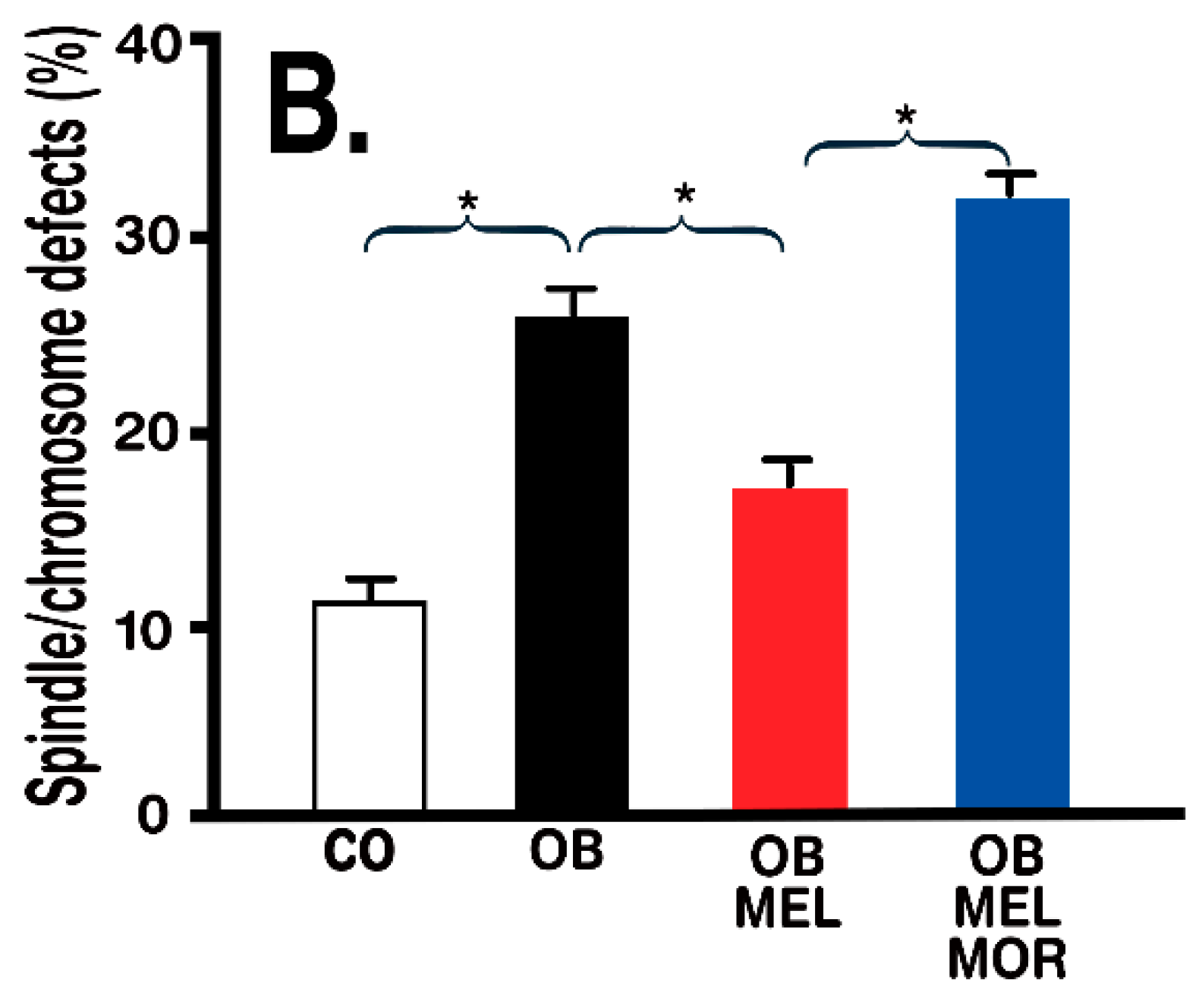
- Han, L.; Wang, H.; Li, L.; Li, X.; Ge, J.; Reiter, R.J.; Wang, Q. Melatonin protects against maternal obesity-associated oxidative stress and meiotic defects in oocytes via the SIRT3-SOD2-dependent pathway. J. Pineal Res. 2017, 63, 12431. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Peng, W.; Yin, S.; Zhao, J.; Fu, B.; Zhang, J.; Mao, T.; Wu, H.; Zhang, Y. Melatonin improves age-induced fertility decline and attenuates ovarian mitochondrial oxidative stress in mice. Sci. Rep. 2016, 6, 35165. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Kawamoto, M.; Sato, S.; Tamura, I.; Maekawa, R.; Taketani, T.; Aasada, H.S.; Takaki, E.; Nakai, A.; Reiter, R.J.; et al. Long term melatonin treatment delays ovarian aging. J. Pineal Res. 2017, 62, e12381. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Y.; Feng, Y.; Yan, J.; Fan, C.; Jiang, S.; Qu, Y. A review of melatonin in hepatic ischemia/reperfusion injury and clinical liver disease. Ann. Med. 2014, 46, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Halladin, N.L. Oxidative and inflammatory biomarkers of ischemia and reperfusion injuries. Dan. Med. J. 2015, 62, B5054. [Google Scholar] [PubMed]
- Paterniti, I.; Cordaro, M.; Esposito, E.; Cuzzocrea, S. The antioxidant property of melatonin against brain ischemia. Exp. Rev. Neurother. 2016, 16, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gong, B.; Duan, W.; Fan, C.; Zhang, J.; Li, Z.; Xue, X.; Xu, Y.; Meng, D.; Li, B.; et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: Role of AMPK-PGC-1α-SIRT3 signaling. Sci. Rep. 2017, 7, 41337. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.; Li, B.; Duan, W.; Jing, L.; Zhang, B.; Zhang, M.; Yu, L.; Liu, Z.; Yu, B.; Ren, K.; et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J. Pineal Res. 2017, 63, e12419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lin, J.; Wang, S.; Cheng, Z.; Hu, J.; Wang, T.; Man, W.; Yin, T.; Guo, W.; Gao, E.; et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt 3 signaling. J. Pineal Res. 2017, 63, e12418. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Arthrosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.K.; Sun, C.K.; Chang, L.T.; Wu, C.J. Strong correlation between serum levels of inflammatory mediators and their distribution in infarct-related coronary artery. Circ. J. 2006, 70, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Kim, S.J.; Manchester, L.C.; Qi, W.; Garcia, J.J.; Cabrera, J.C.; El-Sokkary, G.; Rouvier-Garay, V. Augmentation of indices of oxidative damage in life-long melatonin-deficient rats. Mech. Ageing Rev. 1999, 110, 157–173. [Google Scholar] [CrossRef]
- Garcia, J.J.; Lopez-Pingarron; Almeida-Souza, P.; Tres, A.; Escudero, P.; Garcia-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramirez, J.M.; Bernal-Perez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.Y.; Sun, C.K.; Sung, P.H.; Chen, K.H.; Chua, S.; Sheu, J.J.; Chung, S.Y.; Chai, H.T.; Chen, Y.L.; Huang, T.H.; et al. Daily melatonin protects the endothelial lineage and functions integrity against the aging process, oxidative stress, and toxic environment and restores blood flow in critical limb ischemia area in mice. J. Pineal Res. 2018, 64, e12489. [Google Scholar] [CrossRef] [PubMed]
- Pereira, H.A.; Leite Ade, S.; Charone, S.; Lobo, J.G.; Cestari, T.M.; Peres-Buzalaf, C.; Buzalaf, M.A. Proteomic analysis of liver in rats chronically exposed to fluoride. PLoS ONE 2013, 8, e75343. [Google Scholar] [CrossRef]
- Bharti, V.K.; Srivastava, R.S.; Kumar, S.; Bag, S.; Majumdar, A.C.; Singh, G.; Pandi-Perumal, S.R.; Brown, G.M. Effects of melatonin and epiphyseal proteins on fluoride-induced adverse changes in antioxidant status of heart, liver, and kidney of rat. Adv. Pharmacol. Sci. 2014, 2014, 532969. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhao, J.; Fu, B.; Li, D.; Mao, T.; Peng, W.; Wu, H.; Zhang, Y. Melatonin-mediated upregulation of Sirt3 attenuates sodium fluoride-induced hepatotoxicity by activating the MT1-PI3K/AKT-PGC-1α signaling pathway. Free Radic. Biol. Med. 2017, 112, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qing, W.; Sun, M.; Lv, L.; Guo, D.; Jiang, Y. Melatonin protects hepatocytes against bile-acid-induced mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway. Free Radic. Res. 2015, 49, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Kongo, M.; Kishikawa, T. Melatonin exerts a therapeutic effect on cholestatic liver injury in rats with bile duct ligation. J. Pineal Res. 2003, 34, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Kongo-Nishimura, M.; Imai, Y.; Matsura, T.; Kitagawa, A.; Yamada, K. Alpha-tocopherol protects against alpha-naphthylisothiocyanate-induced hepatotoxicity in rats less effectively than melatonin. Chem. Biol. Interact. 2006, 161, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Leon, J.; Carazo, A.; Khaldy, H. Mitochondrial regulation by melatonin and its metabolites. Adv. Exp. Med. Biol. 2003, 527, 549–557. [Google Scholar] [PubMed]
- Acuna-Castroviejo, D.; Rahim, I.; Acuna-Fernandez, C.; Fernandez-Ortiz, M.; Solera-Marin, J.; Sayed, R.K.A.; Diaz-Casado, M.E.; Rusanova, I.; Lopez, L.C.; Escames, G. Melatonin, clock genes and mitochondria in sepsis. Cell. Mol. Life Sci. 2017, 74, 3965–3987. [Google Scholar] [CrossRef] [PubMed]
- Bordo, D. Structure and evolution of human sirtuins. Curr. Drug Targets 2013, 14, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Park, J.E.; Jang, C.Y. Sirt3 controls chromosome alignment by regulating spindle dynamics during mitosis. Biochem. Biophys. Res. Commun. 2014, 444, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Zwaans, B.M. SIRT3: As simple as it seems. Gerontology 2014, 60, 56–64. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, E.; Peterson, B.S.; Bomze, H.M.; Hirschey, M.D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 2015, 26, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Perico, L.; Macconi, D. Mitochondrial dynamics are linked to longevity and protect from end-organ injury: The emerging role of sirtuin 3. Antioxid. Redox Signal. 2016, 25, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.F.; Chen, H.Z.; Lui, D.P. Mitochondrial sirtuins in cardiometabolic diseases. Clin. Sci. 2017, 131, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging. Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Proietti, S.; Cucina, A.; Minini, M.; Bizzarri, M. Melatonin, mitochondria and the cancer cell. Cell. Mol. Life Sci. 2017, 74, 4015–4025. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to the post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64, 12447. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.; Zhou, X.; Tan, D.X. Role of SIRT3/SOD2 signaling in mediating the antioxidant actions of melatonin in mitochondria. Curr. Trends Endocrinol. 2017, 9, 45–49. [Google Scholar]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics decay in aging: Beneficial effect of melatonin. Cell. Mol. Life Sci. 2017, 74, 3897–3911. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin/Process | Tissue Protected | Reference |
|---|---|---|
| Carbon tetrachloride | Liver | [69] |
| Hyperbaric oxygen | Lung and brain | [70] |
| 2-Nitropropane | Lipid and DNA | [71] |
| Cerulein | Pancreas | [72] |
| Quinolinic acid | Brain | [73] |
| δ-Aminolevulinic acid | Lipid and DNA | [74] |
| Neonatal asphyxia (human) | Lipid | [75] |
| Ochratoxin A | Liver and kidney | [76] |
| Ultraviolet radiation | Leucocytes | [77] |
| Respiratory distress syndrome (human) | Lung | [78] |
| 1-Methyl-4-phenyl-1,2,3,6-tetrahydro pyridine | Brain | [79] |
| Organ transplantation | Liver | [80] |
| Diquat | Lipid | [81] |
| Cerebral hypoperfusion | Brain | [82] |
| Aspirin (human) | Gastrointestinal tract | [83] |
| Opisthorchis viverrini (liver fluke) | Liver | [84] |
| Methamphetamine | Brain | [85] |
| Doxorubicin | Heart | [86] |
| Cobra toxin | Multiple tissues | [87] |
| Strenuous exercise | Skeletal muscle | [88] |
| Ionizing radiation | Oral mucosa | [89] |
| Ischemia/reperfusion | Heart | [90] |
| Cadmium | Brain | [91] |
| Phosphine | Heart | [92] |
| Neonatal sepsis (human) | Neonate | [93] |
| ST-segment elevation, myocardial infarction (human) | Heart | [94] |
| Ischemia/reperfusion | Brain | [95] |
| p-Cresol | Mesenchymal stem cells | [96] |
| Arsenic trioxide | Liver | [97] |
| Type 2 diabetes | Cardiovascular system | [98] |
| Hexavalent chromium | Spermatogonia | [99] |
| Ischemia/reperfusion | Lung | [100] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Jou, M.-J.; Acuna-Castroviejo, D. Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. Int. J. Mol. Sci. 2018, 19, 2439. https://doi.org/10.3390/ijms19082439
Reiter RJ, Tan DX, Rosales-Corral S, Galano A, Jou M-J, Acuna-Castroviejo D. Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. International Journal of Molecular Sciences. 2018; 19(8):2439. https://doi.org/10.3390/ijms19082439
Chicago/Turabian StyleReiter, Russel J., Dun Xian Tan, Sergio Rosales-Corral, Annia Galano, Mei-Jie Jou, and Dario Acuna-Castroviejo. 2018. "Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3" International Journal of Molecular Sciences 19, no. 8: 2439. https://doi.org/10.3390/ijms19082439
APA StyleReiter, R. J., Tan, D. X., Rosales-Corral, S., Galano, A., Jou, M.-J., & Acuna-Castroviejo, D. (2018). Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. International Journal of Molecular Sciences, 19(8), 2439. https://doi.org/10.3390/ijms19082439