Novel Insights from Comparative In Silico Analysis of Green Microalgal Cellulases

 ,
, 

 ,
, 


Abstract
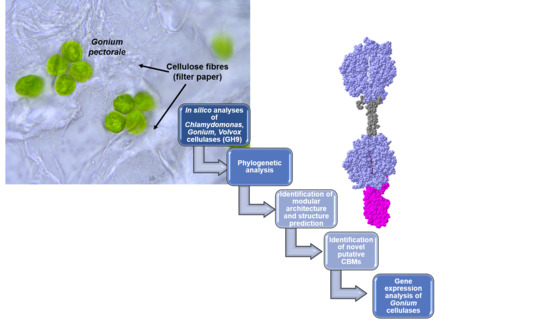
1. Introduction
2. Results and Discussion
2.1. Algal Cellulases Belong to Glycosyl Hydrolase Family 9
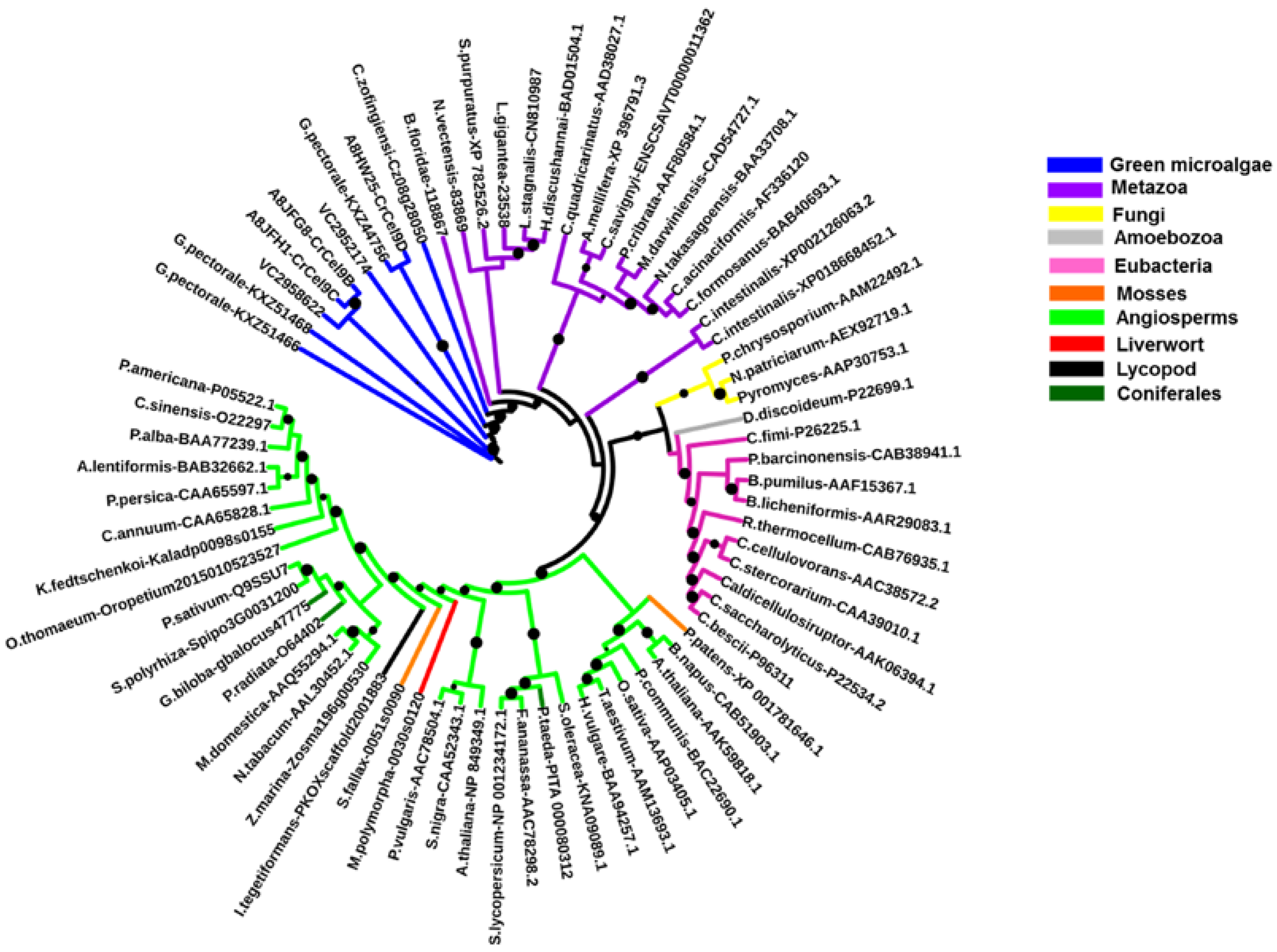
2.2. Algal Cellulases Are Closest to Invertebrate Metazoan GH9 Enzymes
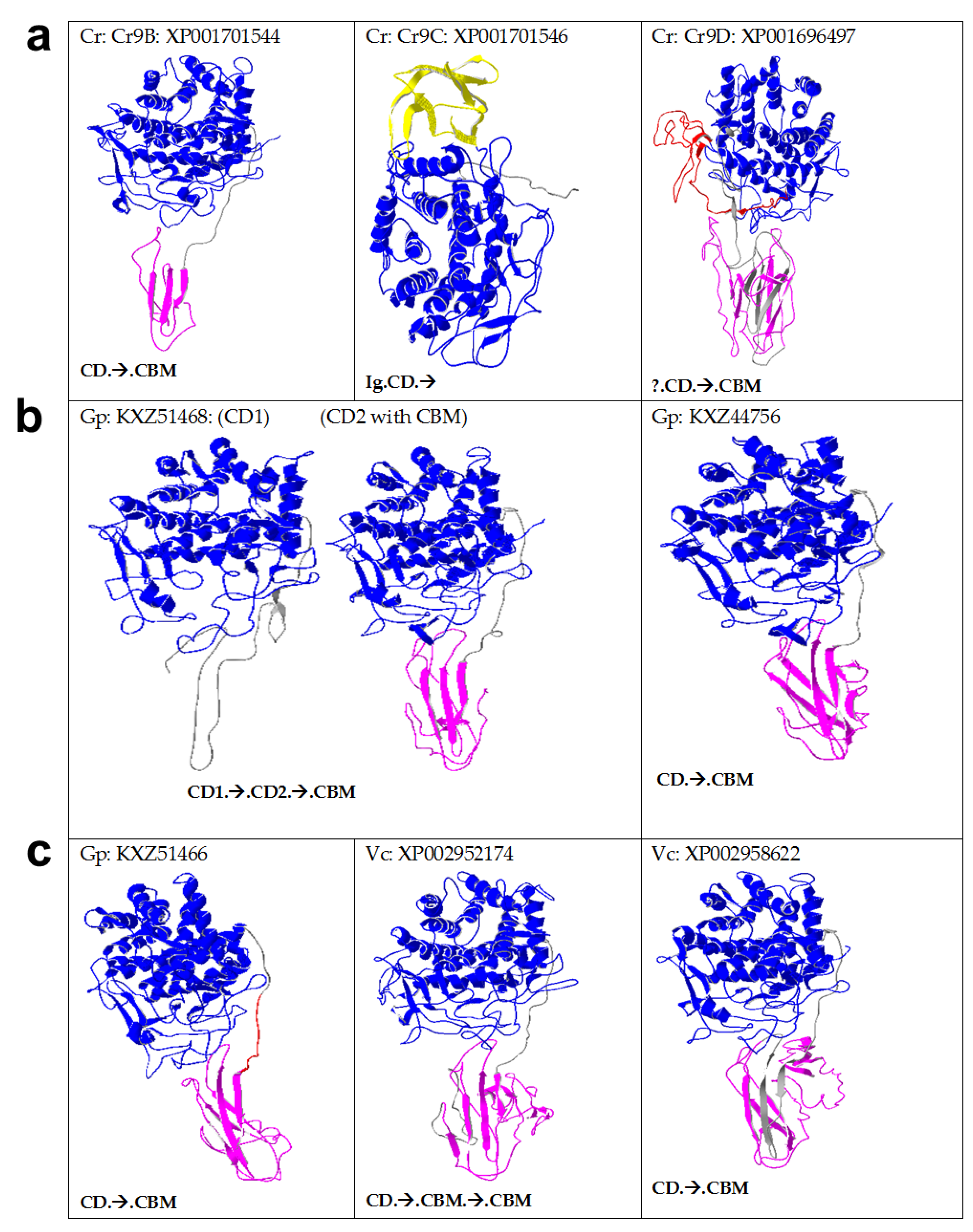
2.3. Algal Cellulases Are Multimodular
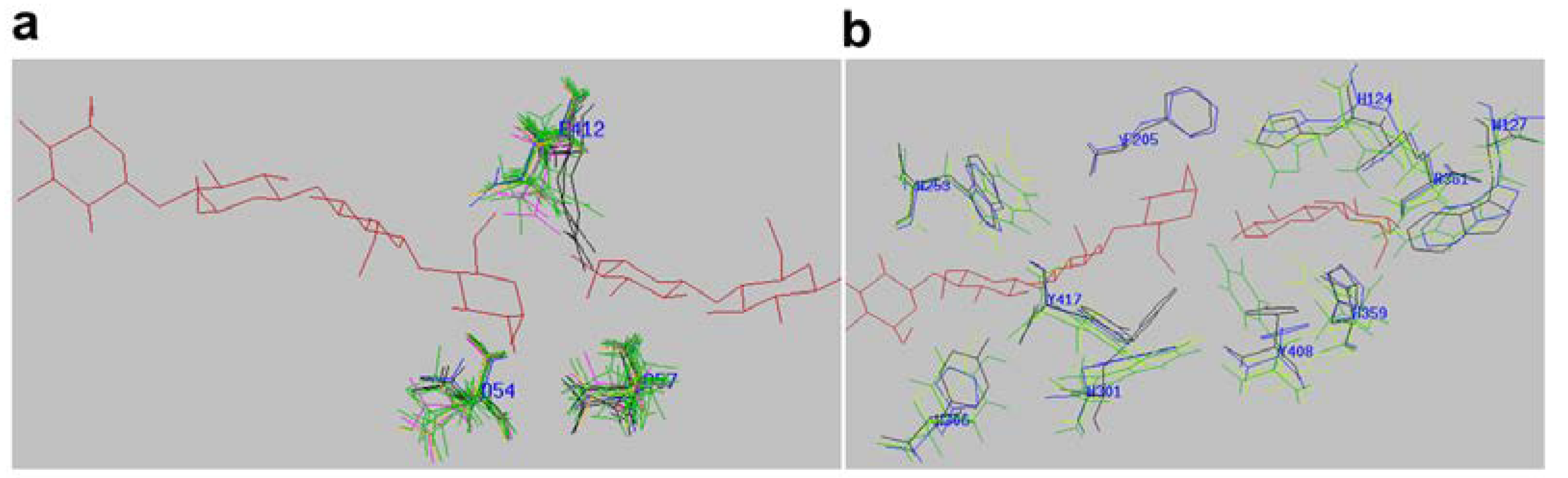
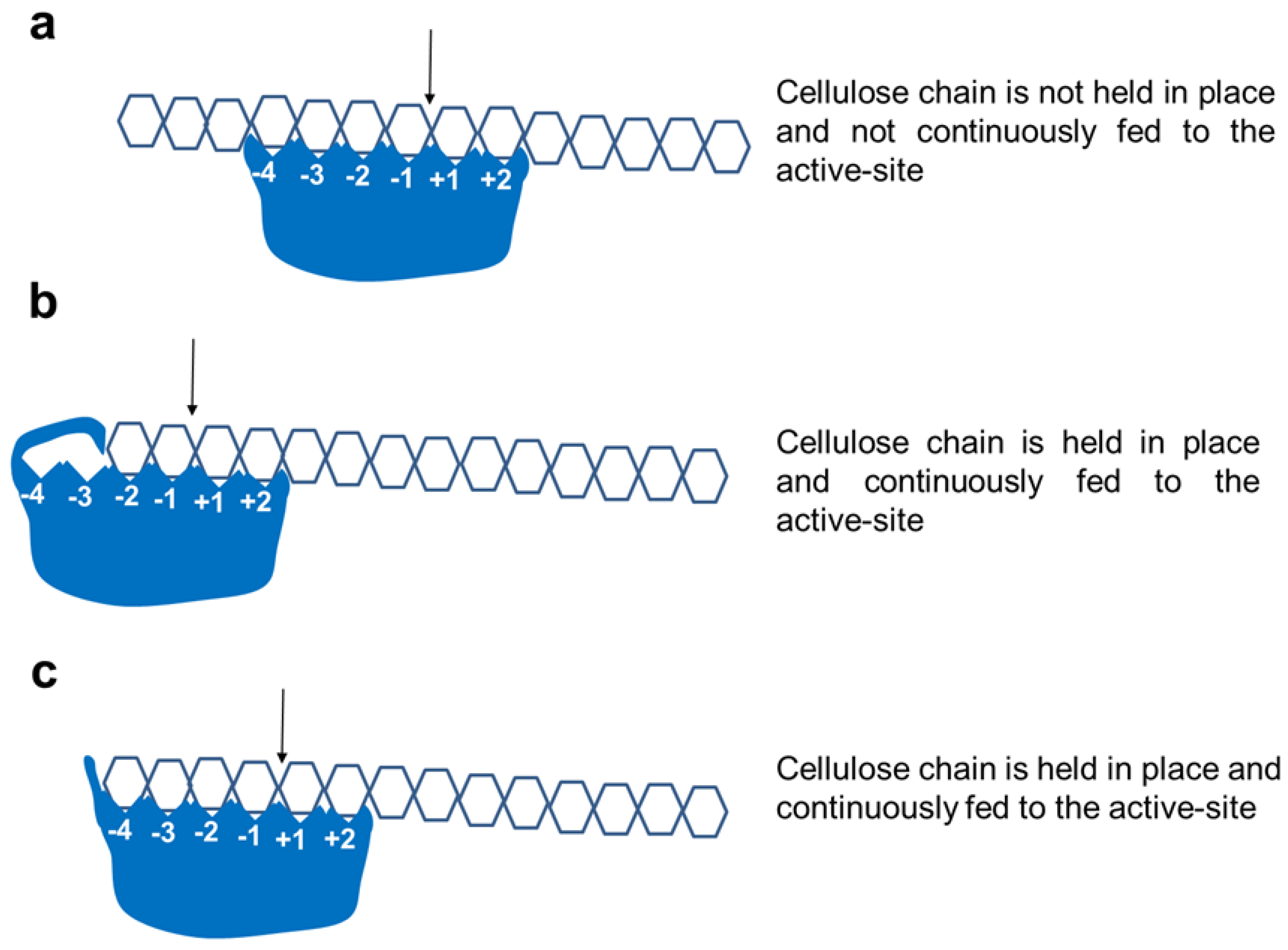
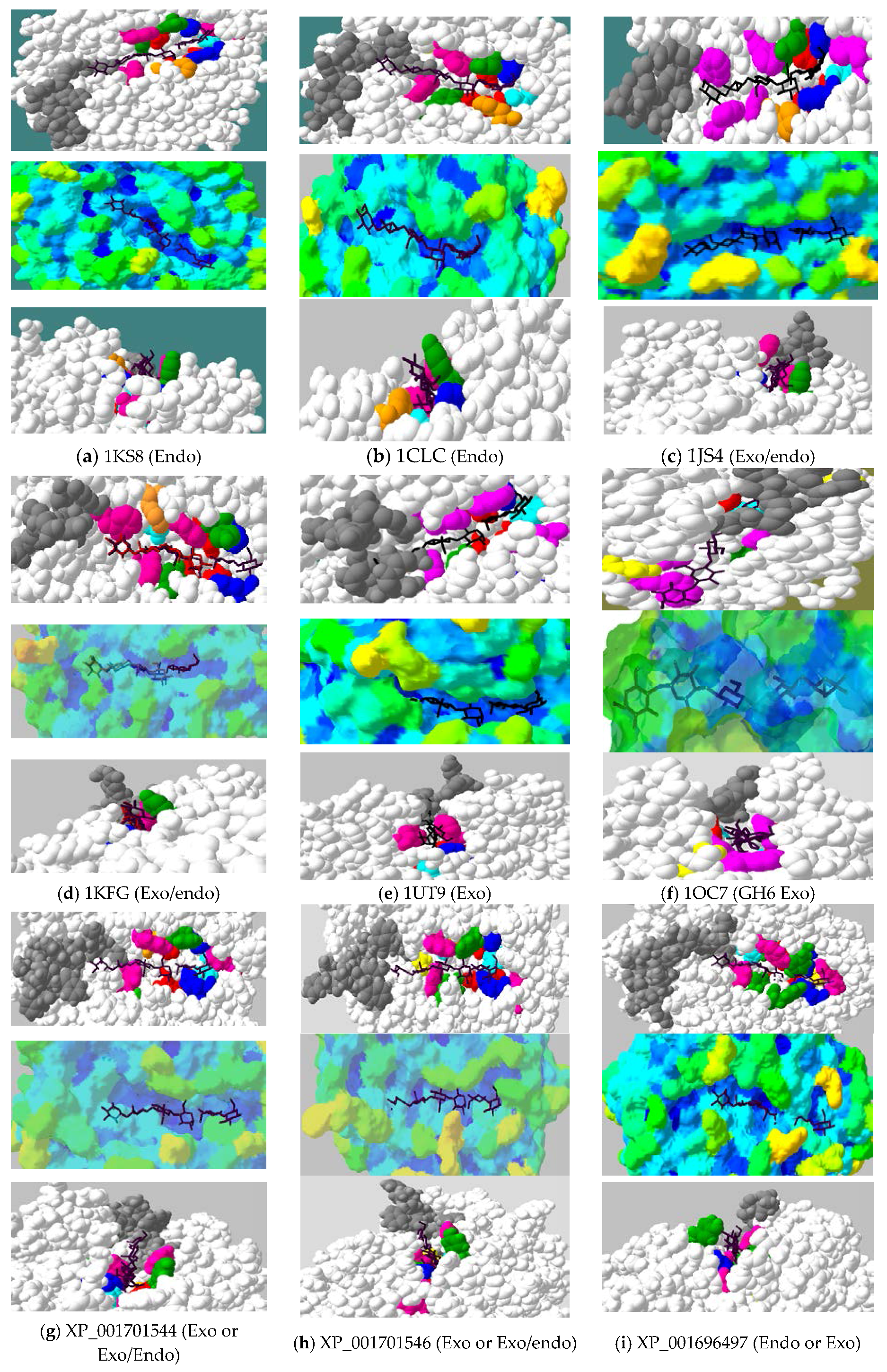
2.4. Active-Site Architecture Shows Different Types of Cellulolytic Activities in Algal GH9 Cellulases
2.5. Novel Cysteine-Rich CBM in Algal Cellulases
2.6. Algal Cellulases Have PS-Rich Linkers
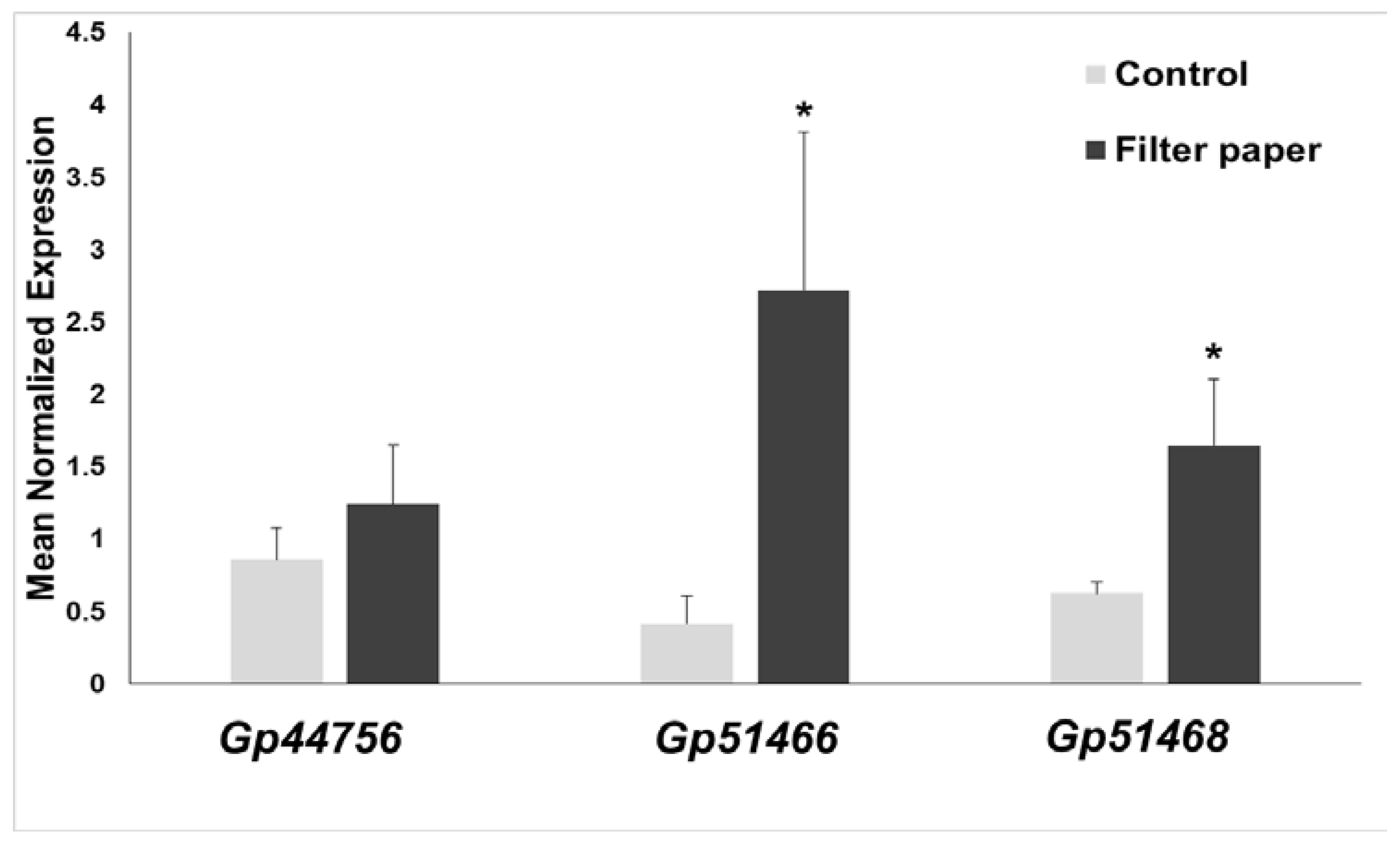
2.7. Expression of Cellulases in Gonium Pectorale (Gp)
3. Materials and Methods
3.1. Computational Methods
3.2. Growth of G. pectorale, RNA Extraction, cDNA Synthesis and RT-qPCR
4. Conclusions and Future Direction
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Guerriero, G.; Hausman, J.F.; Strauss, J.; Ertan, H.; Siddiqui, K.S. Lignocellulosic biomass: Biosynthesis, degradation, and industrial utilization. Eng. Life Sci. 2016, 16, 1–16. [Google Scholar] [CrossRef]
- Guerriero, G.; Siddiqui, K.S. Modular and Multifunctional Enzyme Systems for Plant Cell Wall Degradation: Diversity, Synergy, Chimeras and Magnetic-Glycosidases. Ref. Modul. Life Sci 2017. [Google Scholar] [CrossRef]
- Li, Y.; Irwin, D.C.; Wilson, D.B. Processivity, substrate binding, and mechanism of cellulose hydrolysis by Thermobifida fusca Cel9A. Appl. Environ. Microbiol. 2007, 73, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.J.; Zhang, H.; Longmire, A.G.; Moon, Y.H.; Hutcheson, S.W. Processive endoglucanases mediate degradation of cellulose by Saccharophagus degradans. J. Bacteriol. 2009, 191, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Henrissat, B. Structure and mechanism of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucl. Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, G.; Hausman, J.F.; Strauss, J.; Ertan, H.; Siddiqui, K.S. Destructuring plant biomass: Focus on fungal and extremophilic cell wall hydrolases. Plant. Sci. 2015, 234, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Schweiger-Hufnagel, U.; Ono, T.; Izumi, K.; Hufnagel, P.; Morita, N.; Kaga, H.; Morita, M.; Hoshino, T.; Yumoto, I.; Matsumoto, N.; et al. Identification of the extracellular polysaccharide produced by the snow mold fungus Microdochium Nivale. Biotechnol. Lett. 2000, 22, 183–187. [Google Scholar] [CrossRef]
- Brummell, D.A.; Lashbrook, C.C.; Bennett, A.B. Plant endo-1, 4-b-d-glucanases. In Enzymatic Conversion of Biomass for Fuels Production; ACS Symp: Washington DC, WA, USA, 1994; Volume 566, pp. 100–129.4-b-d-glucanases. In Enzymatic Conversion of Biomass for Fuels Production; ACS Symp: Washington DC, WA, USA, 1994; Volume 566, pp. 100–129. [Google Scholar]
- Weimer, P.J. Cellulose Degradation by Ruminal Microorganisms. Crit. Rev. Biotechnol. 1992, 12, 189–233. [Google Scholar] [CrossRef]
- Watanabe, H.; Tokuda, G. Animal cellulases. Cell. Mol. Life Sci. 2001, 58, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Blifernez-Klassen, O.; Klassen, V.; Doebbe, A.; Kersting, K.; Grimm, P.; Wobbe, L.; Kruse, O. Cellulose degradation and assimilation by the unicellular phototrophic eukaryote Chlamydomonas reinhardtii. Nat. Commun. 2012, 3, 1214. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, E.R.; Marriage, T.N.; Ferris, P.J.; Hamaji, T.; Toyoda, A.; Fujiyama, A.; Neme, R. The Gonium pectorale genome demonstrates co-option of cell cycle regulation during the evolution of multicellularity. Nat. Commun. 2016, 7, 11370. [Google Scholar] [CrossRef] [PubMed]
- Domozych, D.S.; Ciancia, M.; Fangel, J.U.; Mikkelsen, M.D.; Ulvskov, P.; Willats, W.G. The cell walls of green algae: A journey through evolution and diversity. Front. Plant. Sci. 2012, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.; Blaxter, M. Ancient origin of glycosyl hydrolase family 9 cellulase genes. Mol. Biol. Evol. 2005, 22, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Sakon, J.; Irwin, D.; Wilson, D.B.; Karplus, P.A. Structure and mechanism of exo/endocellulase E4 from Thermomonospora fusca. Nat. Struct. Biol. 1997, 4, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Khademi, S.; Guarino, L.A.; Watanabe, H.; Tokuda, G.; Meyer, E.F. Structure of an endoglucanase from termite, Nasutitermes takasagoensis. Acta Cryst. D Biol. Cryst. 2002, 58 Pt 4, 653–659. [Google Scholar] [CrossRef]
- Zhou, W.; Irwin, D.C.; Escovar-Kousen, J.; Wilson, D.B. Kinetic studies of Thermobifida fusca Cel9A active site mutant enzymes. Biochemistry 2004, 43, 9655–9663. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucl. Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef] [PubMed]
- Parsiegla, G.; Belaïch, A.; Belaïch, J.P.; Haser, R. Crystal structure of the cellulase Cel9M enlightens structure/function relationships of the variable catalytic modules in glycoside hydrolases. Biochemistry 2002, 41, 11134–11142. [Google Scholar] [CrossRef] [PubMed]
- Schubot, F.D.; Kataeva, I.A.; Chang, J.; Shah, A.K.; Ljungdahl, L.G.; Rose, J.P.; Wang, B.C. Structural basis for the exocellulase activity of the cellobiohydrolase CbhA from Clostridium thermocellum. Biochemistry 2004, 43, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Ravachol, J.; Borne, R.; Tardif, C.; de Philip, P.; Fierobe, H.P. Characterization of all family-9 glycoside hydrolases synthesized by the cellulosome-producing bacterium Clostridium cellulolyticum. J. Biol. Chem. 2014, 289, 7335–7348. [Google Scholar] [CrossRef] [PubMed]
- Mandelman, D.; Belaich, A.; Belaich, J.P.; Aghajari, N.; Driguez, H.; Haser, R. X-Ray crystal structure of the multidomain endoglucanase Cel9G from Clostridium cellulolyticum complexed with natural and synthetic cello-oligosaccharides. J. Bacteriol. 2003, 185, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Fontes, C.M.G.A.; Gilbert, H.J. Cellulosomes: Highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Ann. Rev. Biochem. 2010, 79, 655–681. [Google Scholar] [CrossRef] [PubMed]
- Sammond, D.W.; Payne, C.M.; Brunecky, R.; Himmel, M.E.; Crowley, M.F.; Beckham, G.T. Cellulase linkers are optimized based on domain type and function: Insights from sequence analysis, biophysical measurements, and molecular simulation. PLoS ONE 2012, 7, e48615. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.M.; Resch, M.G.; Chen, L.; Crowley, M.F.; Himmel, M.E.; Taylor, L.E.; Sandgren, M.; Ståhlberg, J.; Stals, I.; Tan, Z.; et al. Glycosylated linkers in multimodular lignocellulose-degrading enzymes dynamically bind to cellulose. Proc. Natl. Acad. Sci. USA 2013, 110, 14646–14651. [Google Scholar] [CrossRef] [PubMed]
- Teo, V.S.; Saul, D.J.; Bergquist, P.L. Cel A, Another Gene Coding for A Multidomain Cellulase from the Extreme Thermophile Caldocellum Saccharolyticum. Appl. Microbiol. Biotechnol. 1995, 43, 291–296. [Google Scholar] [CrossRef]
- Zverlov, V.V.; Velikodvorskaya, G.V.; Schwarz, W.H.; Bronnenmeier, K.; Kellermann, J.; Staudenbauer, W.L. Multidomain structure and cellulosomal localization of the Clostridium thermocellum cellobiohydrolase CbhA. J. Bacteriol. 1998, 180, 3091–3099. [Google Scholar] [PubMed]
- Tian, L.; Liu, S.; Wang, S.; Wang, L. Ligand-binding specificity and promiscuity of the main lignocellulolytic enzyme families as revealed by active-site architecture analysis. Sci. Rep. 2016, 6, 23605. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, N.R.; Henrissat, B.; Kilburn, D.G.; Miller, R.C., Jr.; Warren, R.A. Domains in microbial beta-1, 4-glycanases: Sequence conservation, function, and enzyme families. Microbiol. Rev. 1991, 155, 303–315. [Google Scholar]
- Wolfrom, M.L.; Thompson, A.; Timberlake, C.E. Comparative hydrolysis rates of the reducing disaccharides of d-glucopyranose. Cereal. Chem. 1963, 40, 82–86. [Google Scholar]
- Behera, B.C.; Sethi, B.R.; Mishra, R.R.; Dutta, S.K.; Thatoi, H.N. Microbial cellulases-Diversity and biotechnology with reference to mangrove environment: A review. J. Gen. Eng. Biotechnol. 2017, 15, 197–210. [Google Scholar] [CrossRef]
- Varrot, A.; Frandsen, T.P.; von Ossowski, I.; Boyer, V.; Cottaz, S.; Driguez, H.; Schülein, M.; Davies, G.J. Structural basis for ligand binding and processivity in cellobiohydrolase Cel6A from Humicola insolens. Structure 2003, 11, 855–864. [Google Scholar] [CrossRef]
- Tomme, P.; Kwan, E.; Gilkes, N.R.; Kilburn, D.G.; Warren, R.A. Characterization of CenC, an enzyme from Cellulomonas fimi with both endo-and exoglucanase activities. J. Bacteriol. 1996, 178, 4216–4223. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.; Shin, D.H.; Zhang, S.; Barr, B.K.; Sakon, J.; Karplus, P.A.; Wilson, D.B. Roles of the catalytic domain and two cellulose binding domains of Thermomonospora fusca E4 in cellulose hydrolysis. J. Bacteriol. 1998, 180, 1709–1714. [Google Scholar] [PubMed]
- Pages, S.; Kester, H.C.M.; Visser, J.; Benen, J.A.E. Changing a single amino acid residue switches processive and non-processive behavior of Aspergillus niger endopolygalacturonase I and II. J. Biol. Chem. 2001, 276, 33652–562001. [Google Scholar] [CrossRef] [PubMed]
- Guérin, D.M.; Lascombe, M.B.; Costabel, M.; Souchon, H.; Lamzin, V.; Béguin, P.; Alzari, P.M. Atomic (0.94 A) resolution structure of an inverting glycosidase in complex with substrate. J. Mol. Biol. 2002, 316, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.J.; Chen, G.J.; Wang, T.H.; Zhang, Y.S.; Liu, J.; Sheng, W. Non-hydrolytic Disruption of Crystalline Structure of Cellulose by Cellulose Binding Domain and Linker Sequence of Cellobiohydrolase I from Penicillium janthinellum. Acta Biochim. Biophys. Sin. 2001, 33, 13–18. [Google Scholar] [PubMed]
- Petkun, S.; Rozman Grinberg, I.; Lamed, R.; Jindou, S.; Burstein, T.; Yaniv, O.; Shoham, Y.; Shimon, L.J.; Bayer, E.A.; Frolow, F. Reassembly and co-crystallization of a family 9 processive endoglucanase from its component parts: Structural and functional significance of the intermodular linker. Peer. J. 2015, 15, e1126. [Google Scholar] [CrossRef] [PubMed]
- Tormo, J.; Lamed, R.; Chirino, A.J.; Morag, E.; Bayer, E.A.; Shoham, Y.; Steitz, T.A. Crystal structure of a bacterial family-III cellulose-binding domain: A general mechanism for attachment to cellulose. EMBO J. 1996, 15, 5739–5751. [Google Scholar] [PubMed]
- Popolo, L.; Ragni, E.; Carotti, C.; Palomares, O.; Aardema, R.; Back, J.W.; Dekker, H.L.; de Koning, L.J.; de Jong, L.; de Koster, C.G. Disulfide bond structure and domain organization of yeast beta(1, 3)-glucanosyltransferases involved in cell wall biogenesis. J. Biol. Chem. 2008, 283, 18553–18565. [Google Scholar] [CrossRef] [PubMed]
- Receveur, V.; Czjzek, M.; Schülein, M.; Panine, P.; Henrissat, B. Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle X-ray scattering. J. Biol. Chem. 2002, 277, 40887–40892. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.D.; Lao, G.; Irwin, D.; Barr, B.K.; Benjamin, A.; Wilson, D.B. DNA sequences and expression in Streptomyces lividans of an exoglucanase gene and an endoglucanase gene from Thermomonospora fusca. Appl. Environ. Microbiol. 1993, 59, 3032–3043. [Google Scholar] [PubMed]
- Rabinovich, M.L.; Melnick, M.S.; Bolobova, A.V. The structure and mechanism of action of cellulolytic enzymes. Biochemistry 2002, 67, 850–871. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ojima, T.; Nishita, K. Purification and cDNA cloning of a cellulase from abalone Haliotis discus hannai. Eur. J. Biochem. 2003, 270, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Lis, H.; Sharon, N. Protein glycosylation. structural and functional aspects. Eur. J. Biochem. 1993, 218, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Elodie, M.-R.; Marie-Christine, K.-M.; Gaëtan, V.; Clément, O.; Carole, B.; Patrice, L.; Muriel, B. Protein N-glycosylation in eukaryotic microalgae and its impact on the production of nuclear expressed biopharmaceuticals. Front. Plant. Sci. 2014, 5, 359. [Google Scholar] [CrossRef]
- Hui, J.P.; Lanthier, P.; White, T.C.; McHugh, S.G.; Yaguchi, M.; Roy, R.; Thibault, P. Characterization of cellobiohydrolase I (Cel7A) glycoforms from extracts of Trichoderma reesei using capillary isoelectric focusing and electrospray mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 349–368. [Google Scholar] [CrossRef]
- Corzana, F.; Busto, J.H.; Jiménez-Osés, G.; de Luis, M.G.; Asensio, J.L.; Jiménez-Barbero, J.; Peregrina, J.M.; Avenoza, A. Serine versus threonine glycosylation: The methyl group causes a drastic alteration on the carbohydrate orientation and on the surrounding water shell. J. Am. Chem. Soc. 2007, 129, 9458–9467. [Google Scholar] [CrossRef] [PubMed]
- Boze, H.; Marlin, T.; Durand, D.; Perez, J.; Vernhet, A.; Canon, F.; Sarni-Manchado, P.; Cheynier, V.; Cabane, B. Proline-rich salivary proteins have extended conformations. Biophys. J. 2010, 99, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, D.M.; Turowski, V.R.; Murakami, M.T. Effects of the linker region on the structure and function of modular GH5 cellulases. Sci. Rep. 2016, 6, 28504. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Wang, L.; Tong, H.; Bu, M.; Gao, G.; Han, W.; Zhang, Z. The PT/S-Box of modular cellulase AcCel12B plays a key role in the hydrolysis of insoluble cellulose. Catalysts 2018, 8, 123. [Google Scholar] [CrossRef]
- Sonan, G.K.; Receveur-Brechot, V.; Duez, C.; Aghajari, N.; Czjzek, M.; Haser, R.; Gerday, C. The linker region plays a key role in the adaptation to cold of the cellulase from an Antarctic bacterium. Biochem. J. 2007, 407, 293–302. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucl. Acids Res. 2013, 4, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Meth. 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Prot. Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer. An environment for comparative protein modeling. Electrophor 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucl. Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Gen. Biol. 2002, 3. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E-Value/Cys | Motifs | Proteins |
|---|---|---|
| Motif 1 6-C 5.4e-30 |  | Cr9B, Gp51466, Gp51468, Vc2958622 |
| Motif 2 4-C 7.8e-10 |  | Cr9B, Gp51466, Gp51468, Vc2958622 |
| Motif 3 2-C 4.1e-7 |  | Cr9B, Cr9D, all three Gp, Vc2958622 |
| Hevein Motif: |  [CG]X5–7[C]X4[CCS]X4[C]X6[C]X3 [C] [CG]X8[C]X5[CCS]X4[C]X7[C] | General CBM18 motif |
| 7 C | Plants (MS), Fungus (BD) Diatom (Fc) | |
| 6 C |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerriero, G.; Sergeant, K.; Legay, S.; Hausman, J.-F.; Cauchie, H.-M.; Ahmad, I.; Siddiqui, K.S. Novel Insights from Comparative In Silico Analysis of Green Microalgal Cellulases. Int. J. Mol. Sci. 2018, 19, 1782. https://doi.org/10.3390/ijms19061782
Guerriero G, Sergeant K, Legay S, Hausman J-F, Cauchie H-M, Ahmad I, Siddiqui KS. Novel Insights from Comparative In Silico Analysis of Green Microalgal Cellulases. International Journal of Molecular Sciences. 2018; 19(6):1782. https://doi.org/10.3390/ijms19061782
Chicago/Turabian StyleGuerriero, Gea, Kjell Sergeant, Sylvain Legay, Jean-Francois Hausman, Henry-Michel Cauchie, Irshad Ahmad, and Khawar Sohail Siddiqui. 2018. "Novel Insights from Comparative In Silico Analysis of Green Microalgal Cellulases" International Journal of Molecular Sciences 19, no. 6: 1782. https://doi.org/10.3390/ijms19061782
APA StyleGuerriero, G., Sergeant, K., Legay, S., Hausman, J.-F., Cauchie, H.-M., Ahmad, I., & Siddiqui, K. S. (2018). Novel Insights from Comparative In Silico Analysis of Green Microalgal Cellulases. International Journal of Molecular Sciences, 19(6), 1782. https://doi.org/10.3390/ijms19061782