Modulating Chemosensitivity of Tumors to Platinum-Based Antitumor Drugs by Transcriptional Regulation of Copper Homeostasis

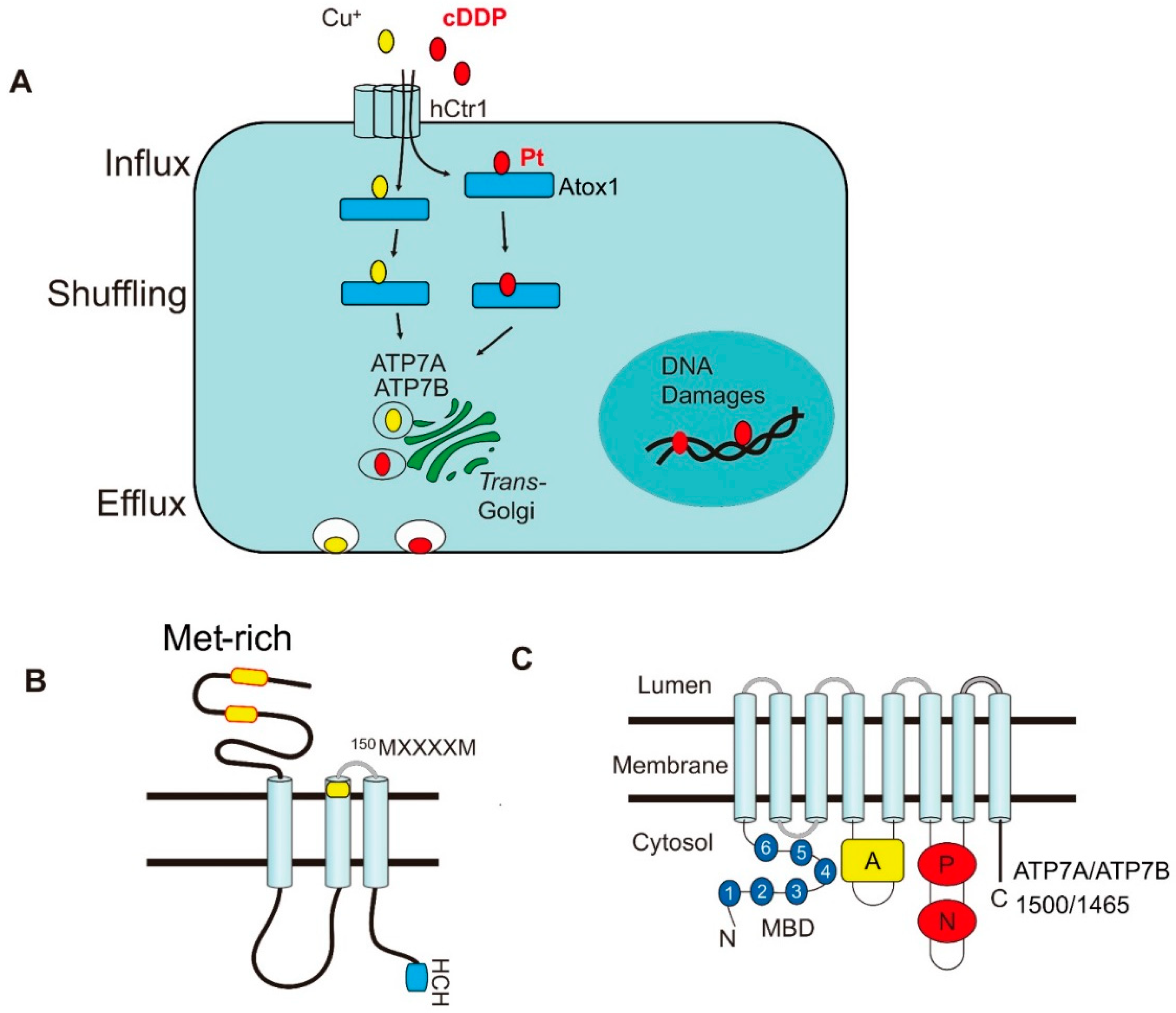{kind=link}
{kind=link}
{kind=link}
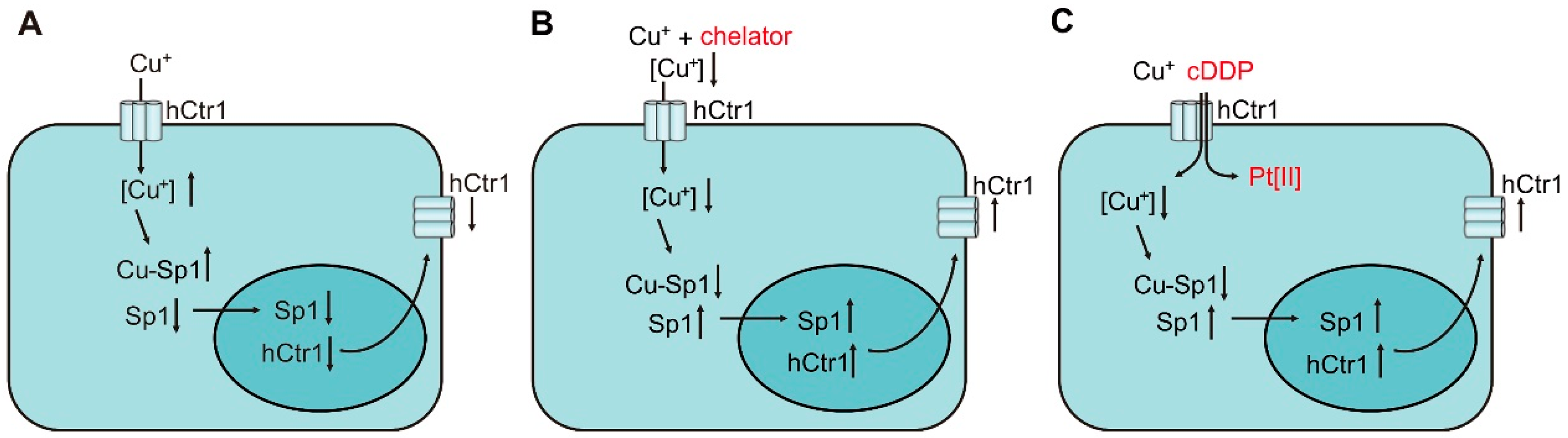{kind=link}
Abstract
1. Introduction
2. Roles of the Copper Transporters in Pt Drug Transport Mechanism
2.1. Cisplatin Importers
2.2. Cisplatin Chaperone
2.3. Role of ATP7A and ATP7B in cDDP Efflux
3. Modulating cDDP Sensitivity through Redox Regulation of Cu Homeostasis
4. Modulating cDDP Sensitivity through Transcriptional Regulation of hCtr1 Expression
4.1. Regulation of Ctr1 Internalization by Cu Bioavailability
4.2. Transcriptional Regulation of Ctr1 Expression by Cu Bioavailability
4.3. The Sensing Mechanisms of Cu Bioavailability by Sp1
4.4. The Capacity of hCtr1 Regulation and Cellular Cu Bioavailability
5. Modulation of hCtr1 Transcriptional Regulation for Overcoming cDDP Resistance in Cancer Chemotherapy
6. Conclusions and Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Atox1 | antioxidant 1 |
| BSO | buthionine sulfoximine |
| Cbp | carboplatin |
| Ctr1 | the high-affinity copper transporter 1 (SLC31A1) |
| GCS | γ-glutamylcysteine synthetase |
| GGT | γ-glutamyltransferase |
| GS | glutathione synthetase |
| GSH | Glutathione |
| MTF-1 | metal responsive transcription factor 1 |
| D-pen | D-pencillamide |
| Oxl | oxaliplatin |
| ROS | reactive oxygen species |
| Sp1 | specific protein |
| TM | tetrathiomolybdate |
| TGN | trans-Golgi network |
| xCT | cystine glutamate antipolar transporter |
References
- Muggia, F.M.; Bonetti, A.; Hoeschele, J.D.; Rozencweig, M.; Howell, S.B. Platinum Antitumor Complexes: 50 Years Since Barnett Rosenberg’s Discovery. J. Clin. Oncol. 2015, 33, 4219–4226. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Chen, W.C.; Liang, Z.D.; Tsai, W.B.; Long, Y.; Aiba, I.; Fu, S.; Broaddus, R.; Liu, J.; Feun, L.G.; et al. Targeting drug transport mechanisms for improving platinum-based cancer chemotherapy. Expert Opin. Ther. Targets 2015, 19, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Cassinelli, G.; Zaffaroni, N.; Lanzi, C.; Perego, P. New mechanisms for old drugs: Insights into DNA-unrelated effects of platinum compounds and drug resistance determinants. Drug Resist. Updates 2015, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Lee, J.J.; He, G.; Chow, C.W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Wistuba, I.I.; Stewart, D.J.; Siddik, Z.H. Tissue platinum concentration and tumor response in non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3345–3352. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Okabe, M.; Shen, D.W.; Liang, X.J.; Gottesman, M.M. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 495–535. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.A.; Howell, S.B. Cellular pharmacology of cisplatin: Perspectives on mechanisms of acquired resistance. Cancer Cells 1990, 2, 35–43. [Google Scholar] [PubMed]
- Sun, S.; Cai, J.; Yang, Q.; Zhao, S.; Wang, Z. The association between copper transporters and the prognosis of cancer patients undergoing chemotherapy: A meta-analysis of literatures and datasets. Oncotarget 2017, 8, 16036–16051. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Tsai, W.B.; Chang, J.T.; Estecio, M.; Wangpaichitr, M.; Savaraj, N.; Feun, L.G.; Chen, H.H.; Kuo, M.T. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1alpha, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget 2016, 7, 82658–82670. [Google Scholar] [CrossRef] [PubMed]
- Gately, D.P.; Howell, S.B. Cellular accumulation of the anticancer agent cisplatin: A review. Br. J. Cancer 1993, 67, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Ivy, K.D.; Kaplan, J.H. A re-evaluation of the role of hCtr1, the human high-affinity copper transporter, in platinum-drug entry into human cells. Mol. Pharmacol. 2013, 83, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Ohrvik, H.; Thiele, D.J. The role of Ctr1 and Ctr2 in mammalian copper homeostasis and platinum-based chemotherapy. J. Trace Elem. Med. Biol. 2015, 31, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Martelli, L.; Di Mario, F.; Ragazzi, E.; Apostoli, P.; Leone, R.; Perego, P.; Fumagalli, G. Different accumulation of cisplatin, oxaliplatin and JM216 in sensitive and cisplatin-resistant human cervical tumour cells. Biochem. Pharmacol. 2006, 72, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Buss, I.; Hamacher, A.; Sarin, N.; Kassack, M.U.; Kalayda, G.V. Relevance of copper transporter 1 and organic cation transporters 1–3 for oxaliplatin uptake and drug resistance in colorectal cancer cells. Metall. Integr. Biomet. Sci. 2018, 10, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3, 1543–1549. [Google Scholar] [PubMed]
- Beretta, G.L.; Gatti, L.; Tinelli, S.; Corna, E.; Colangelo, D.; Zunino, F.; Perego, P. Cellular pharmacology of cisplatin in relation to the expression of human copper transporter CTR1 in different pairs of cisplatin-sensitive and -resistant cells. Biochem. Pharmacol. 2004, 68, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Maryon, E.B.; Kasza, K.; Shafer, J.T.; Bartnik, C.M.; Dolan, M.E. Role of copper transporters in resistance to platinating agents. Cancer Chemother. Pharmacol. 2009, 64, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.D.; Stockton, D.; Savaraj, N.; Tien Kuo, M. Mechanistic comparison of human high-affinity copper transporter 1-mediated transport between copper ion and cisplatin. Mol. Pharmacol. 2009, 76, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Li, H.; Sun, H. Comparison between copper and cisplatin transport mediated by human copper transporter 1 (hCtr1). Metall. Integr. Biomet. Sci. 2012, 4, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Unger, V.M. Projection structure of the human copper transporter CTR1 at 6-A resolution reveals a compact trimer with a novel channel-like architecture. Proc. Natl. Acad. Sci. USA 2006, 103, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Pottier, A.; Borghi, E.; Levy, L. New use of metals as nanosized radioenhancers. Anticancer Res. 2014, 34, 443–453. [Google Scholar] [PubMed]
- Logeman, B.L.; Wood, L.K.; Lee, J.; Thiele, D.J. Gene duplication and neo-functionalization in the evolutionary and functional divergence of the metazoan copper transporters Ctr1 and Ctr2. J. Biol. Chem. 2017, 292, 11531–11546. [Google Scholar] [CrossRef] [PubMed]
- Ohrvik, H.; Nose, Y.; Wood, L.K.; Kim, B.E.; Gleber, S.C.; Ralle, M.; Thiele, D.J. Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proc. Natl. Acad. Sci. USA 2013, 110, E4279–E4288. [Google Scholar] [CrossRef] [PubMed]
- Ohrvik, H.; Logeman, B.; Turk, B.; Reinheckel, T.; Thiele, D.J. Cathepsin Protease Controls Copper and Cisplatin Accumulation via Cleavage of the Ctr1 Metal-binding Ectodomain. J. Biol. Chem. 2016, 291, 13905–13916. [Google Scholar] [CrossRef] [PubMed]
- Bompiani, K.M.; Tsai, C.Y.; Achatz, F.P.; Liebig, J.K.; Howell, S.B. Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity. Metall. Integr. Biomet. Sci. 2016, 8, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Choi, C.H.; Do, I.G.; Song, S.Y.; Lee, W.; Park, H.S.; Song, T.J.; Kim, M.K.; Kim, T.J.; Lee, J.W.; et al. Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemotherapy. Gynecol. Oncol. 2011, 122, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Teramae, M.; Yamauchi, M.; Fukuda, T.; Yasui, T.; Sumi, T.; Honda, K.; Ishiko, O. Association of copper transporter expression with platinum resistance in epithelial ovarian cancer. Anticancer Res. 2013, 33, 1409–1414. [Google Scholar] [PubMed]
- Robinson, N.J.; Winge, D.R. Copper metallochaperones. Annu. Rev. Biochem. 2010, 79, 537–562. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.G.; Unger, V.M. Atox1 contains positive residues that mediate membrane association and aid subsequent copper loading. J. Membr. Biol. 2013, 246, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Kahra, D.; Kovermann, M.; Wittung-Stafshede, P. The C-Terminus of Human Copper Importer Ctr1 Acts as a Binding Site and Transfers Copper to Atox1. Biophys. J. 2016, 110, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yuan, S.; Wang, E.; Tong, Y.; Ma, G.; Wei, K.; Liu, Y. Platinum transfer from hCtr1 to Atox1 is dependent on the type of platinum complex. Metall. Integr. Biomet. Sci. 2017, 9, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Kozyreva, T.; Zovo, K.; Palumaa, P. Affinity gradients drive copper to cellular destinations. Nature 2010, 465, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Huffman, D.L.; Lamb, A.L.; O'Halloran, T.V.; Rosenzweig, A.C. Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat. Struct. Biol. 2000, 7, 766–771. [Google Scholar] [PubMed]
- Boal, A.K.; Rosenzweig, A.C. Crystal structures of cisplatin bound to a human copper chaperone. J. Am. Chem. Soc. 2009, 131, 14196–14197. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Gunther, V.; Georgiev, O.; Schaffner, W. Distorted copper homeostasis with decreased sensitivity to cisplatin upon chaperone Atox1 deletion in Drosophila. Biometals 2011, 24, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Maktabi, M.H.; Blair, B.G.; Larson, C.A.; Howell, S.B. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J. Inorg. Biochem. 2009, 103, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Ozumi, K.; Kim, H.W.; Nakagawa, O.; McKinney, R.D.; Folz, R.J.; Zelko, I.N.; Ushio-Fukai, M.; Fukai, T. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: Role of antioxidant-1. Free Radic. Biol. Med. 2009, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Celauro, E.; Mukaj, A.; Fierro-Gonzalez, J.C.; Wittung-Stafshede, P. Copper chaperone ATOX1 regulates pluripotency factor OCT4 in preimplantation mouse embryos. Biochem. Biophys. Res. Commun. 2017, 491, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Inesi, G.; Pilankatta, R.; Tadini-Buoninsegni, F. Biochemical characterization of P-type copper ATPases. Biochem. J. 2014, 463, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Jayakanthan, S.; Braiterman, L.T.; Hasan, N.M.; Unger, V.M.; Lutsenko, S. Human Copper Transporter Atp7b (Wilson Disease Protein) Forms Stable Dimers in Vitro and in Cells. J. Biol. Chem. 2017, 292, 18760–18774. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.W.; Moore, S.D. Copper transporting P-type ATPases and human disease. J. Bioenergy Biomembr. 2002, 34, 333–338. [Google Scholar] [CrossRef]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Banci, L.; Bertini, I.; Thompsett, A.R. Solution structure of CopC: A cupredoxin-like protein involved in copper homeostasis. Structure 2002, 10, 1337–1347. [Google Scholar] [CrossRef]
- Hussain, F.; Olson, J.S.; Wittung-Stafshede, P. Conserved residues modulate copper release in human copper chaperone Atox1. Proc. Natl. Acad. Sci. USA 2008, 105, 11158–11163. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Shi, C.; Tian, C.; Liu, Y. Conserved residue modulates copper-binding properties through structural dynamics in human copper chaperone Atox1. Metall. Integr. Biomet. Sci. 2013, 5, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Yang, N.; Bothe, J.; Tonelli, M.; Nokhrin, S.; Dolgova, N.V.; Braiterman, L.; Lutsenko, S.; Dmitriev, O.Y. The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics. J. Biol. Chem. 2017, 292, 18169–18177. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.; Pilankatta, R.; Inesi, G.; Bartolommei, G.; Moncelli, M.R.; Tadini-Buoninsegni, F. Distinctive features of catalytic and transport mechanisms in mammalian sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) and Cu+ (ATP7A/B) ATPases. J. Biol. Chem. 2012, 287, 32717–32727. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Guerrero, M.; Arguello, J.M. Mechanism of Cu+-transporting ATPases: Soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc. Natl. Acad. Sci. USA 2008, 105, 5992–5997. [Google Scholar] [CrossRef] [PubMed]
- Pilankatta, R.; Lewis, D.; Inesi, G. Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J. Biol. Chem. 2011, 286, 7389–7396. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Adams, P.L.; Maktabi, M.H.; Mathews, R.A.; Howell, S.B. The CXXC motifs in the metal binding domains are required for ATP7B to mediate resistance to cisplatin. J. Inorg. Biochem. 2012, 110, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Inesi, G.; Galliani, A.; Sinisi, M.; Losacco, M.; Natile, G.; Arnesano, F. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew. Chem. 2014, 53, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Palm, M.E.; Weise, C.F.; Lundin, C.; Wingsle, G.; Nygren, Y.; Bjorn, E.; Naredi, P.; Wolf-Watz, M.; Wittung-Stafshede, P. Cisplatin binds human copper chaperone Atox1 and promotes unfolding in vitro. Proc. Natl. Acad. Sci. USA 2011, 108, 6951–6956. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Inoue, Y.; Kodama, H.; Yamazaki, H.; Kawai, K.; Suemizu, H.; Masuda, R.; Iwazaki, M.; Yamada, S.; Ueyama, Y.; et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) correlates with cisplatin resistance in human non-small cell lung cancer xenografts. Oncol. Rep. 2008, 20, 265–270. [Google Scholar] [PubMed]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar] [PubMed]
- Safaei, R.; Howell, S.B. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit. Rev. Oncol. Hematol. 2005, 53, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, K.; Gebhardt, R.; Mossner, J.; Lutsenko, S.; Huster, D. Functional interactions of Cu-ATPase ATP7B with cisplatin and the role of ATP7B in the resistance of cells to the drug. J. Biol. Chem. 2009, 284, 7793–7802. [Google Scholar] [CrossRef] [PubMed]
- Samimi, G.; Safaei, R.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Goodman, M.; Howell, S.B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Balibrea, E.; Martinez-Cardus, A.; Musulen, E.; Gines, A.; Manzano, J.L.; Aranda, E.; Plasencia, C.; Neamati, N.; Abad, A. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer 2009, 124, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Brozovic, A.; Ambriovic-Ristov, A.; Osmak, M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met.-Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Song, I.S.; Hossain, A.; Choi, M.K.; Yamane, Y.; Liang, Z.D.; Lu, J.; Wu, L.Y.; Siddik, Z.H.; Klomp, L.W.; et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol. Pharmacol. 2008, 74, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radic. Biol. Med. 2017, 104, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, D.J.; Kim, J.H.; Jeong, E.Y.; Jung, M.H.; Kim, T.H.; Yang, J.I.; Lee, G.W.; Chung, H.J.; Chang, S.H. Glutamine protects against cisplatin-induced nephrotoxicity by decreasing cisplatin accumulation. J. Pharmacol. Sci. 2015, 127, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Franzini, M.; Corti, A.; Lorenzini, E.; Paolicchi, A.; Pompella, A.; De Cesare, M.; Perego, P.; Gatti, L.; Leone, R.; Apostoli, P.; et al. Modulation of cell growth and cisplatin sensitivity by membrane gamma-glutamyltransferase in melanoma cells. Eur. J. Cancer 2006, 42, 2623–2630. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cheng, Q.; Wang, Z.; Xi, Z.; Xu, D.; Liu, Y. Cisplatin binds to human copper chaperone Cox17: The mechanistic implication of drug delivery to mitochondria. Chem. Commun. 2014, 50, 2667–2669. [Google Scholar] [CrossRef] [PubMed]
- Narindrasorasak, S.; Zhang, X.; Roberts, E.A.; Sarkar, B. Comparative analysis of metal binding characteristics of copper chaperone proteins, Atx1 and ATOX1. Bioinorg. Chem. Appl. 2004, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Tanchou, V.; Gas, F.; Urvoas, A.; Cougouluegne, F.; Ruat, S.; Averseng, O.; Quemeneur, E. Copper-mediated homo-dimerisation for the HAH1 metallochaperone. Biochem. Biophys. Res. Commun. 2004, 325, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Dolgova, N.V.; Yu, C.; Cvitkovic, J.P.; Hodak, M.; Nienaber, K.H.; Summers, K.L.; Cotelesage, J.J.H.; Bernholc, J.; Kaminski, G.A.; Pickering, I.J.; et al. Binding of Copper and Cisplatin to Atox1 Is Mediated by Glutathione through the Formation of Metal-Sulfur Clusters. Biochemistry 2017, 56, 3129–3141. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Guo, W.; Tian, C.; Wang, F.; Liu, Y. Copper binding promotes the interaction of cisplatin with human copper chaperone Atox1. Chem. Commun. 2013, 49, 11197–11199. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. The ATP-dependent glutathione S-conjugate export pump. Trends Biochem. Sci. 1992, 17, 463–468. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ali-Osman, F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J. Biol. Chem. 1993, 268, 20116–20125. [Google Scholar] [PubMed]
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M.; et al. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer 2011, 104, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Dancis, A.; Yuan, D.S.; Haile, D.; Askwith, C.; Eide, D.; Moehle, C.; Kaplan, J.; Klausner, R.D. Molecular characterization of a copper transport protein in S. cerevisiae: An unexpected role for copper in iron transport. Cell 1994, 76, 393–402. [Google Scholar] [CrossRef]
- Kuo, M.T.; Fu, S.; Savaraj, N.; Chen, H.H. Role of the human high-affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012, 72, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol. Pharmacol. 2010, 77, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.E.; Rabinovich, E.; Dancis, A.; Bonifacino, J.S.; Klausner, R.D. Copper-dependent degradation of the Saccharomyces cerevisiae plasma membrane copper transporter Ctr1p in the apparent absence of endocytosis. EMBO J. 1996, 15, 3515–3523. [Google Scholar] [PubMed]
- Guo, Y.; Smith, K.; Lee, J.; Thiele, D.J.; Petris, M.J. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J. Biol. Chem. 2004, 279, 17428–17433. [Google Scholar] [CrossRef] [PubMed]
- Molloy, S.A.; Kaplan, J.H. Copper-dependent recycling of hCtr1, the human high affinity copper transporter. J. Biol. Chem. 2009, 284, 29704–29713. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.T.; Posewitz, M.C.; Srinivasan, C.; Winge, D.R. Mapping of the DNA binding domain of the copper-responsive transcription factor Mac1 from Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 23805–23811. [Google Scholar] [CrossRef] [PubMed]
- Furst, P.; Hu, S.; Hackett, R.; Hamer, D. Copper activates metallothionein gene transcription by altering the conformation of a specific DNA binding protein. Cell 1988, 55, 705–717. [Google Scholar] [CrossRef]
- Keller, G.; Bird, A.; Winge, D.R. Independent metalloregulation of Ace1 and Mac1 in Saccharomyces cerevisiae. Eukaryot Cell 2005, 4, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Thiele, D.J. ACE1 regulates expression of the Saccharomyces cerevisiae metallothionein gene. Mol. Cell. Biol. 1988, 8, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, A.; Balamurugan, K.; Yepiskoposyan, H.; Zhou, H.; Egli, D.; Georgiev, O.; Thiele, D.J.; Schaffner, W. Metal-responsive transcription factor (MTF-1) handles both extremes, copper load and copper starvation, by activating different genes. Genes Dev. 2005, 19, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Strenkert, D.; Schmollinger, S.; Sommer, F.; Schulz-Raffelt, M.; Schroda, M. Transcription factor-dependent chromatin remodeling at heat shock and copper-responsive promoters in Chlamydomonas reinhardtii. Plant Cell 2011, 23, 2285–2301. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Molina, A.; Xing, S.; Huijser, P. Functional characterisation of Arabidopsis SPL7 conserved protein domains suggests novel regulatory mechanisms in the Cu deficiency response. BMC Plant Biol. 2014, 14, 231. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Hayashi, M.; Fukazawa, M.; Kobayashi, Y.; Shikanai, T. SQUAMOSA Promoter Binding Protein-Like7 Is a Central Regulator for Copper Homeostasis in Arabidopsis. Plant Cell 2009, 21, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H. Overcoming platinum drug resistance with copper-lowering agents. Anticancer Res. 2013, 33, 4157–4161. [Google Scholar]
- Liang, Z.D.; Tsai, W.B.; Lee, M.Y.; Savaraj, N.; Kuo, M.T. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol. Pharmacol. 2012, 81, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Chen, H.H.; Aiba, I.; Hossain, A.; Liang, Z.D.; Klomp, L.W.; Kuo, M.T. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol. Pharmacol. 2008, 74, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Smirnova, I.V.; Andrews, G.K. Functional heterogeneity in the zinc fingers of metalloregulatory protein metal response element-binding transcription factor-1. J. Biol. Chem. 2000, 275, 37194–37201. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Aiba, I.; Chen, H.H.; Kuo, M.T. Effects of Cu(II) and cisplatin on the stability of Specific protein 1 (Sp1)-DNA binding: Insights into the regulation of copper homeostasis and platinum drug transport. J. Inorg. Biochem. 2016, 161, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chen, S.; Xi, Z.; Liu, Y. Copper-finger protein of Sp1: The molecular basis of copper sensing. Metall. Integr. Biomet. Sci. 2017, 9, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I. Sp1: Emerging roles—Beyond constitutive activation of TATA-less housekeeping genes. Biochem. Biophys. Res. Commun. 2008, 372, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef] [PubMed]
- Anzellotti, A.I.; Liu, Q.; Bloemink, M.J.; Scarsdale, J.N.; Farrell, N. Targeting retroviral Zn finger-DNA interactions: A small-molecule approach using the electrophilic nature of trans-platinum-nucleobase compounds. Chem. Biol. 2006, 13, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Maurmann, L.; Bose, R.N. Unwinding of zinc finger domain of DNA polymerase I by cis-diamminedichloroplatinum(II). Dalton Trans. 2010, 39, 7968–7979. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, D.; Jiang, H.; Xi, Z.; Zhu, P.; Liu, Y. Trans-platinum/thiazole complex interferes with Sp1 zinc-finger protein. Angew. Chem. 2012, 51, 12258–12262. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; de Paiva, R.E.; Qu, Y.; Farrell, N. Tuning the reactivity of Sp1 zinc fingers with platinum complexes. Dalton Trans. 2016, 45, 8712–8716. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.D.; Long, Y.; Chen, H.H.; Savaraj, N.; Kuo, M.T. Regulation of the high-affinity copper transporter (hCtr1) expression by cisplatin and heavy metals. J. Biol. Inorg. Chem. 2014, 19, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Wegner, S.V.; Sun, F.; Hernandez, N.; He, C. The tightly regulated copper window in yeast. Chem. Commun. 2011, 47, 2571–2573. [Google Scholar] [CrossRef] [PubMed]
- Nose, Y.; Kim, B.E.; Thiele, D.J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Tang, X.; Peterson, D.R.; Kilari, D.; Chow, C.W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Stewart, D.J.; Wistuba, I.I.; et al. Copper transporter CTR1 expression and tissue platinum concentration in non-small cell lung cancer. Lung Cancer 2014, 85, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.D.; Long, Y.; Tsai, W.B.; Fu, S.; Kurzrock, R.; Gagea-Iurascu, M.; Zhang, F.; Chen, H.H.; Hennessy, B.T.; Mills, G.B.; et al. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Mol. Cancer Ther. 2012, 11, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Brem, S.; Grossman, S.A.; Carson, K.A.; New, P.; Phuphanich, S.; Alavi, J.B.; Mikkelsen, T.; Fisher, J.D.; New Approaches to Brain Tumor Therapy CNS Consortium. Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro-Oncology 2005, 7, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAF(V600E)-Driven Melanomagenesis and Counters Resistance to BRAF(V600E) and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [PubMed]
- Garber, K.; BIOMEDICINE. Targeting copper to treat breast cancer. Science 2015, 349, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hou, M.M.; Wheler, J.; Hong, D.; Naing, A.; Tsimberidou, A.; Janku, F.; Zinner, R.; Piha-Paul, S.; Falchook, G.; et al. Exploratory study of carboplatin plus the copper-lowering agent trientine in patients with advanced malignancies. Investig. New Drugs 2014, 32, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Naing, A.; Fu, C.; Kuo, M.T.; Kurzrock, R. Overcoming platinum resistance through the use of a copper-lowering agent. Mol. Cancer Ther. 2012, 11, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Cossa, G.; Gatti, L.; Zunino, F.; Perego, P. Strategies to improve the efficacy of platinum compounds. Curr. Med. Chem. 2009, 16, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Chen, S.J.; Kuo, C.C.; Pan, H.Y.; Tsou, T.C.; Yeh, S.C.; Chang, J.Y. Desferal regulates hCtr1 and transferrin receptor expression through Sp1 and exhibits synergistic cytotoxicity with platinum drugs in oxaliplatin-resistant human cervical cancer cells in vitro and in vivo. Oncotarget 2016, 7, 49310–49321. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. The promise of copper lowering therapy with tetrathiomolybdate in the cure of cancer and in the treatment of inflammatory disease. J. Trace Elem. Med. Biol. 2014, 28, 372–378. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, Y.-H.; Kuo, C.; Kuo, M.T.; Chen, H.H.W. Modulating Chemosensitivity of Tumors to Platinum-Based Antitumor Drugs by Transcriptional Regulation of Copper Homeostasis. Int. J. Mol. Sci. 2018, 19, 1486. https://doi.org/10.3390/ijms19051486
Lai Y-H, Kuo C, Kuo MT, Chen HHW. Modulating Chemosensitivity of Tumors to Platinum-Based Antitumor Drugs by Transcriptional Regulation of Copper Homeostasis. International Journal of Molecular Sciences. 2018; 19(5):1486. https://doi.org/10.3390/ijms19051486
Chicago/Turabian StyleLai, Yu-Hsuan, Chin Kuo, Macus Tien Kuo, and Helen H. W. Chen. 2018. "Modulating Chemosensitivity of Tumors to Platinum-Based Antitumor Drugs by Transcriptional Regulation of Copper Homeostasis" International Journal of Molecular Sciences 19, no. 5: 1486. https://doi.org/10.3390/ijms19051486
APA StyleLai, Y.-H., Kuo, C., Kuo, M. T., & Chen, H. H. W. (2018). Modulating Chemosensitivity of Tumors to Platinum-Based Antitumor Drugs by Transcriptional Regulation of Copper Homeostasis. International Journal of Molecular Sciences, 19(5), 1486. https://doi.org/10.3390/ijms19051486