Design, Synthesis, and Biological Evaluation of Benzimidazole-Derived Biocompatible Copper(II) and Zinc(II) Complexes as Anticancer Chemotherapeutics


 ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
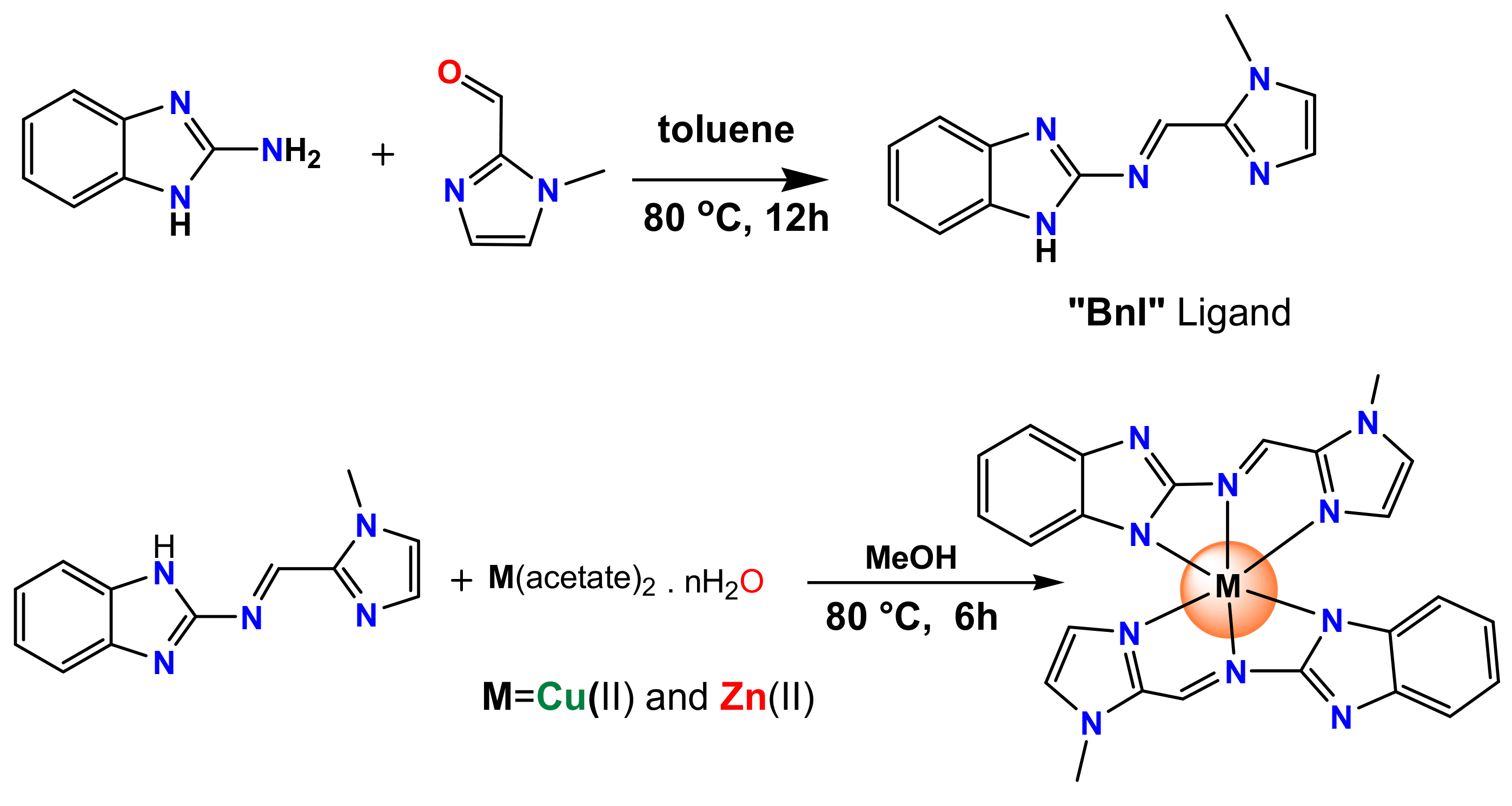
2.1. Synthesis and Characterization
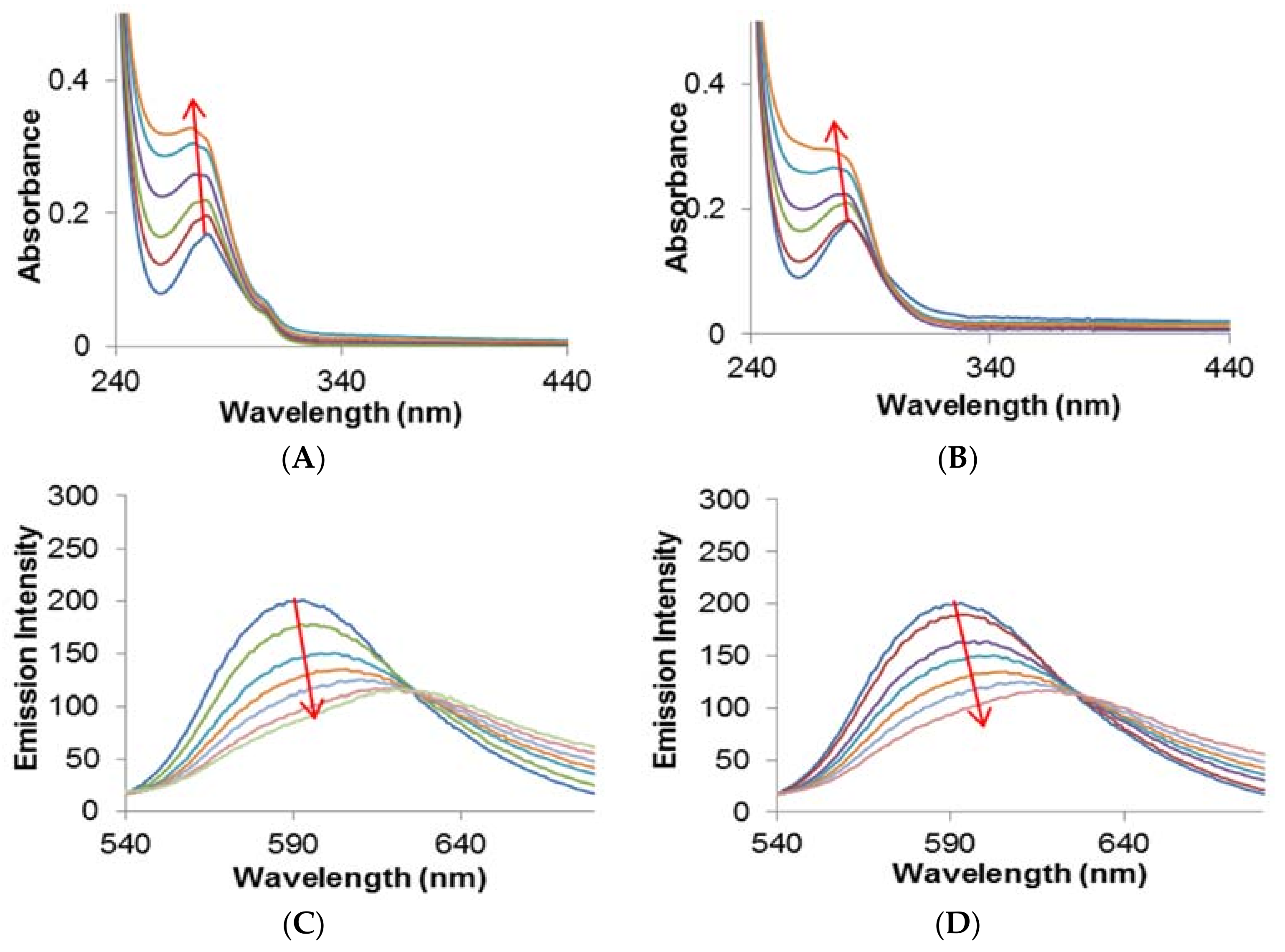
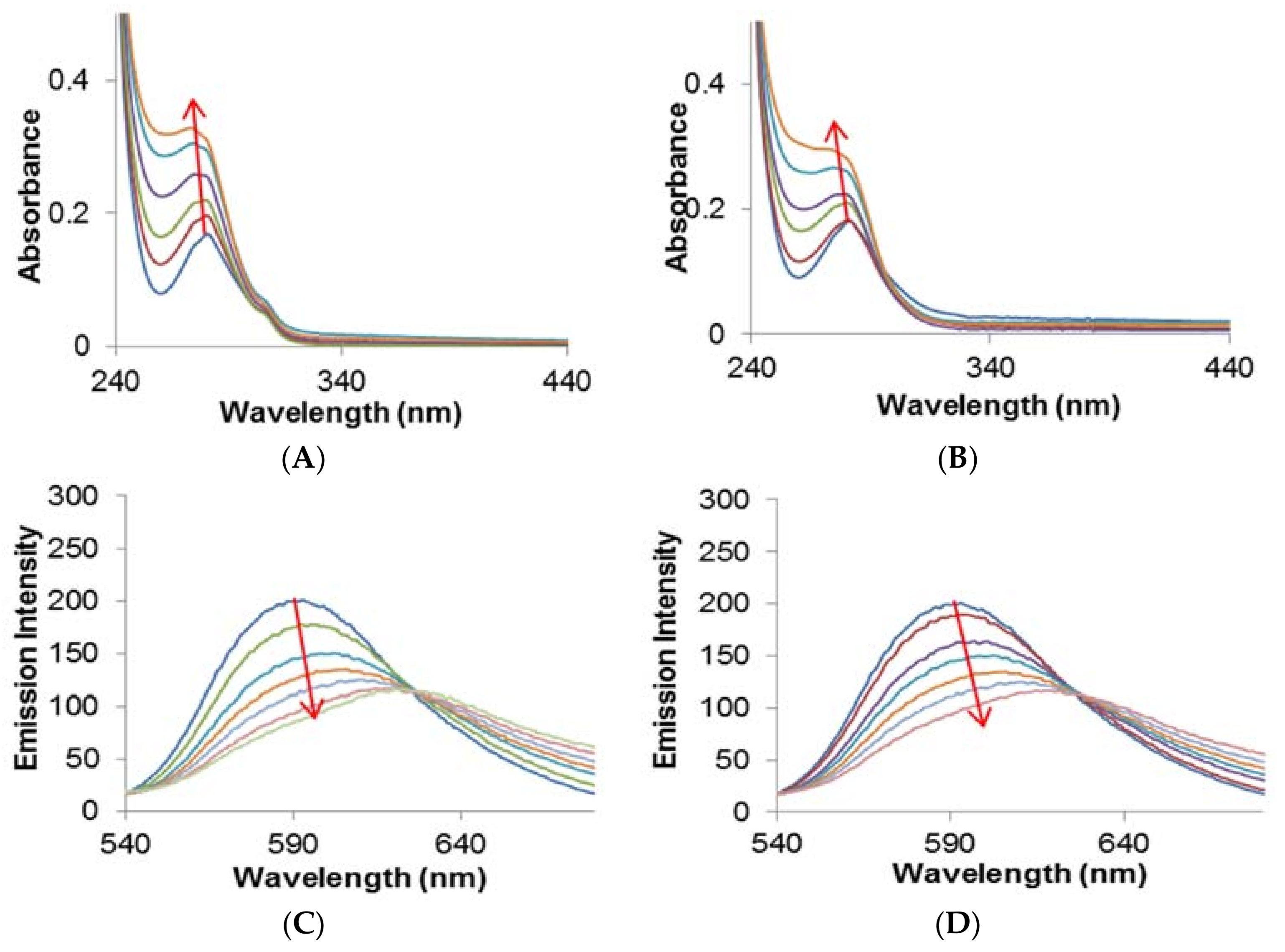
2.2. HSA–Metal Complex Interaction by Spectroscopy
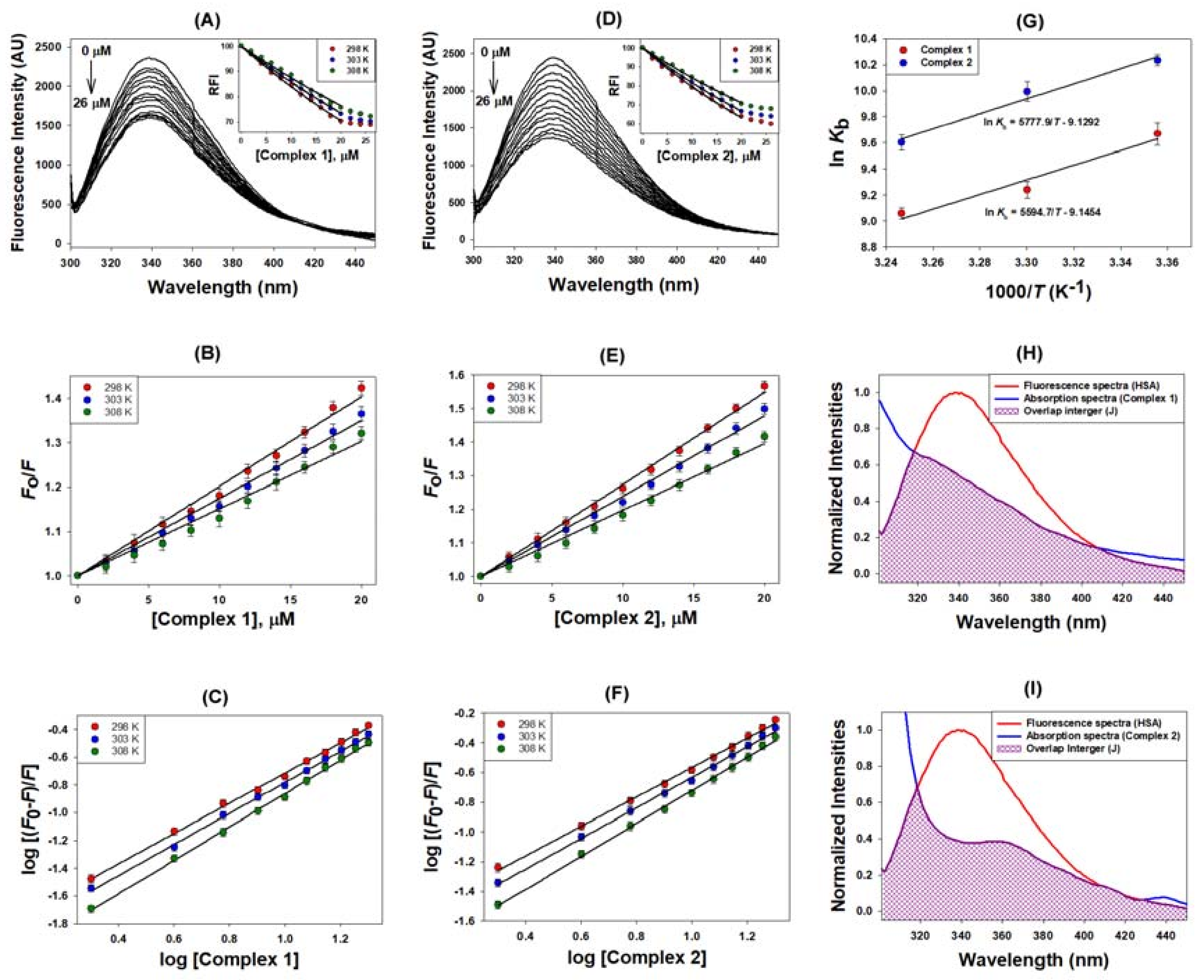
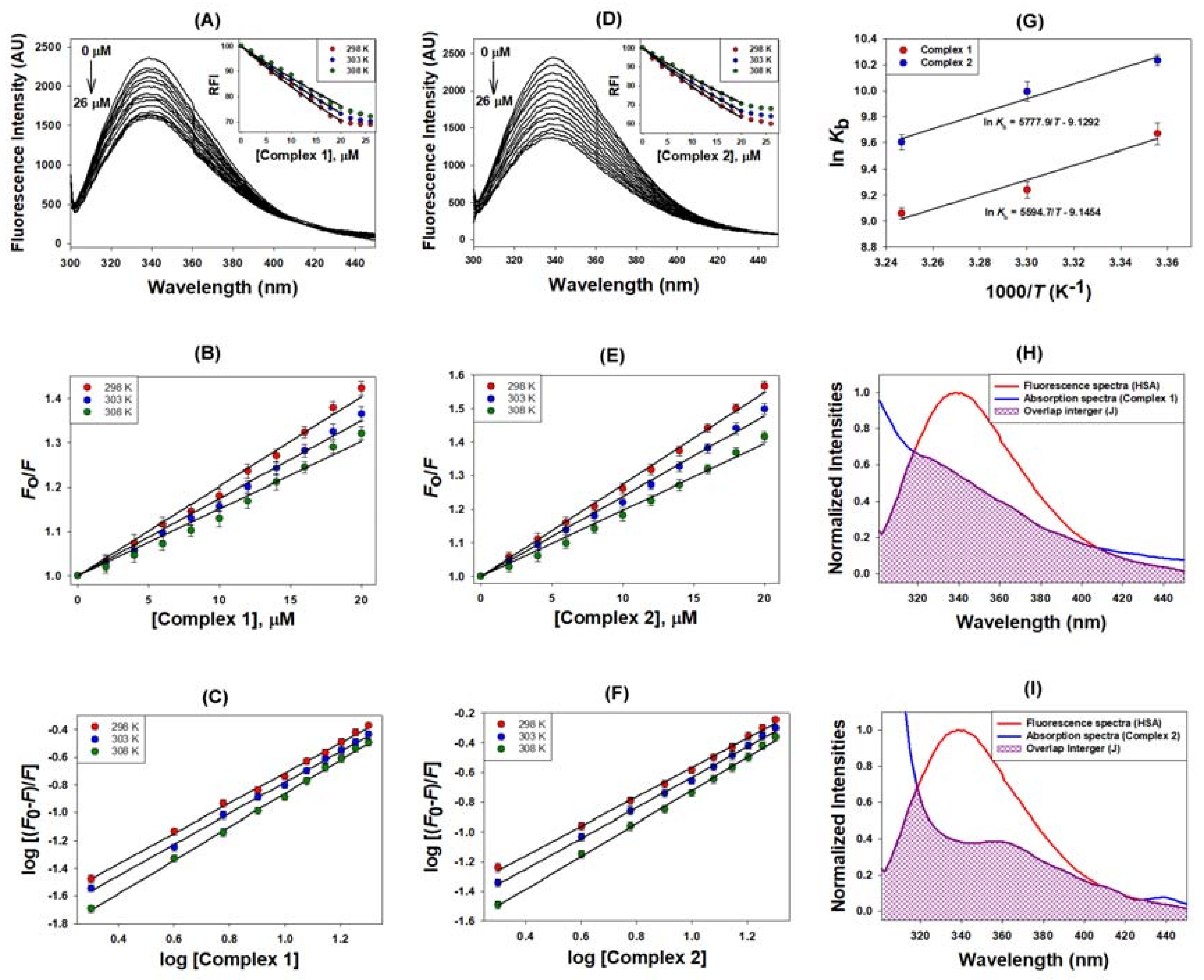
2.2.1. Quenching Experiment
2.2.2. Förster Resonance Energy Transfer (FRET) between HSA and Metal Complexes
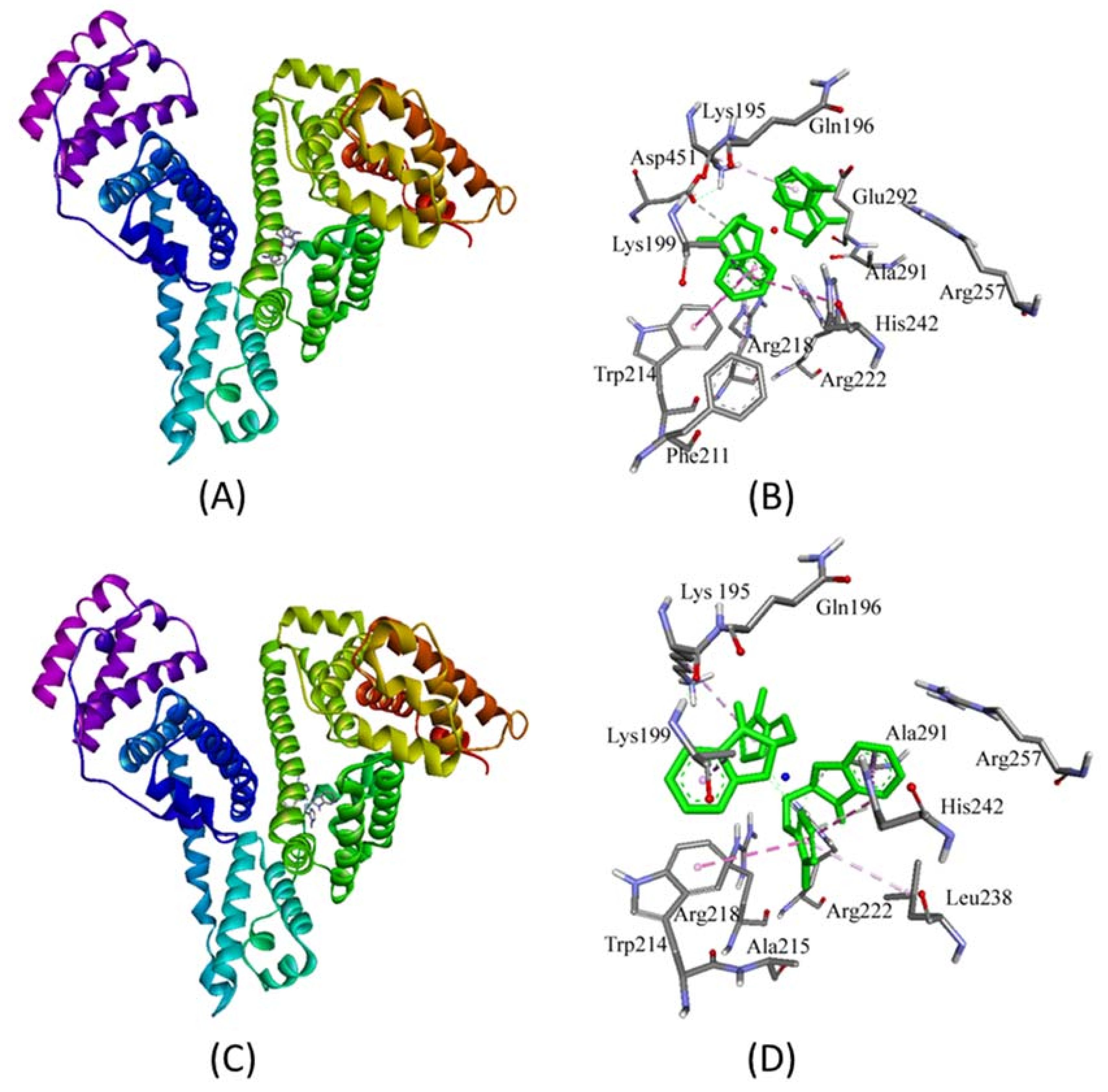
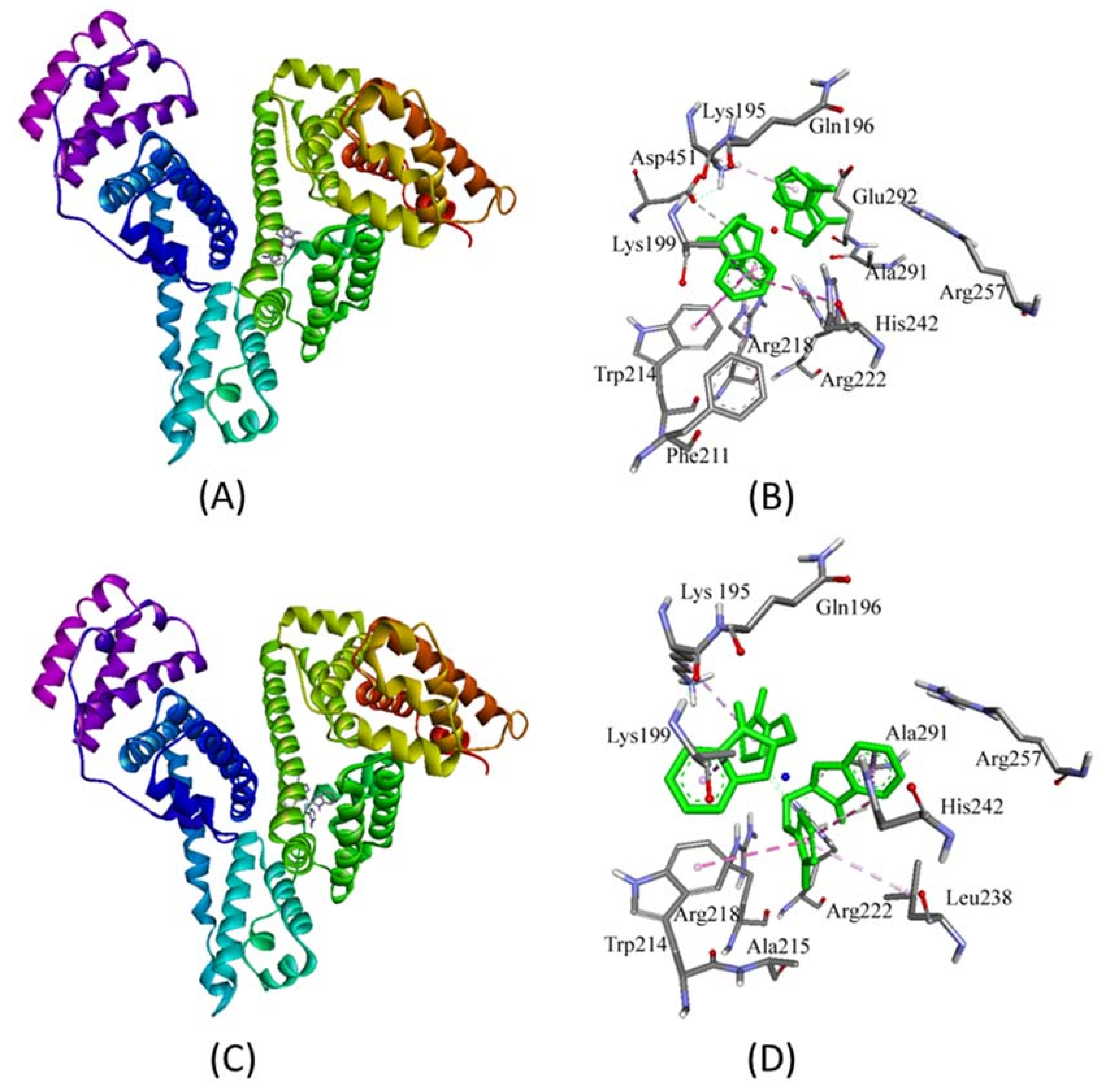
2.3. Molecular Docking Studies with HSA by Autodock
Prediction of Binding Sites of Metal Complexes on HSA
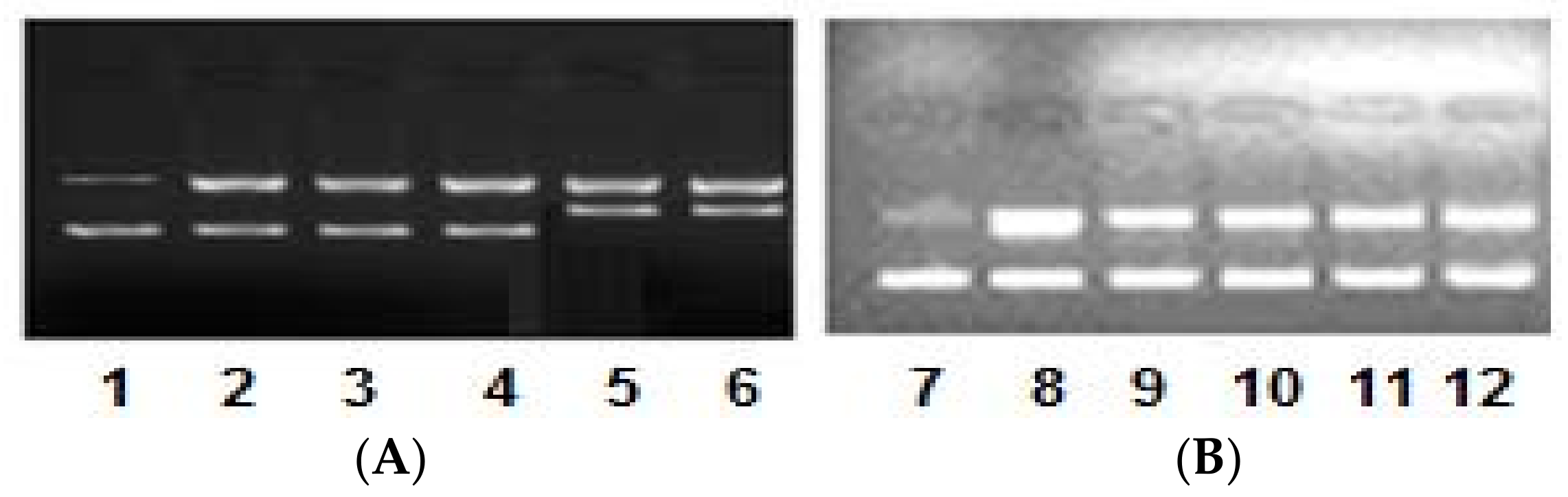
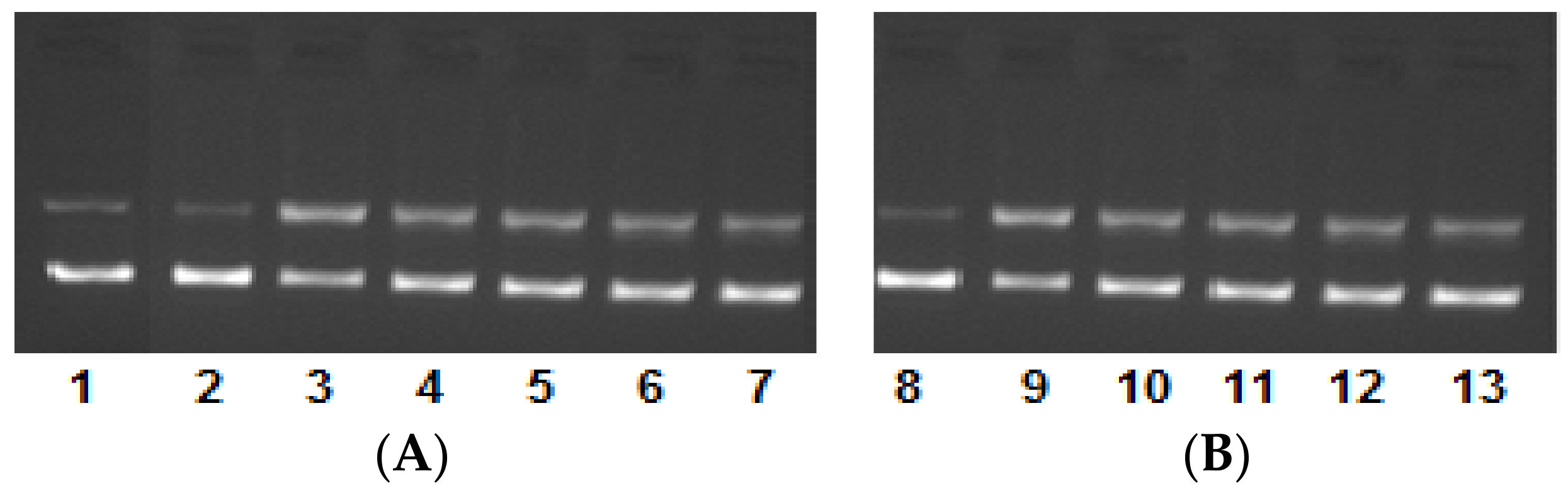
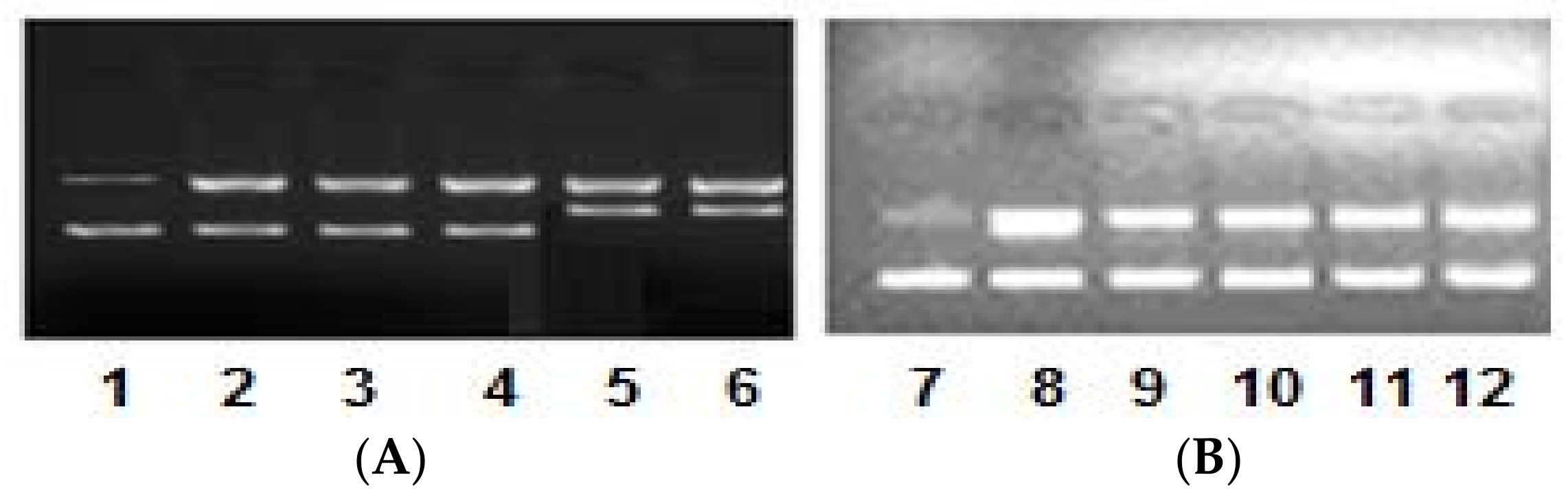
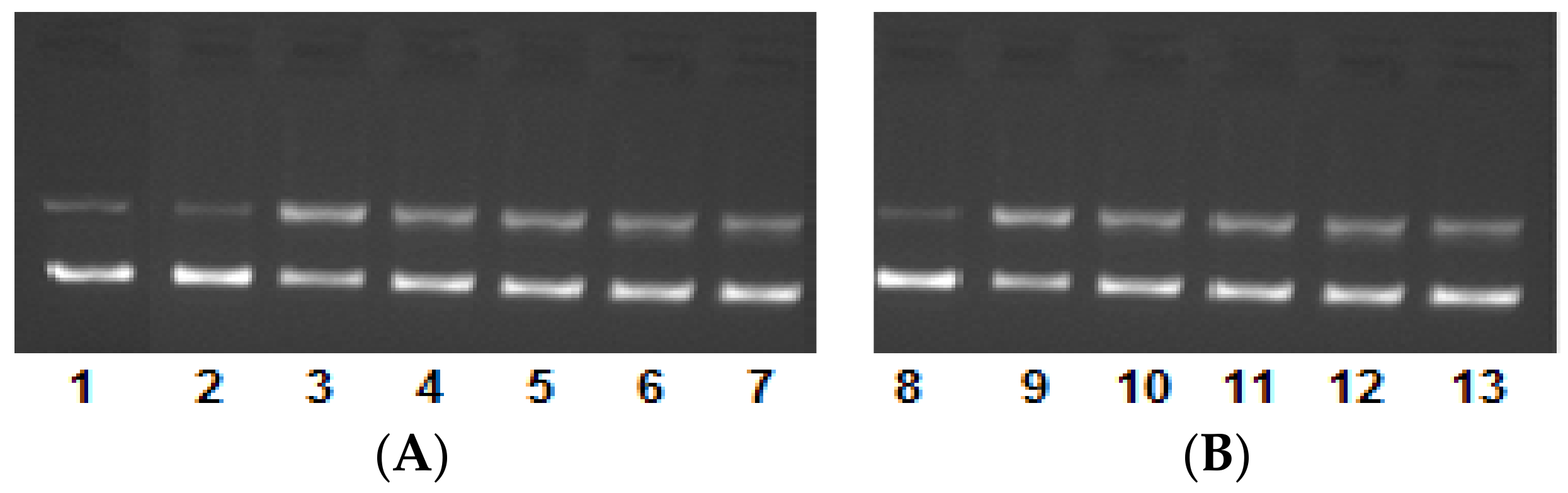
2.4. DNA Binding/Nuclease Activity
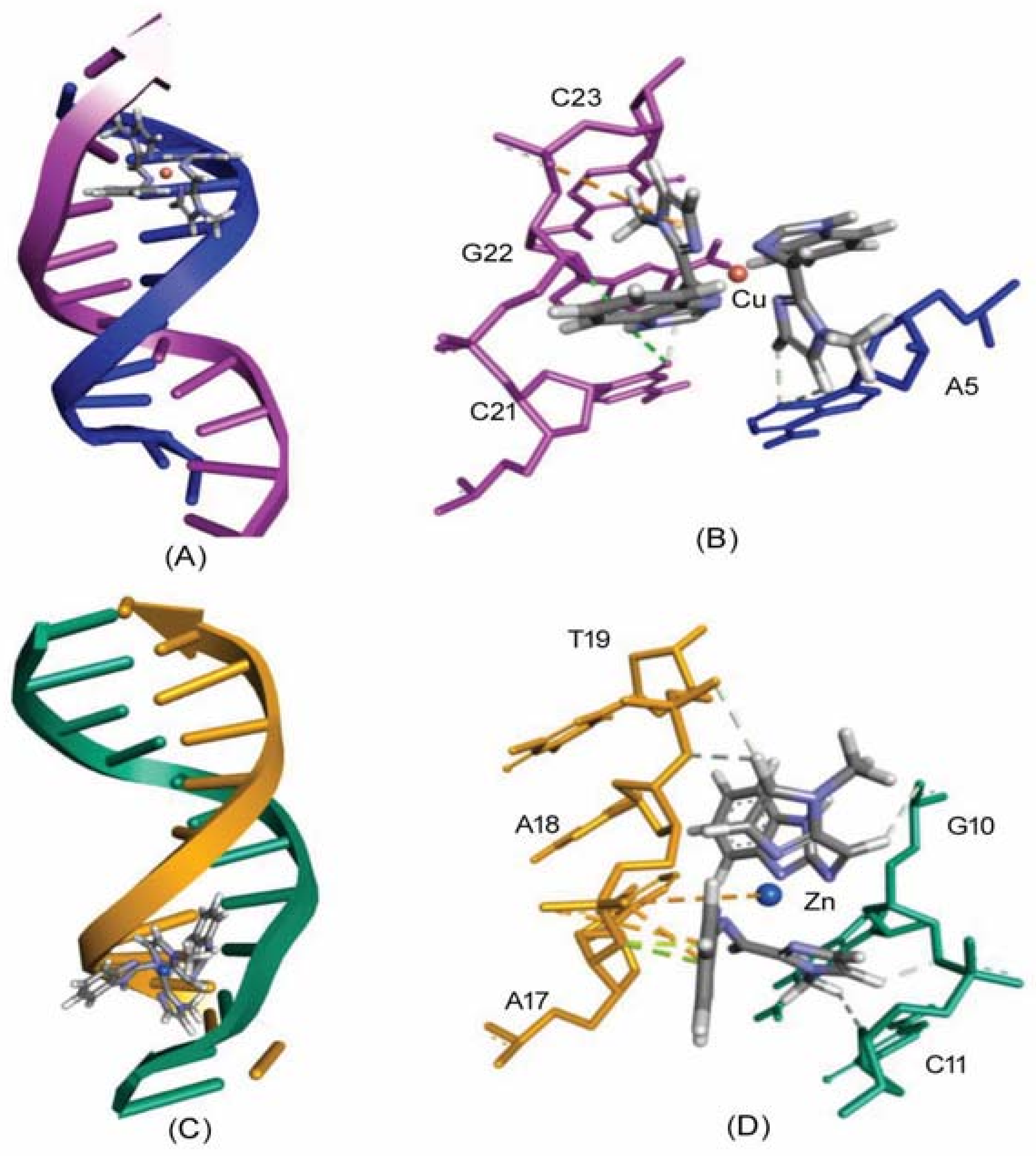
2.5. Molecular Docking Studies with DNA by Hex
Prediction of Binding Sites of Metal Complexes on DNA
2.6. In Vitro Cytotoxicity Assays
2.6.1. Analysis of Growth Inhibition Using 5-Diphenyltetrazolium Bromide (MTT) Assay
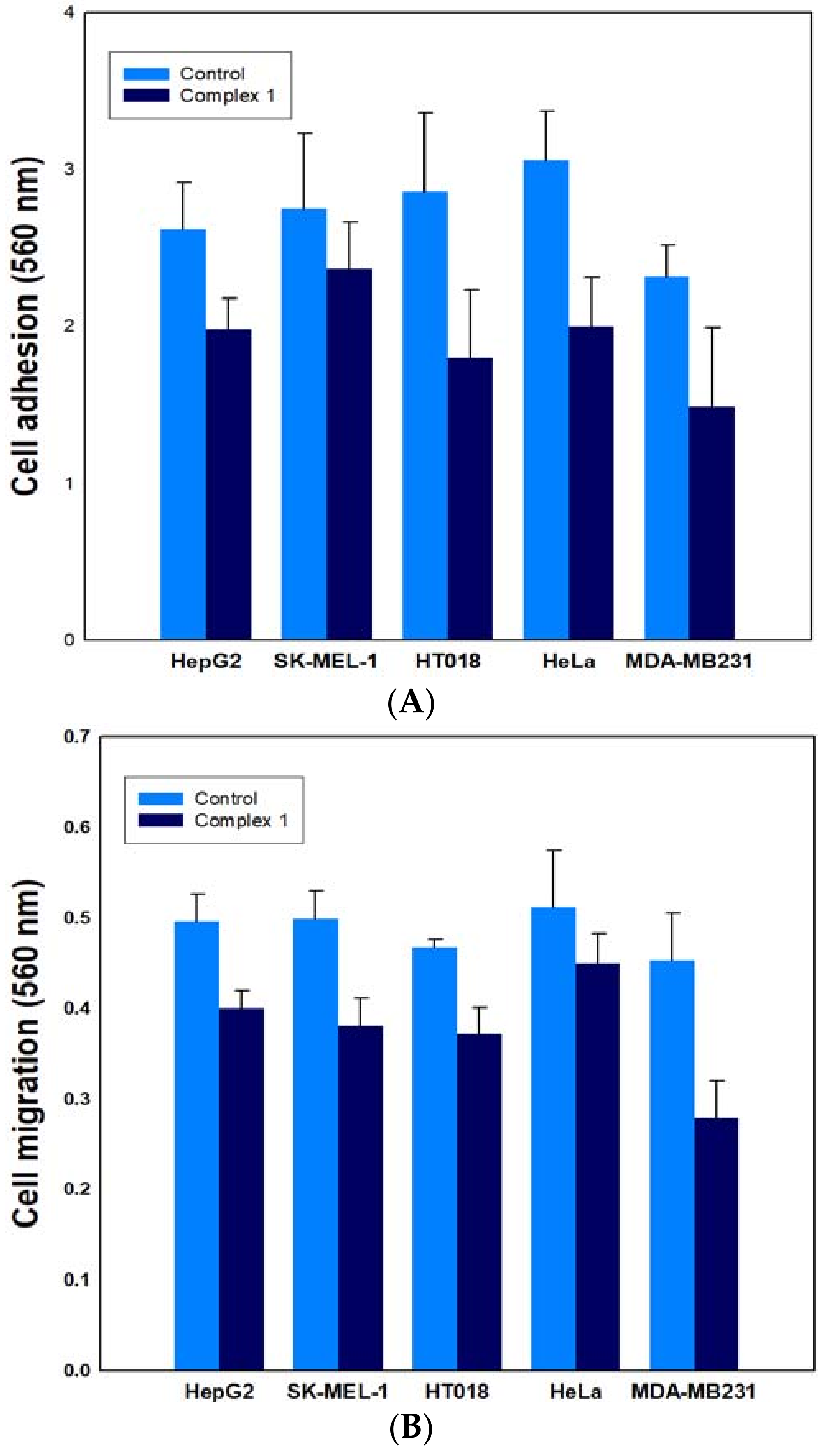
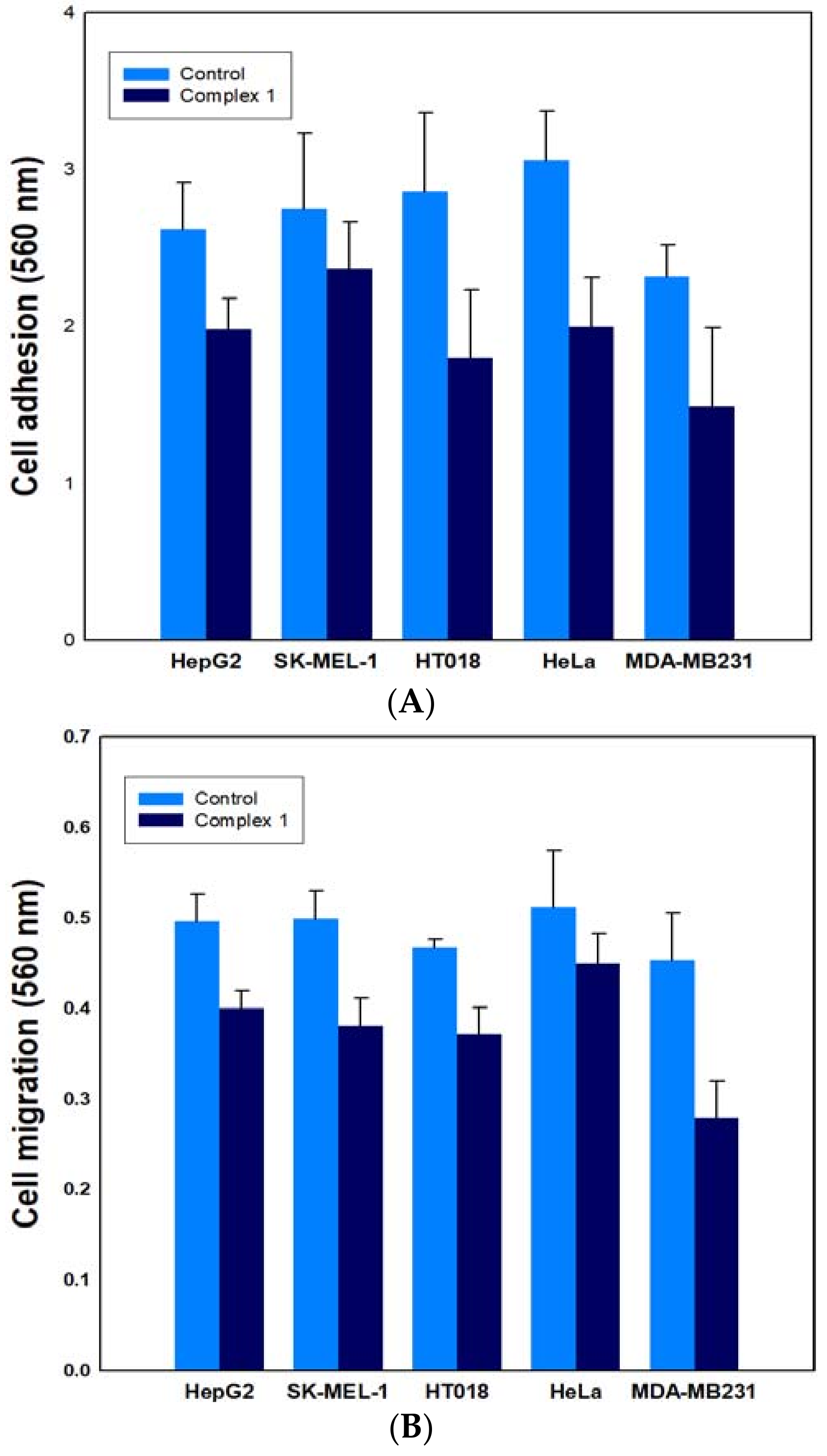
2.6.2. Effect on Cancer Cell Adhesion/Migration
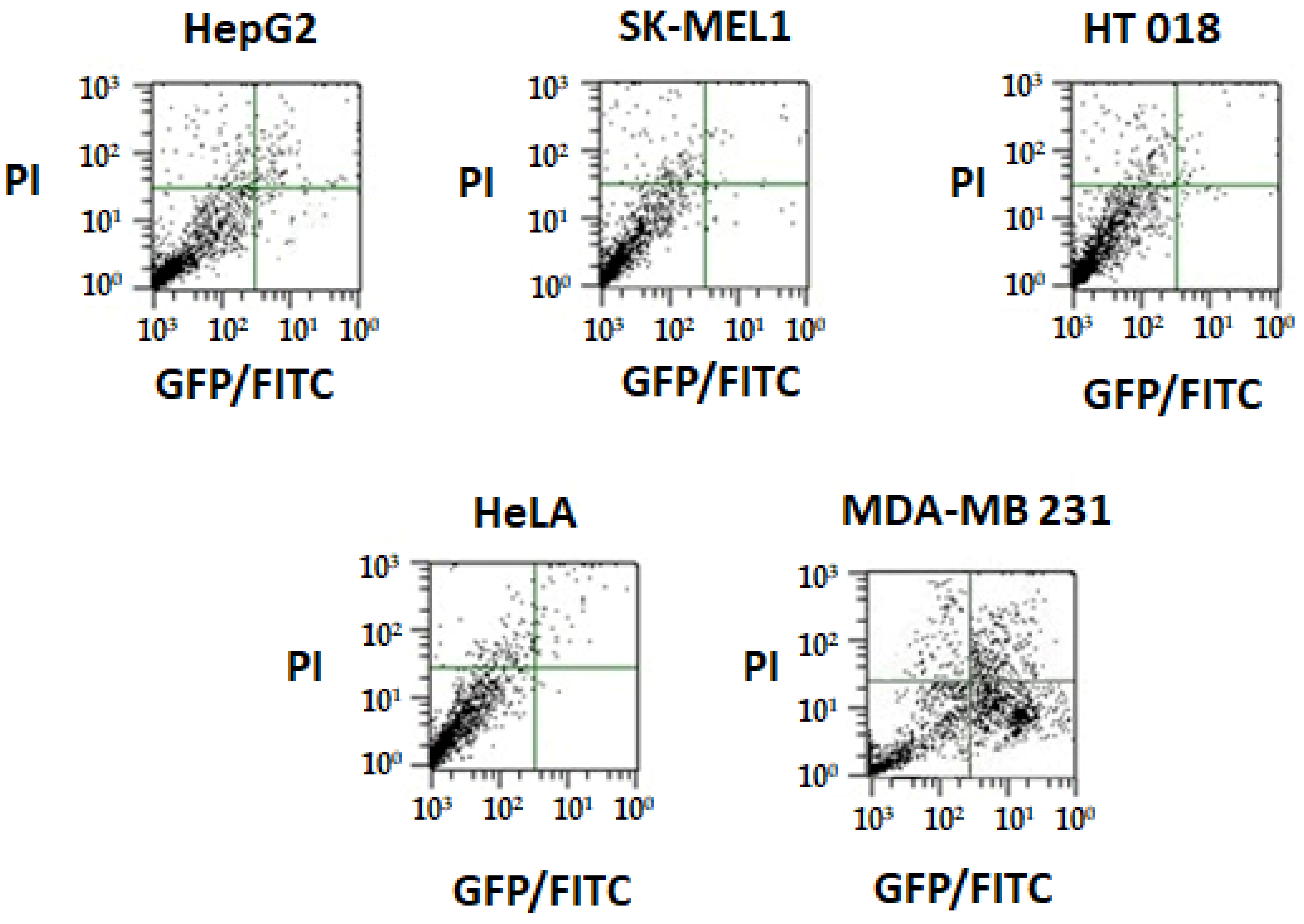
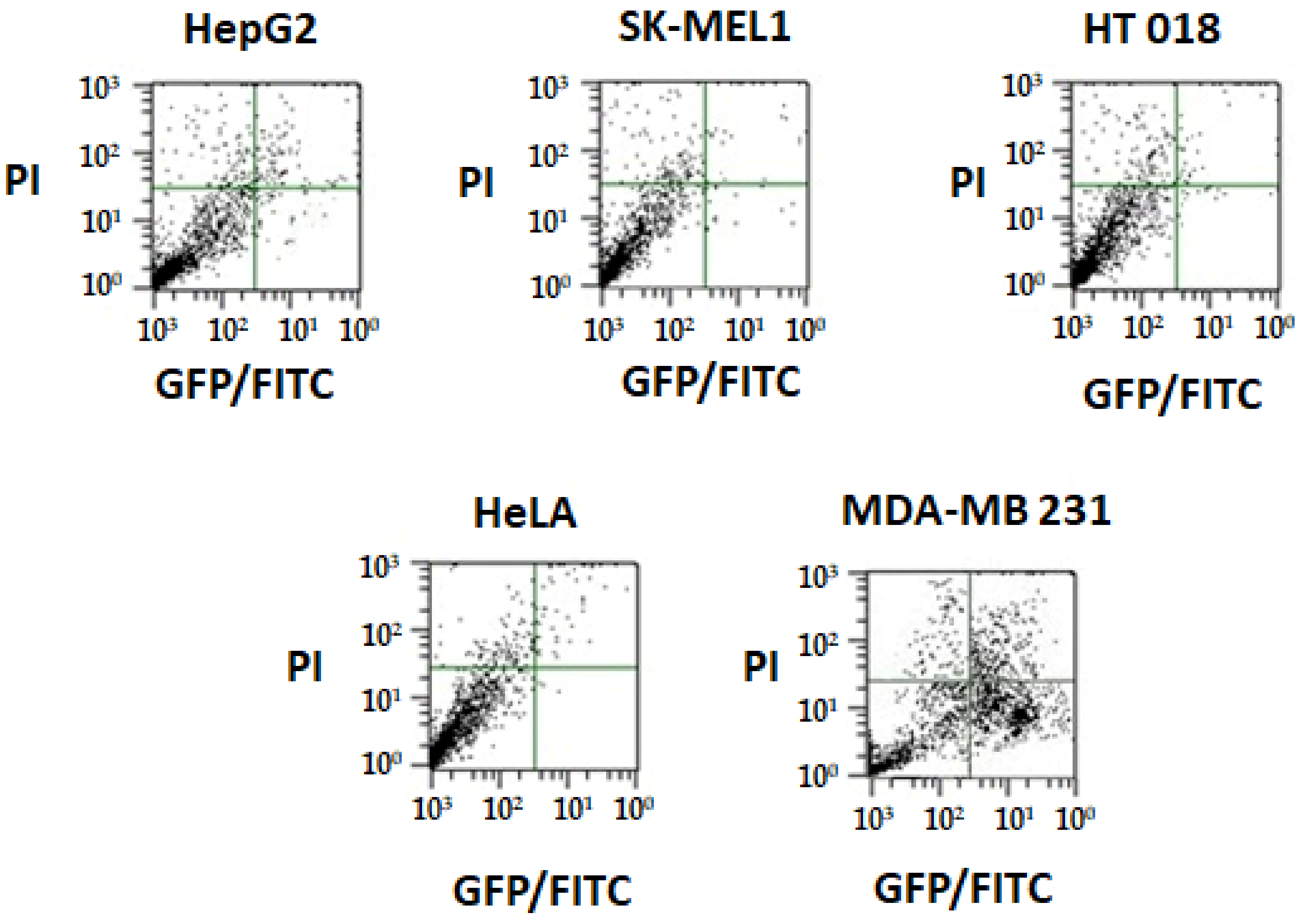
2.6.3. Annexin V Apoptosis Detection Assay
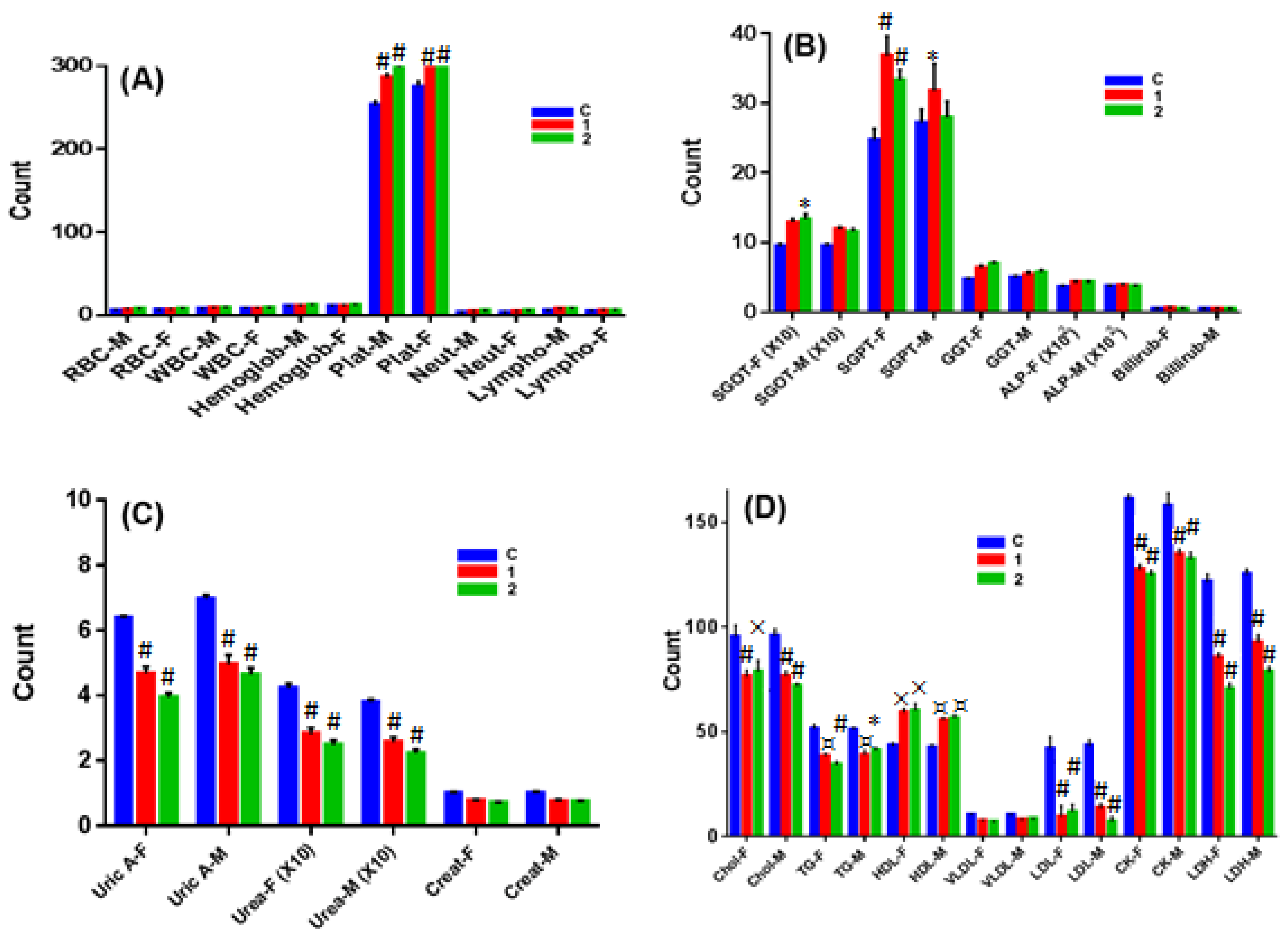
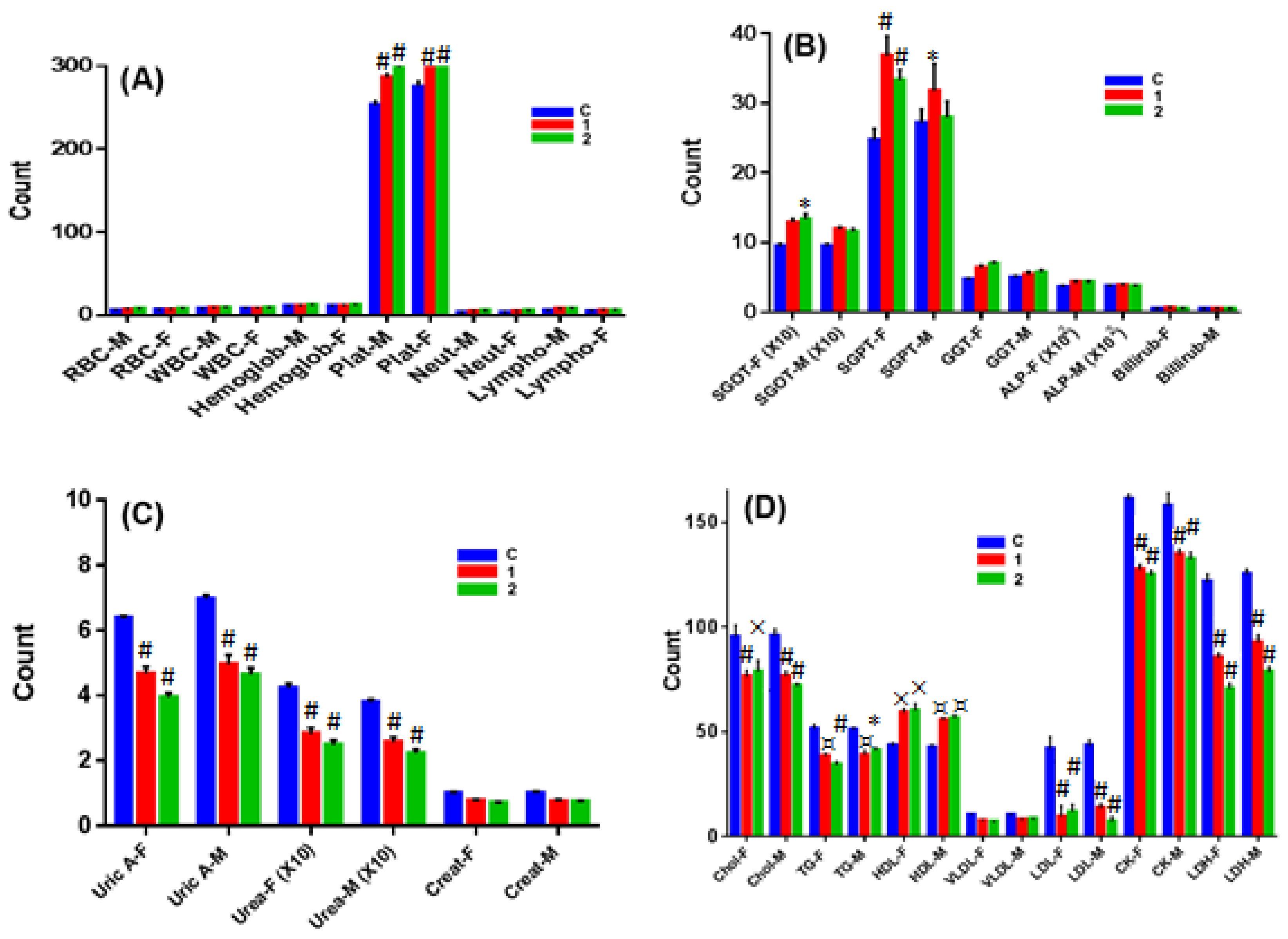
2.7. In Vivo Chronic Toxicity Studies of Complex 1 vs. 2
3. Experimental Section
3.1. Material
3.2. Synthesis Procedure of Schiff Base
3.3. Copper Complex: [Cu(BnI)2], (1)
3.4. Zinc Complex, [Zn(BnI)2], (2)
3.5. Fluorescence Quenching Measurements
3.6. FRET Measurements
3.7. DNA Binding and Nuclease Activity
3.8. Molecular Docking Studies
3.8.1. Preparation of Ligands and Receptors
3.8.2. Molecular Docking of Complexes 1 and 2 with DNA
3.8.3. Molecular Docking of Complexes 1 and 2 with HSA
3.9. Cytotoxicity
3.9.1. Cell Lines and Culture Conditions
3.9.2. MTT Assay
3.9.3. Measurement of Cancer Cell Adhesion
3.9.4. Measurement of Cancer Cell Migration
3.9.5. Analysis of Annexin-V Binding by Flow Cytometry
3.10. In Vivo Toxicity
3.10.1. Toxicity Study Design
3.10.2. Chronic Toxicity Study
3.10.3. Hematological Studies
3.10.4. Serum Analysis of Biochemical Parameters
3.10.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B.; VanCamp, L.; Grimley, E.B.; Thomson, A.J. The inhibition of growth or cell division in Escherichia coli by different ionic species of platinum (IV) complexes. J. Biol. Chem. 1967, 242, 1347–1352. [Google Scholar] [PubMed]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B. Noble metal complexes in cancer chemotherapy. Adv. Exp. Med. Biol. 1977, 91, 129–150. [Google Scholar] [PubMed]
- Rosenberg, B. Platinum Complexes for the Treatment of Cancer. Interdiscip. Sci. Rev. 1978, 3, 134–147. [Google Scholar] [CrossRef]
- Wang, C.H.; Shih, W.C.; Chang, H.C.; Kuo, Y.Y.; Hung, W.C.; Ong, T.G.; Li, W.S. Preparation and Characterization of Amino-Linked Heterocyclic Carbene Palladium, Gold, and Silver Complexes and Their Use as Anticancer Agents That Act by Triggering Apoptotic Cell Death. J. Med. Chem. 2011, 54, 5245–5249. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Khan, A.U. Understanding the interaction between human serum albumin and anti-bacterial/anti-cancer compounds. Curr. Pharm. Des. 2015, 21, 1785–1799. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Guo, Z. Metal-based anticancer chemotherapeutic agents. Curr. Opin. Chem. Biol. 2014, 19, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Ma, Z.Y.; Xie, C.Z.; Xue, F.; Zhang, Y.W.; Xu, J.Y.; Qiang, Z.Y.; Lou, J.S.; Chen, G.J.; Yan, S.P. Study on potential antitumor mechanism of a novel Schiff base copper(II)complex: Synthesis, crystal structure, DNA binding, cytotoxicity and apoptosis induction activity. J. Inorg. Biochem. 2011, 105, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, X.Y.; Hu, M.; Zhu, C.C.; Guo, Z.J. A mitochondrion-targeting copper complex exhibits potent cytotoxicity against cisplatin-resistant tumor cells through a multiple mechanism of action. Chem. Sci. 2014, 5, 2761–2770. [Google Scholar] [CrossRef]
- Khodade, V.S.; Dharmaraja, A.T.; Chakrapani, H. Synthesis, reactive oxygen species generation and copper-mediated nuclease activity profiles of 2-aryl-3-amino-1, 4-naphthoquinones. Bioorg. Med. Chem. Lett. 2012, 22, 3766–3769. [Google Scholar] [CrossRef] [PubMed]
- Palanimuthu, D.; Shinde, S.V.; Somasundaram, K.; Samuelson, A.G. In Vitro and in Vivo Anticancer Activity of Copper Bis(thiosemicarbazone) Complexes. J. Med. Chem. 2013, 56, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Frei, E.; Straubb, J.; Breuerb, A.; Wiesslerb, M. Induction of metallothionein by zinc protects from daunorubicin toxicity in rats. Toxicology 2002, 85, 85–93. [Google Scholar] [CrossRef]
- Pushie, M.J.; Pickering, I.J.; Korbas, M.; Hackett, M.J.; George, G.N. Elemental and Chemically Specific X-ray Fluorescence Imaging of Biological Systems. Chem. Rev. 2014, 114, 8499. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Sharma, G.; Pandey, R.A.; Kumar, A.; Koch, B.; Li, P.-Z.; Xu, Q.; Pandey, D.S. DNA/Protein Binding, Molecular Docking, and in Vitro Anticancer Activity of Some Thioether-Dipyrrinato Complexes. Inorg. Chem. 2013, 52, 13984–13996. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.A.; Li, S.S.; Zhao, D.D.; Chen, S.F.; Hu, J.Y. Metal and structure tuned in vitro antitumor activity of benzimidazole-based copper and zinc complexes. J. Coord. Chem. 2013, 66, 1650–1660. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Mentese, E.; Yilmaz, F.; Ülker, S.; Kahveci, B. Microwave-assisted synthesis of some new coumarin derivatives including 1,2,4-triazol-3-one and investigation of their biological activities. Chem. Heterocycl. Compd. 2015, 50, 447–456. [Google Scholar]
- Sridevi, C.H.; Balaji, K.; Naidu, A.; Sudhakaran, R. Synthesis of Some Phenylpyrazolo Benzimidazolo Quinoxaline Derivatives as Potent Antihistaminic Agents. E-J. Chem. 2010, 7, 234–238. [Google Scholar] [CrossRef]
- Pan, T.; He, X.; Chen, B.; Chen, H.; Geng, G.; Luo, H.; Zhang, H.; Bai, C. Development of benzimidazole derivatives to inhibit HIV-1 replication through protecting APOBEC3G protein. Eur. J. Med. Chem. 2015, 95, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Hiremathad, A.; Budagumpi, S.; Nagaraja, B.M. Comprehensive Review in Current Developments of Benzimidazole-Based Medicinal Chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Narasimhan, B.; Kaur, H. Perspectives of Benzimidazole Derivatives as Anticancer Agents in the New Era. Anticancer Agents Med. Chem. 2016, 16, 1403–1425. [Google Scholar] [CrossRef] [PubMed]
- Ware, W.R. Oxygen quenching of fluorescence in solution: AN experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Kang, J.; Liu, Y.; Xie, M.X.; Li, S.; Jiang, M.; Wang, Y.D. Interactions of human serum albumin with chlorogenic acid and ferulic acid. Biochim. Biophys. Acta 2004, 1674, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, D.; Bourassa, P.; Bruneau, J.; Bérubé, G.; Asselin, E.; Tajmir-Riahi, H.A. Probing the binding sites of antibiotic drugs doxorubicin and N-(trifluoroacetyl) doxorubicin with human and bovine serum albumins. PLoS ONE 2012, 7, e43814. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the Binding Mechanism of Imipenem to Human Serum Albumin by Spectroscopic and Computational Approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Ahmed, S.; Khan, A.U. Interaction of meropenem with ‘N’ and ‘B’ isoforms of human serum albumin: A spectroscopic and molecular docking study. J. Biomol. Struct. Dyn. 2016, 34, 1849–1864. [Google Scholar] [CrossRef] [PubMed]
- Kamtekar, N.; Pandey, A.; Agrawal, N.; Pissurlenkar, R.R.S.; Borana, M.; Ahmad, B. Interaction of multimicrobial synthetic inhibitor 1,2-bis(2-benzimidazolyl)-1,2-ethanediol with serum albumin: Spectroscopic and computational studies. PLoS ONE 2013, 8, e53499. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Intermolecular energy migration and fluorescence. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.; Shimer, G.H.; Meehan, T. Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, L.-J.; Sun, Y.-N.; Qiu, P.-H.; Liang, G. Syntheses, characterization and antitumor activities of transition metal complexes with isoflavone. J. Inorg. Biochem. 2010, 104, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Galal, S.A.; Hegab, K.H.; Kassab, A.S.; Rodriguez, M.L.; Kerwin, S.M.; El-Khamry, A.-M.A.; El Diwani, H.I. New transition metal ion complexes with benzimidazole-5-carboxylic acid hydrazides with antitumor activity. Eur. J. Med. Chem. 2009, 44, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Jabeen, M.; Chauhan, A.; Owais, M. Synthesis and characterization of novel n-9 fatty acid conjugates possessing antineoplastic properties. Lipids 2012, 47, 973–86. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Faheem, M.; Khan, A.U. Insignificant β-lactamase activity of human serum albumin: No panic to nonmicrobial-based drug resistance. Lett. Appl. Microbiol. 2013, 57, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Zaki, M.; Khan, R.A.; Alsalme, A.; Ahmad, M.; Tabassum, S. Coumarin centered copper(II) complex with appended-imidazole as cancer chemotherapeutic agents against lung cancer: Molecular insight via DFT-based vibrational analysis. RSC Adv. 2017, 7, 36056–36071. [Google Scholar] [CrossRef]
- Tabassum, S.; Asim, A.; Khan, R.A.; Arjmand, F.; Divya, R.; Balaji, P.; Akbarsha, M.A. A multifunctional molecular entity CuII–SnIV heterobimetallic complex as a potential cancer chemotherapeutic agent: DNA binding/cleavage, SOD mimetic, topoisomerase Iα inhibitory and in vitro cytotoxic activities. RSC Adv. 2015, 5, 47439–47450. [Google Scholar] [CrossRef]
- Usman, M.; Arjmand, F.; Khan, R.A.; Alsalme, A.; Ahmad, M.; Tabassum, S. Biological evaluation of dinuclear copper complex/dichloroacetic acid cocrystal against human breast cancer: Design, synthesis, characterization, DFT studies and cytotoxicity assays. RSC Adv. 2017, 7, 47920–47932. [Google Scholar] [CrossRef]
- Khan, R.A.; Usman, M.; Rajakumar, D.; Balaji, P.; Alsalme, A.; Arjmand, F.; AlFarhan, K.; Akbarsha, M.A.; Marchetti, F.; Pettinari, C.; et al. Heteroleptic Copper(I) Complexes of “Scorpionate” Bis-pyrazolyl Carboxylate Ligand with Auxiliary Phosphine as Potential Anticancer Agents: An Insight into Cytotoxic Mode. Sci. Rep. 2017, 7, 45229. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 4.0; Accelrys Software Inc.: San Diego, CA, USA, 2013. [Google Scholar]
- Khan, A.A.; Alanazi, A.M.; Jabeen, M.; Chauhan, A.; Abdelhameed, A.S. Design, Synthesis and In Vitro Anticancer Evaluation of a Stearic Acid-based Ester Conjugate. Anticancer Res. 2013, 33, 1217–1224. [Google Scholar]
- Khan, A.A. Pro-apoptotic activity of nano-escheriosome based oleic acid conjugate against 7,12-dimethylbenz(a)anthracene (DMBA) induced cutaneous carcinogenesis. Biomed. Pharmacother. 2017, 90, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Qureshi, S.; Tariq, M.; Ageel, A.M. Toxicity studies on six plants used in the traditional Arab system of medicine. Phytother. Res. 1989, 3, 25–29. [Google Scholar] [CrossRef]
- WHO Scientific Group. Principles for Pre-clinical Testing of Drugs Safety; Technical Report Series; World Health Organization: Geneva, Switzerland, 1967; Volume 341, pp. 9–11. [Google Scholar]
- Edwards, C.R.W.; Bouchier, I.A.D. Davidson’s Principles, and Practice Medicine; Churchill Livingstone Press: London, UK, 1991; p. 492. [Google Scholar]
- Daniel, W.W. Biostatistics: A Foundation for Analysis in the Health Sciences, 6th ed.; Wiley: New York, NY, USA, 1995; pp. 273–303. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Parameters | |||||
| Complex | Temp | KSV | kq | n | Kb |
| (K) | ×104 (M−1) | ×1012 (M−1 s−1) | ×104 (M−1) | ||
| 1 | 298 | 2.02 | 3.54 | 1.09 | 1.58 |
| 303 | 1.75 | 3.24 | 1.12 | 1.27 | |
| 308 | 1.51 | 2.64 | 1.20 | 0.86 | |
| 2 | 298 | 2.74 | 4.79 | 0.99 | 2.78 |
| 303 | 2.39 | 4.19 | 1.03 | 2.19 | |
| 308 | 1.97 | 3.45 | 1.11 | 1.48 | |
| Thermodynamic Parameters | |||||
| Complex | Temp | ∆H | ∆S | T∆S (kcal/mol) | ∆G |
| (K) | (kcal/mol) | (cal/mol/K) | (kcal/mol) | ||
| 1 | 298 | −11.12 | −18.17 | −5.41 | −5.71 |
| 303 | −5.50 | −5.62 | |||
| 308 | −5.60 | −5.52 | |||
| 2 | 298 | −11.48 | −18.14 | −5.40 | −6.08 |
| 303 | −5.49 | −5.99 | |||
| 308 | −5.59 | −5.89 | |||
| FRET Parameters | |||||
| Complex | J | Ro | R | ||
| ×10−15 (M−1 cm−1) | (nm) | (nm) | |||
| 1 | 3.26 | 2.04 | 2.35 | ||
| 2 | 4.63 | 2.16 | 2.36 | ||
| Interaction | Nature of Interaction | Bond Length (Å) | Binding Affinity, Kd (M−1) | ΔG (kcal/mol) |
|---|---|---|---|---|
| Complex 1 | ||||
| Unk:C—Asp451:Oδ2 | Hydrogen Bond | 3.71 | 7.8 × 106 | −9.4 |
| Lys199:Cβ—Unk | Hydrophobic (π-σ) | 3.89 | ||
| Trp214—Unk | Hydrophobic (π-π) | 4.79 | ||
| His242—Unk | Hydrophobic (π-π) | 5.25 | ||
| Unk—Trp214 | Hydrophobic (π-π) | 4.91 | ||
| Unk—Arg218 | Hydrophobic (π-alkyl) | 5.23 | ||
| Unk—Lys195 | Hydrophobic (π-alkyl) | 4.92 | ||
| Complex 2 | ||||
| Unk:C—His242:Ne2 | Hydrogen Bond | 3.25 | 4.2 × 107 | −10.4 |
| Unk:C—Ala291:O | Hydrogen Bond | 3.62 | ||
| Lys199:Cβ—Unk | Hydrophobic (π-σ) | 3.68 | ||
| Ala291:Cβ—Unk | Hydrophobic (π-σ) | 3.59 | ||
| His242—Unk | Hydrophobic (π-π) | 4.86 | ||
| His242—Unk | Hydrophobic (π-π) | 4.69 | ||
| Unk—Trp214 | Hydrophobic (π-π) | 5.05 | ||
| Unk—Lys195 | Hydrophobic (π-alkyl) | 5.02 | ||
| Unk—Leu238 | Hydrophobic (π-alkyl) | 5.15 | ||
| Interaction | Bond Distance (Å) | Nature of Interaction | Etotal Value |
|---|---|---|---|
| Complex 1 | |||
| Unk:H—B:d21:O2 | 2.57 | Hydrogen Bond | −238.14 |
| Unk:H—B:dG22:O4′ | 2.25 | Hydrogen Bond | |
| Unk:H—A:dA5:N3 | 2.34 | Hydrogen Bond | |
| Unk:H—A:dA5:N3 | 2.98 | Hydrogen Bond | |
| Unk:H—B:d21:O2 | 2.79 | Hydrogen Bond | |
| B:d23:OP1—Unk | 4.63 | Electrostatic (π-Anion) | |
| Complex 2 | |||
| Zn—B:dA18:OP1 | 5.41 | Electrostatic | −248.34 |
| Unk:H—A:dG10:O3′ | 2.01 | Hydrogen Bond | |
| Unk:H—A:d11:O4′ | 2.89 | Hydrogen Bond | |
| Unk:H—A:dG10:OP1 | 2.93 | Hydrogen Bond | |
| Unk:H—B:dA18:O3′ | 2.62 | Hydrogen Bond | |
| Unk:H—B:dT19:OP1 | 2.81 | Hydrogen Bond | |
| B:dA18:OP1—Unk | 2.88 | Electrostatic (π-Anion) | |
| B:dA18:OP2—Unk | 4.88 | Electrostatic (π-Anion) | |
| B:dA17:O3′—Unk | 2.87 | π-Lone Pair | |
| B:dA17:O3′—Unk | 2.55 | π-Lone Pair | |
| Complex | HepG2 | SK-MEL-1 | HT018 | HeLa | MDA-MB 231 |
|---|---|---|---|---|---|
| (Liver) | (Skin) | (Colon) | (Cervical) | (Breast) | |
| (μM) | (μM) | (μM) | (μM) | (μM) | |
| 1 | 14 ± 2.2 | 17.8 ± 2.7 | 15 ± 2.1 | 13 ± 2.2 | 3.5 ± 2.4 |
| 2 | 19 ± 2.6 | 18 ± 2.3 | 25 ± 4.0 | 24.5 ± 2.2 | 26.7 ± 4.1 |
| Ligand | NA | NA | NA | NA | NA |
| Vehicle control (0.1% DMSO) | NA | NA | NA | NA | NA |
| Cisplatin (Positive control) | 6 ± 0.4 | 5.6 ± 0.8 | 5.7 ± 0.2 | 6 ± 0.6 | 3.1 ± 0.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlAjmi, M.F.; Hussain, A.; Rehman, M.T.; Khan, A.A.; Shaikh, P.A.; Khan, R.A. Design, Synthesis, and Biological Evaluation of Benzimidazole-Derived Biocompatible Copper(II) and Zinc(II) Complexes as Anticancer Chemotherapeutics. Int. J. Mol. Sci. 2018, 19, 1492. https://doi.org/10.3390/ijms19051492
AlAjmi MF, Hussain A, Rehman MT, Khan AA, Shaikh PA, Khan RA. Design, Synthesis, and Biological Evaluation of Benzimidazole-Derived Biocompatible Copper(II) and Zinc(II) Complexes as Anticancer Chemotherapeutics. International Journal of Molecular Sciences. 2018; 19(5):1492. https://doi.org/10.3390/ijms19051492
Chicago/Turabian StyleAlAjmi, Mohamed F., Afzal Hussain, Md. Tabish Rehman, Azmat Ali Khan, Perwez Alam Shaikh, and Rais Ahmad Khan. 2018. "Design, Synthesis, and Biological Evaluation of Benzimidazole-Derived Biocompatible Copper(II) and Zinc(II) Complexes as Anticancer Chemotherapeutics" International Journal of Molecular Sciences 19, no. 5: 1492. https://doi.org/10.3390/ijms19051492
APA StyleAlAjmi, M. F., Hussain, A., Rehman, M. T., Khan, A. A., Shaikh, P. A., & Khan, R. A. (2018). Design, Synthesis, and Biological Evaluation of Benzimidazole-Derived Biocompatible Copper(II) and Zinc(II) Complexes as Anticancer Chemotherapeutics. International Journal of Molecular Sciences, 19(5), 1492. https://doi.org/10.3390/ijms19051492