PIMT/NCOA6IP Deletion in the Mouse Heart Causes Delayed Cardiomyopathy Attributable to Perturbation in Energy Metabolism


 and
and
Abstract
1. Introduction
2. Results
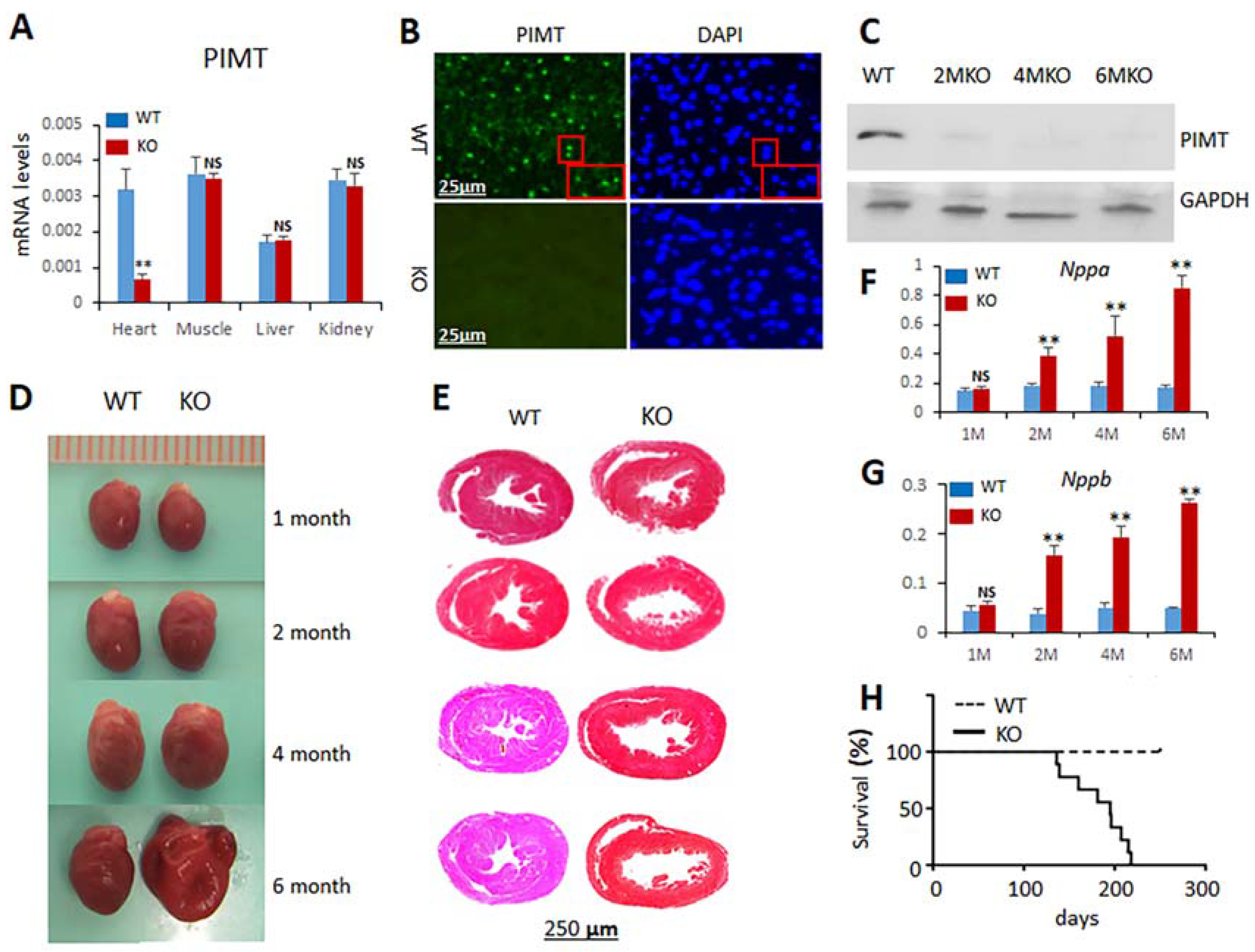
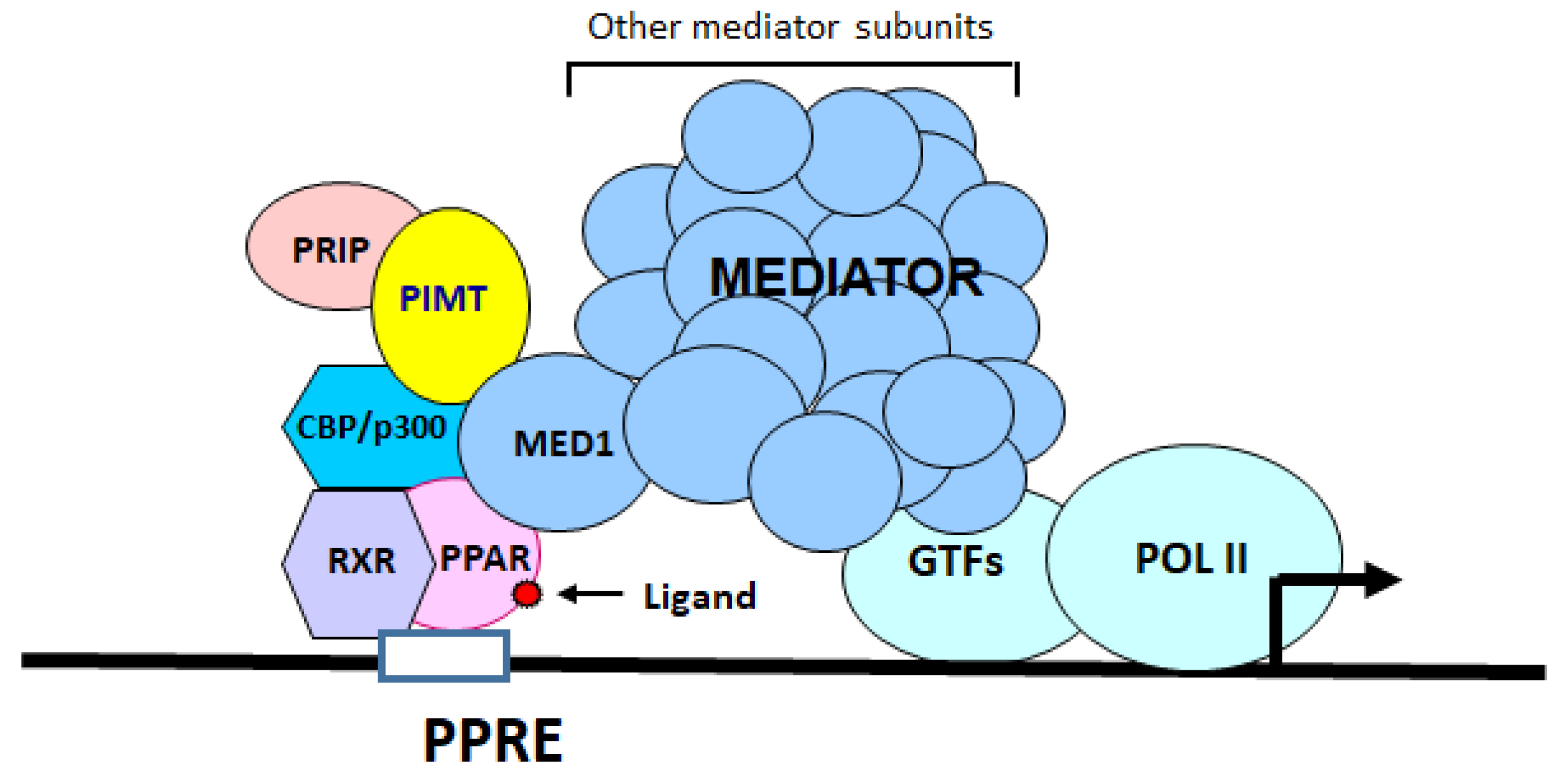
2.1. Generation of Cardiomyocyte-Specific PIMT Heart Knockout Mice
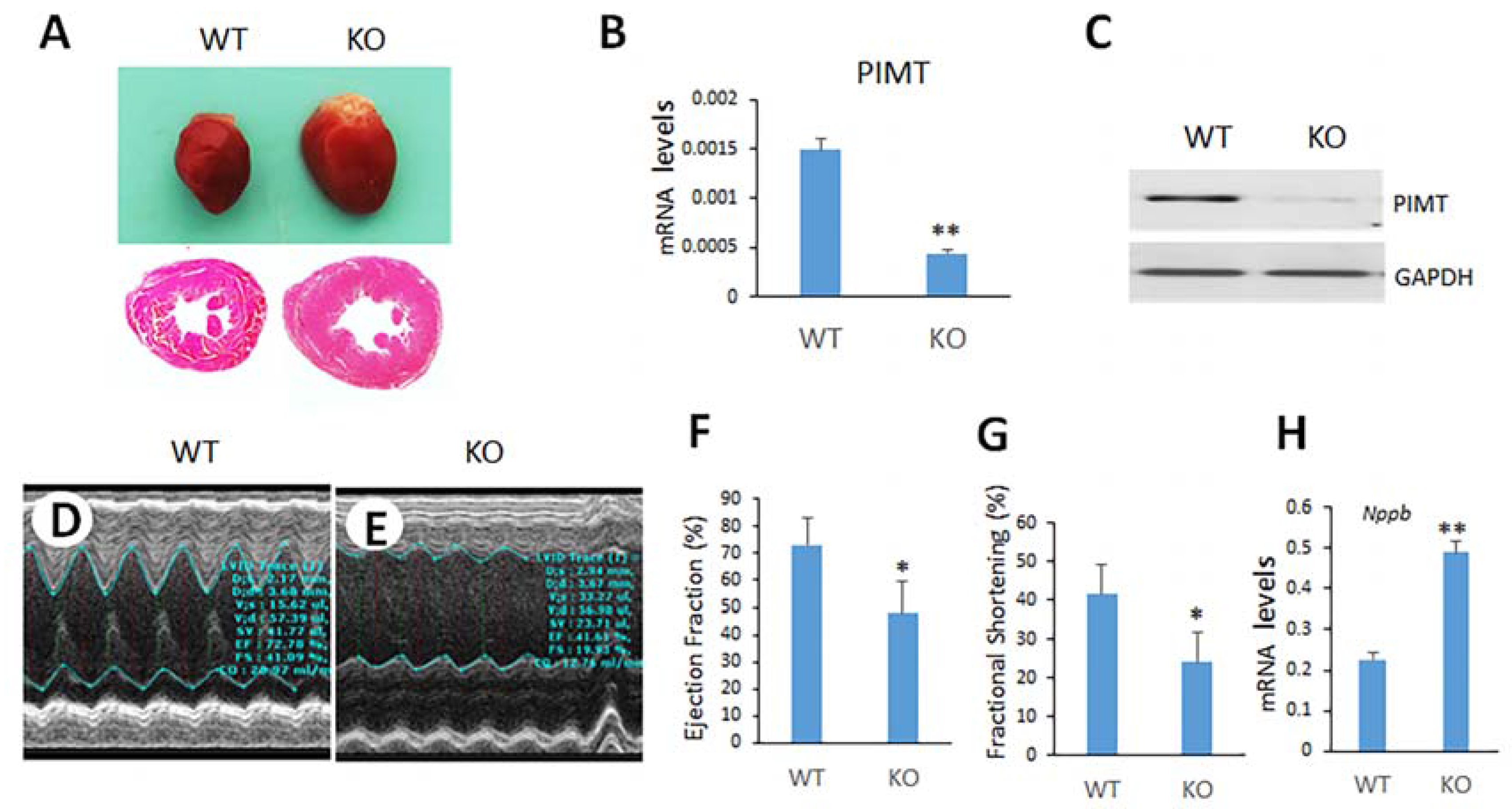
2.2. Cardiomyocyte-Specific Disruption of PIMT Causes Dilated Cardiomyopathy in Mice
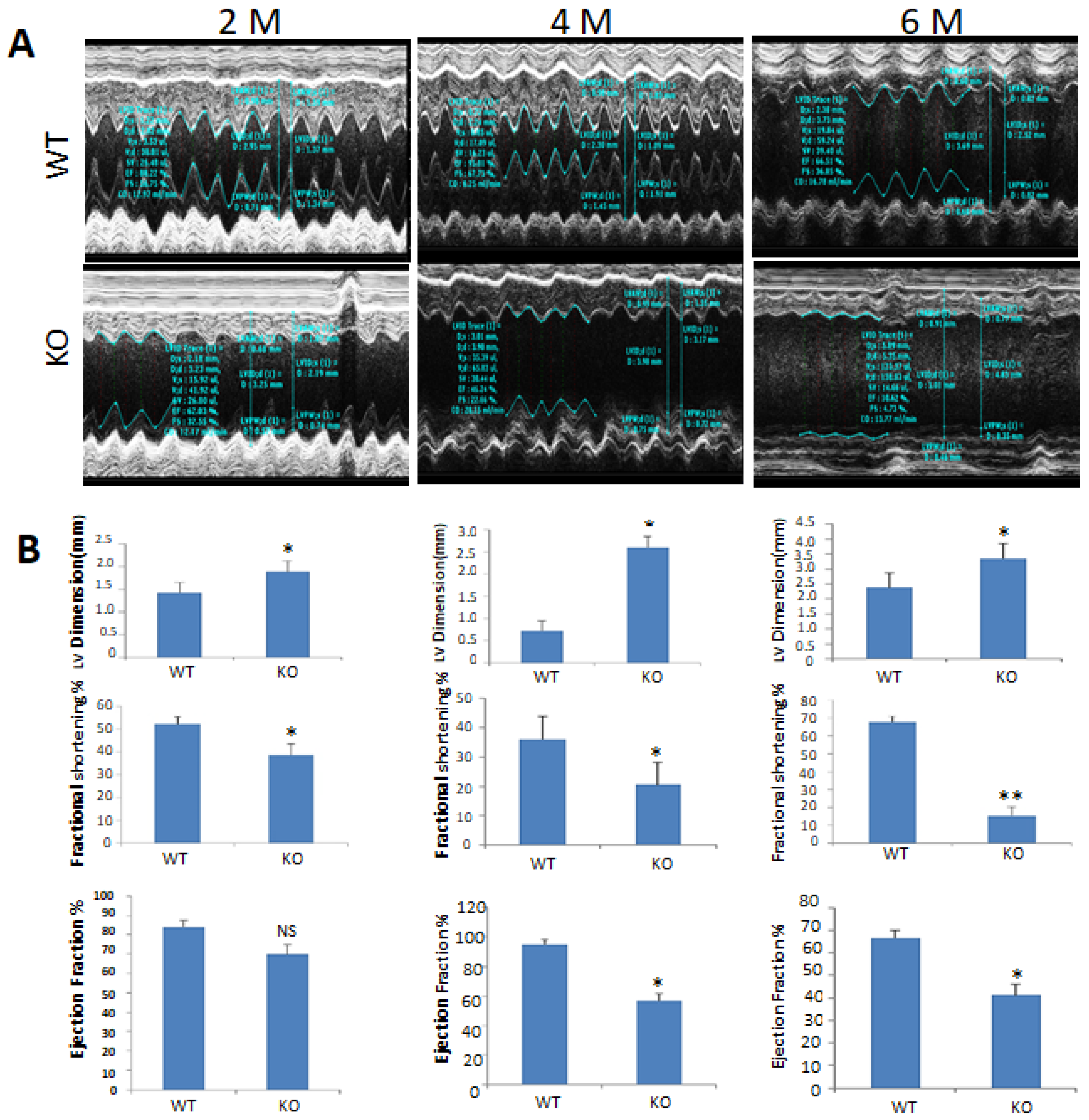
2.3. Echocardiographic Observations of csPIMT−/− Mouse Heart Indicate Poor Contractility
2.4. Global RNA Sequence Analysis of csPIMT−/− Hearts Suggests that Loss of PIMT Affects Multiple Pathways That Are Critical for Heart Function
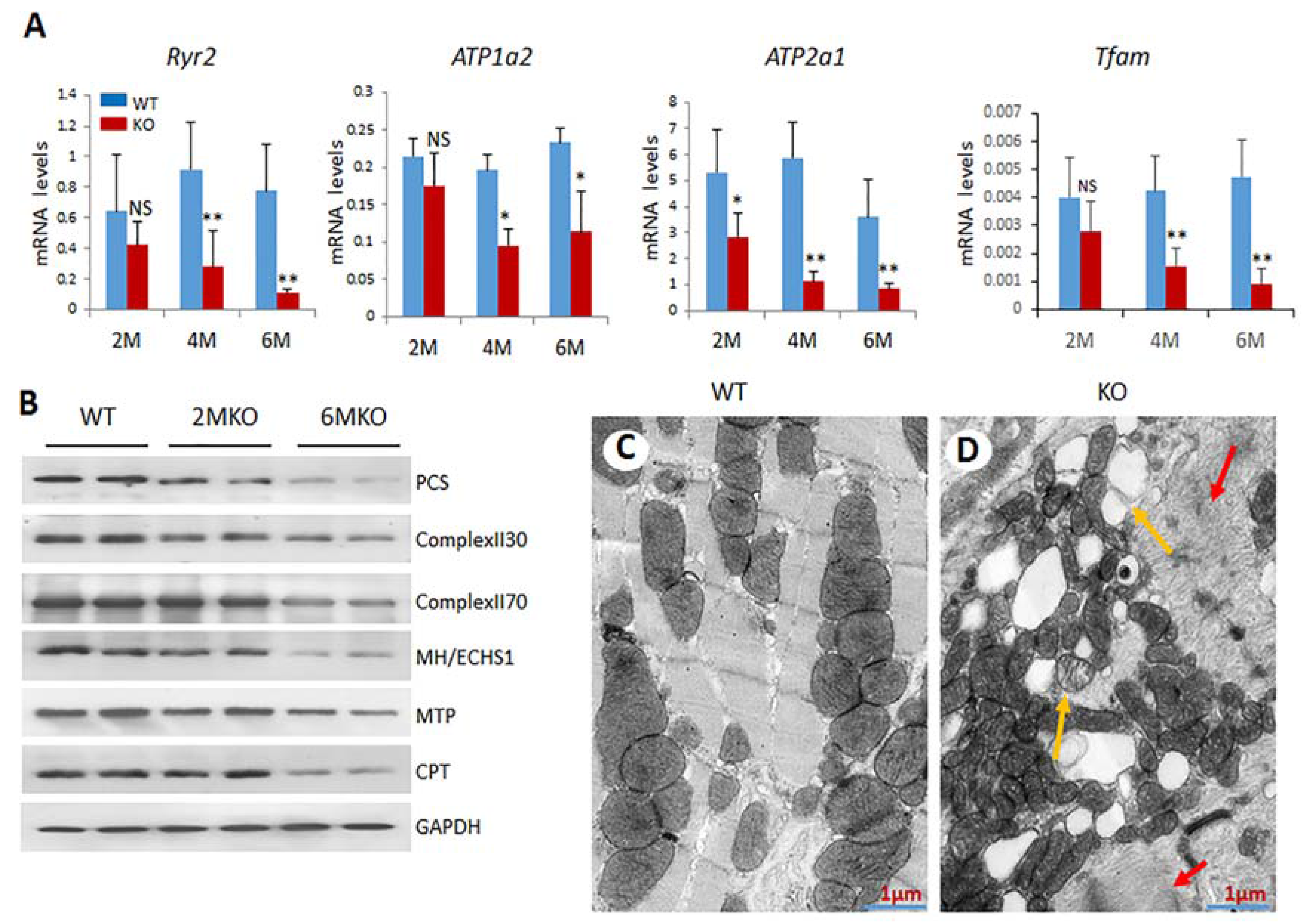
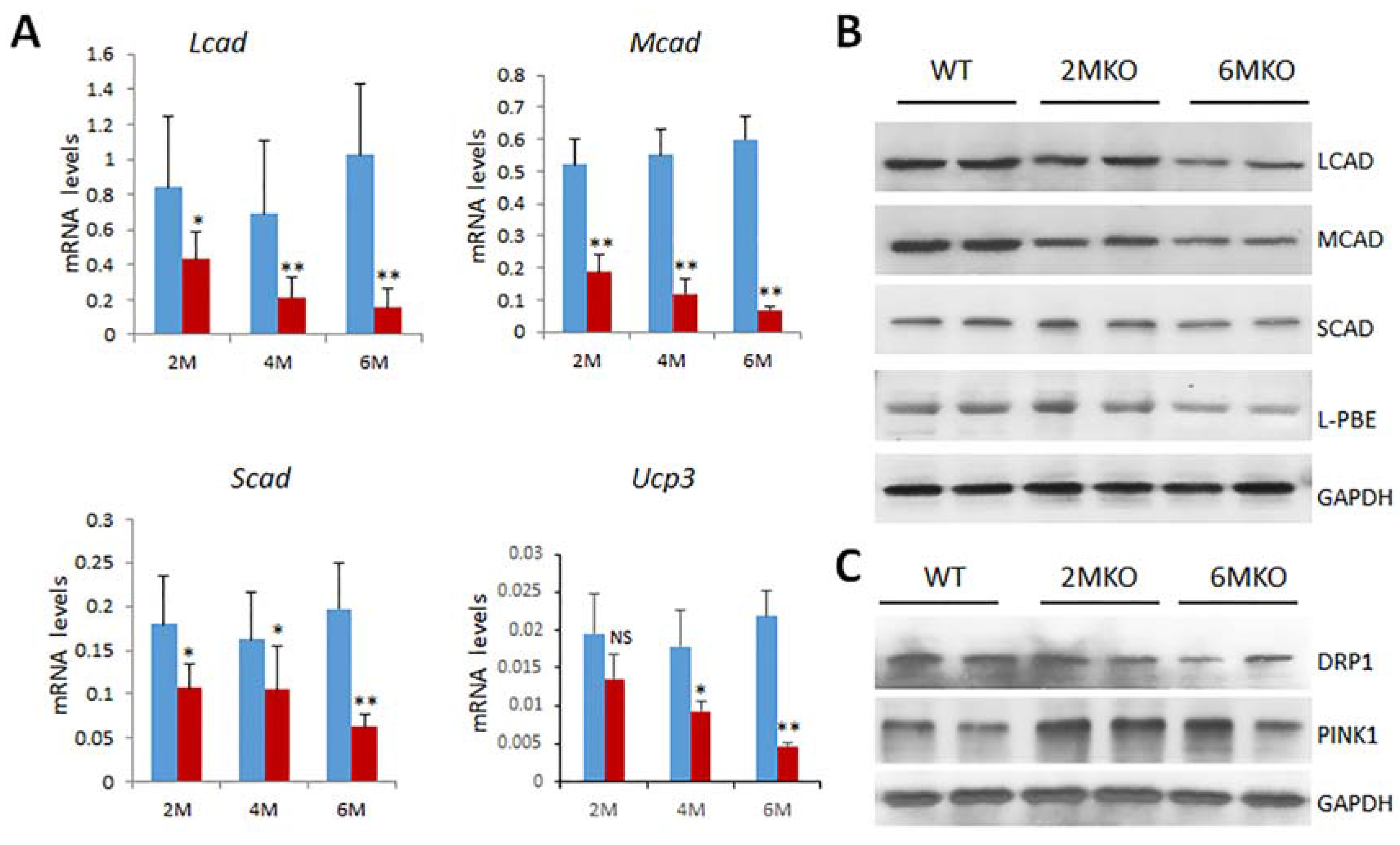
2.5. Reduced Expression of Genes Related to Mitochondrial Functions in csPIMT−/− Hearts
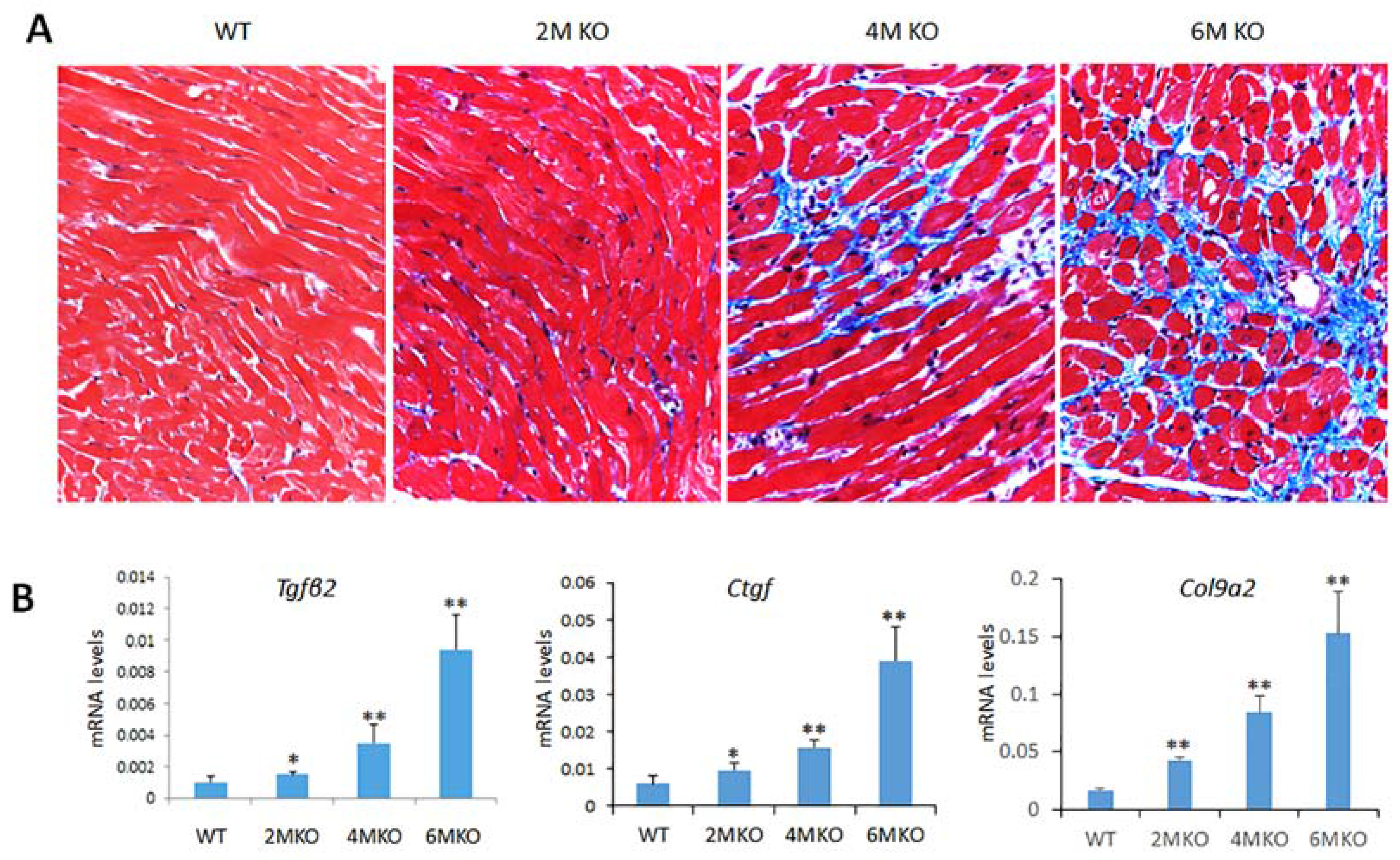
2.6. csPIMT−/− Mice Develop Cardiac Fibrosis
2.7. Genes Related to Glucose Metabolism Are Downregulated in csPIMT−/− Heart Leading to Glycogen Storage
2.8. Tamoxifen-Inducible Heart-Specific Cre-Recombinase to Disrupt PIMT Gene (TmcsPIMT−/−) in Adult Mouse
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Echocardiography
4.3. Histological Analysis
4.4. Electron Microscopy
4.5. Library Construction and Sequencing
4.6. Transcriptome Analysis
4.7. Quantitative Real-Time PCR
4.8. Western Blot Analysis
4.9. Mitochondrial DNA Content
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zhu, Y.; Qi, C.; Cao, W.Q.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Cloning and characterization of PIMT, a protein with a methyltransferase domain, which interacts with and enhances nuclear receptor coactivator PRIP function. Proc. Natl. Acad. Sci. USA 2001, 98, 10380–10385. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, J.O.; Zhu, X.; Gustafsson, C.M. The multitalented Mediator complex. Trends Biochem. Sci. 2013, 38, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Viswakarma, N.; Reddy, J.K. Med1 subunit of the mediator complex in nuclear receptor-regulated energy metabolism, liver regeneration, and hepatocarcinogenesis. Gene Expr. 2014, 16, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARβ/δ and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [PubMed]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Mouaikel, J.; Bujnicki, J.M.; Tazi, J.; Bordonne, R. Sequence-structure-function relationships of Tgs1, the yeast snRNA/snoRNA cap hypermethylase. Nucleic Acids Res. 2003, 31, 4899–4909. [Google Scholar] [CrossRef] [PubMed]
- Mouaikel, J.; Verheggen, C.; Bertrand, E.; Tazi, J.; Bordonne, R. Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol. Cell 2002, 9, 891–901. [Google Scholar] [CrossRef]
- Misra, P.; Qi, C.; Yu, S.; Shah, S.H.; Cao, W.Q.; Rao, M.S.; Thimmapaya, B.; Zhu, Y.; Reddy, J.K. Interaction of PIMT with transcriptional coactivators CBP, p300, and PBP differential role in transcriptional regulation. J. Biol. Chem. 2002, 277, 20011–20019. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chang, H.C.; Schipma, M.J.; Liu, J.; Shete, V.; Liu, N.; Sato, T.; Thorp, E.B.; Barger, P.M.; Zhu, Y.J.; et al. Cardiomyocyte-Specific Ablation of Med1 Subunit of the Mediator Complex Causes Lethal Dilated Cardiomyopathy in Mice. PLoS ONE 2016, 11, e0160755. [Google Scholar]
- Roh, J.I.; Cheong, C.; Sung, Y.H.; Lee, J.; Oh, J.; Lee, B.S.; Lee, J.E.; Gho, Y.S.; Kim, D.K.; Park, C.B.; et al. Perturbation of NCOA6 leads to dilated cardiomyopathy. Cell Rep. 2014, 8, 991–998. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, Y.; Viswakarma, N.; Crawford, S.E.; Sarkar, J.; Sambasiva Rao, M.; Karpus, W.J.; Kanwar, Y.S.; Zhu, Y.J.; Reddy, J.K. Early embryonic lethality of mice with disrupted transcription cofactor PIMT/NCOA6IP/Tgs1 gene. Mech. Dev. 2012, 129, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Nakao, K. Regulation and significance of atrial and brain natriuretic peptides as cardiac hormones. Endocr. J. 2010, 57, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Bar-Yaacov, D.; Blumberg, A.; Mishmar, D. Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim. Biophys. Acta 2012, 1819, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell. Dev. Biol. 2017, 76, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Vonck, J.; Schafer, E. Supramolecular organization of protein complexes in the mitochondrial inner membrane. Biochim. Biophys. Acta 2009, 1793, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [PubMed]
- Ikeda, Y.; Shirakabe, A.; Brady, C.; Zablocki, D.; Ohishi, M.; Sadoshima, J. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J. Mol. Cell. Cardiol. 2015, 78, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, U.A.; Ong, S.B.; Ong, S.G.; Hausenloy, D.J. Parkinson’s disease proteins: Novel mitochondrial targets for cardioprotection. Pharmacol. Ther. 2015, 156, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Shirihai, O.S.; Song, M.; Dorn, G.W., 2nd. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd. Parkin-dependent mitophagy in the heart. J. Mol. Cell. Cardiol. 2016, 95, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires TGF-β. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Byers, M.S.; Howard, C.; Wang, X. Avian and Mammalian Facilitative Glucose Transporters. Microarrays 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Kapadia, B.; Viswakarma, N.; Seshadri, S.; Prajapati, B.; Jena, P.K.; Teja Meda, C.L.; Subramanian, M.; Kaimal Suraj, S.; Kumar, S.T.; et al. Co-activator binding protein PIMT mediates TNF-alpha induced insulin resistance in skeletal muscle via the transcriptional down-regulation of MEF2A and GLUT4. Sci. Rep. 2015, 5, 15197. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Glucose transporters in healthy heart and in cardiac disease. Int. J. Cardiol. 2017, 230, 70–75. [Google Scholar] [CrossRef] [PubMed]
- McCommis, K.S.; Douglas, D.L.; Krenz, M.; Baines, C.P. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J. Am. Heart Assoc. 2013, 2, e000355. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Rider, M.H.; Hue, L. Mechanisms of control of heart glycolysis. Eur. J. Biochem. 1998, 258, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Spitler, K.M.; Ponce, J.M.; Oudit, G.Y.; Hall, D.D.; Grueter, C.E. Cardiac Med1 deletion promotes early lethality, cardiac remodeling, and transcriptional reprogramming. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H768–H780. [Google Scholar] [CrossRef] [PubMed]
- Krebs, P.; Fan, W.; Chen, Y.H.; Tobita, K.; Downes, M.R.; Wood, M.R.; Sun, L.; Li, X.; Xia, Y.; Ding, N.; et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc. Natl. Acad. Sci. USA 2011, 108, 19678–19682. [Google Scholar] [CrossRef] [PubMed]
- Baskin, K.K.; Makarewich, C.A.; DeLeon, S.M.; Ye, W.; Chen, B.; Beetz, N.; Schrewe, H.; Bassel-Duby, R.; Olson, E.N. MED12 regulates a transcriptional network of calcium-handling genes in the heart. JCI Insight 2017, 2, e91920. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, F. Mediating lipid biosynthesis: Implications for cardiovascular disease. Trends Cardiovasc. Med. 2013, 23, 269–273. [Google Scholar]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, A.; Marks, A.R. The ryanodine receptor in cardiac physiology and disease. Adv. Pharmacol. 2010, 59, 1–30. [Google Scholar] [PubMed]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Agah, R.; Frenkel, P.A.; French, B.A.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Investig. 1997, 100, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Tabas-Madrid, D.; Nogales-Cadenas, R.; Pascual-Montano, A. GeneCodis3: A non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012, 40, W478–W483. [Google Scholar] [CrossRef] [PubMed]
- Nogales-Cadenas, R.; Carmona-Saez, P.; Vazquez, M.; Vicente, C.; Yang, X.; Tirado, F.; Carazo, J.M.; Pascual-Montano, A. GeneCodis: Interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 2009, 37, W317–W322. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Chang, H.C.; Khechaduri, A.; Chawla, K.; Tran, M.; Chai, X.; Wagg, C.; Ghanefar, M.; Jiang, X.; Bayeva, M.; et al. Cardiac-specific ablation of ARNT leads to lipotoxicity and cardiomyopathy. J. Clin. Investig. 2014, 124, 4795–4806. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Gene | KO/WT | KO/WT | KO/WT | KO/WT |
|---|---|---|---|---|---|
| 2 M, qPCR | 2 M RNA-Seq | 6 M, qPCR | 6 M, RNA-Seq | ||
| OXPHOS | |||||
| Ndufaf4 | 0.35 * | 0.21 | 0.18 ** | 0.20 | |
| Ndufaf5 | 0.29 * | 0.33 | 0.21 ** | 0.17 | |
| Ndufs4 | 0.51 * | 0.42 | 0.27 ** | 0.24 | |
| Cox7b | 0.32 * | 0.27 | 0.36 * | 0.41 | |
| Cox10 | 0.59 | 0.27 | 0.25 ** | 0.28 | |
| Sdha | 0.63 | 0.52 | 0.41 * | 0.37 | |
| Sucla2 | 0.24 * | 0.12 | 0.12 ** | 0.08 | |
| Energy metabolism/fatty acid | |||||
| Pparα | 0.54 | 0.66 | 0.49 | 0.57 | |
| Ppargc1a | 0.51 | 0.36 | 0.55 | 0.45 | |
| Acadm | 0.36 * | 0.39 | 0.12 ** | 0.27 | |
| Ucp3 | 0.63 | 0.61 | 0.15 ** | 0.24 | |
| Abcc9 | 0.61 | 0.67 | 0.16 * | 0.49 | |
| Glucose metabolism | |||||
| Gck(GK) | 3.93 * | 1.97 | 2.46 * | 3.87 | |
| Pck1 | 1.88 | 4.23 | 0.51 | 0.63 | |
| Pdk4 | 0.38 * | 0.37 | 0.24 * | 0.99 | |
| HK2 | 0.74 | 0.97 | 0.45 * | 0.83 | |
| Glut4 | 0.59 | 0.92 | 0.36 * | 0.78 | |
| Transcription factor, coactivator | |||||
| Med1 | 0.64 | 0.51 | 0.66 | 0.48 | |
| NcoA6 | 0.69 | 0.66 | 0.57 | 0.62 | |
| Tfam | 0.47 | 0.25 | 0.19 ** | 0.48 | |
| Mitophagy/mitochondria fission | |||||
| Pink1 | 2.16 * | 1.93 | 1.57 | 1.65 | |
| Drp1 | 0.58 | 0.41 | 0.39 * | 0.46 | |
| Cardiomyopathy/Fibrosis | |||||
| Tgfb2 | 0.66 | 0.91 | 9.41 ** | 5.23 | |
| Ctgf | 1.16 | 2.83 | 6.42 ** | 7.71 | |
| Col9a2 | 3.24 * | 6.42 | 8.33 ** | 26.06 | |
| Fgf6 | 1.86 | 2.37 | 10.92 ** | 17.65 | |
| Fgf21 | 2.35 * | 3.08 | 5.18 ** | 6.22 | |
| Mmp3 | 3.92 * | 2.37 | 10.86 ** | 7.61 | |
| Timp1 | 4.58 ** | 4.97 | 4.63 ** | 6.18 | |
| Hypertrophy/dilation | |||||
| Atf3 | 1.19 | 1.57 | 3.91 * | 3.02 | |
| Ace | 2.69 * | 2.23 | 3.78 * | 6.48 | |
| Wisp2 | 4.72 ** | 3.62 | 11.61 ** | 8.74 | |
| Thbs4 | 5.91 ** | 6.68 | 9.34 ** | 8.56 | |
| Calcium homeostasis and signaling pathway | |||||
| Atp2b1 | 0.34 * | 0.22 | 0.31 * | 0.25 | |
| Atp2a1 | 0.53 | 0.52 | 0.23 * | 0.43 | |
| Ryr2 | 0.65 | 0.53 | 0.14 ** | 0.54 | |
| Map3k6 | 2.14 | 3.07 | 3.01 * | 8.01 | |
| Cacnb1 | 2.49 * | 1.98 | 3.72 * | 5.12 | |
| Pde1c | 0.38 | 0.21 | 0.21 ** | 0.14 | |
| Cacna1h | 0.29 * | 0.23 | 0.19 ** | 0.01 | |
| Mapk8 | 0.27 * | 0.21 | 0.33 * | 0.18 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Liu, N.; Viswakarma, N.; Sun, R.; Schipma, M.J.; Shang, M.; Thorp, E.B.; Kanwar, Y.S.; Thimmapaya, B.; Reddy, J.K. PIMT/NCOA6IP Deletion in the Mouse Heart Causes Delayed Cardiomyopathy Attributable to Perturbation in Energy Metabolism. Int. J. Mol. Sci. 2018, 19, 1485. https://doi.org/10.3390/ijms19051485
Jia Y, Liu N, Viswakarma N, Sun R, Schipma MJ, Shang M, Thorp EB, Kanwar YS, Thimmapaya B, Reddy JK. PIMT/NCOA6IP Deletion in the Mouse Heart Causes Delayed Cardiomyopathy Attributable to Perturbation in Energy Metabolism. International Journal of Molecular Sciences. 2018; 19(5):1485. https://doi.org/10.3390/ijms19051485
Chicago/Turabian StyleJia, Yuzhi, Ning Liu, Navin Viswakarma, Ruya Sun, Mathew J. Schipma, Meng Shang, Edward B. Thorp, Yashpal S. Kanwar, Bayar Thimmapaya, and Janardan K. Reddy. 2018. "PIMT/NCOA6IP Deletion in the Mouse Heart Causes Delayed Cardiomyopathy Attributable to Perturbation in Energy Metabolism" International Journal of Molecular Sciences 19, no. 5: 1485. https://doi.org/10.3390/ijms19051485
APA StyleJia, Y., Liu, N., Viswakarma, N., Sun, R., Schipma, M. J., Shang, M., Thorp, E. B., Kanwar, Y. S., Thimmapaya, B., & Reddy, J. K. (2018). PIMT/NCOA6IP Deletion in the Mouse Heart Causes Delayed Cardiomyopathy Attributable to Perturbation in Energy Metabolism. International Journal of Molecular Sciences, 19(5), 1485. https://doi.org/10.3390/ijms19051485