Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1


Abstract
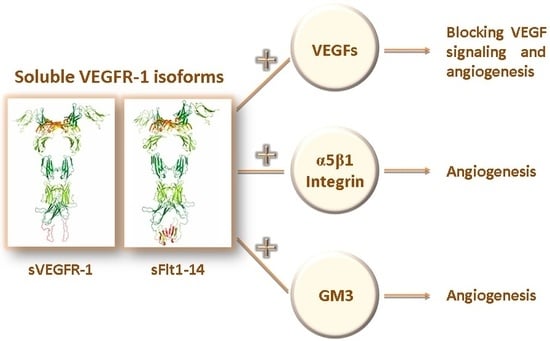
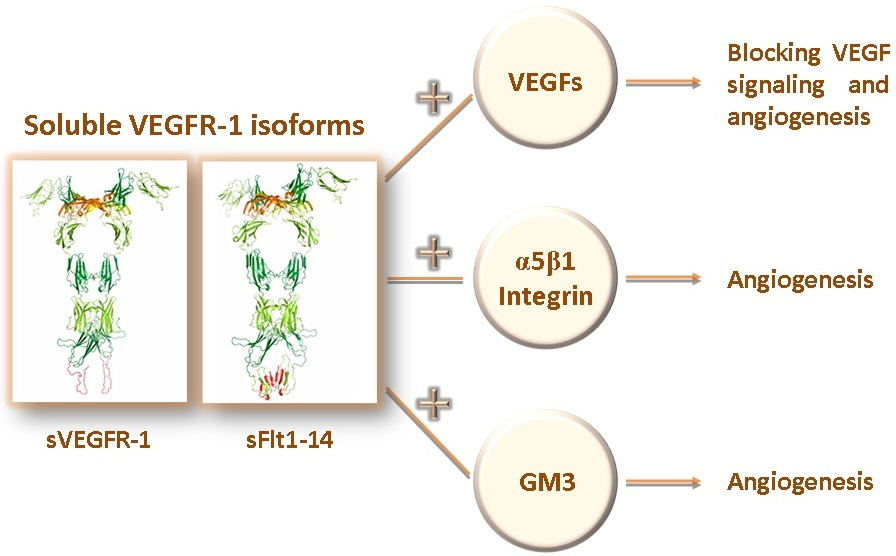
1. Introduction
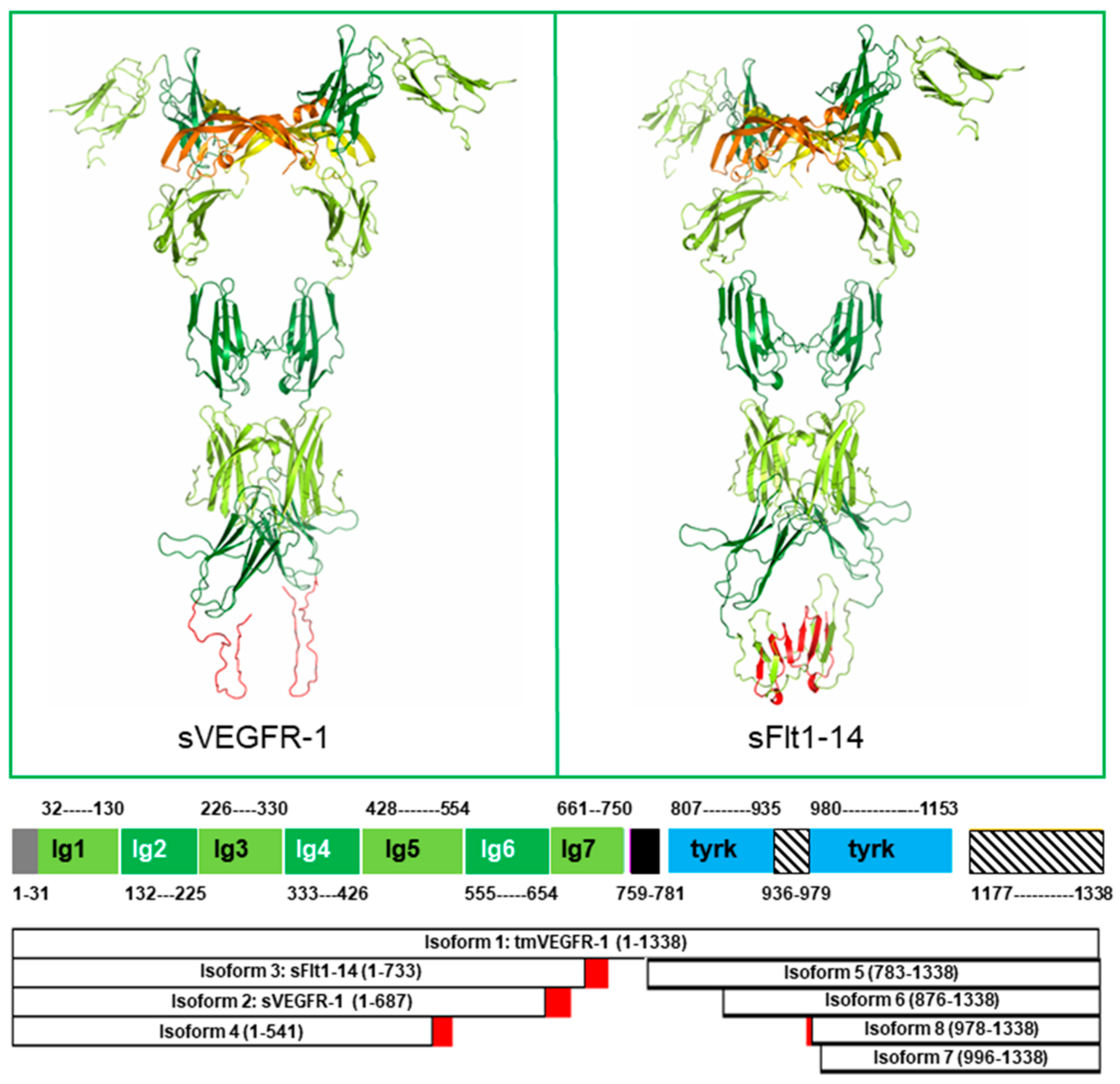
2. Structure of the Soluble Forms of VEGFR-1
3. Expression of Soluble Forms of VEGFR-1
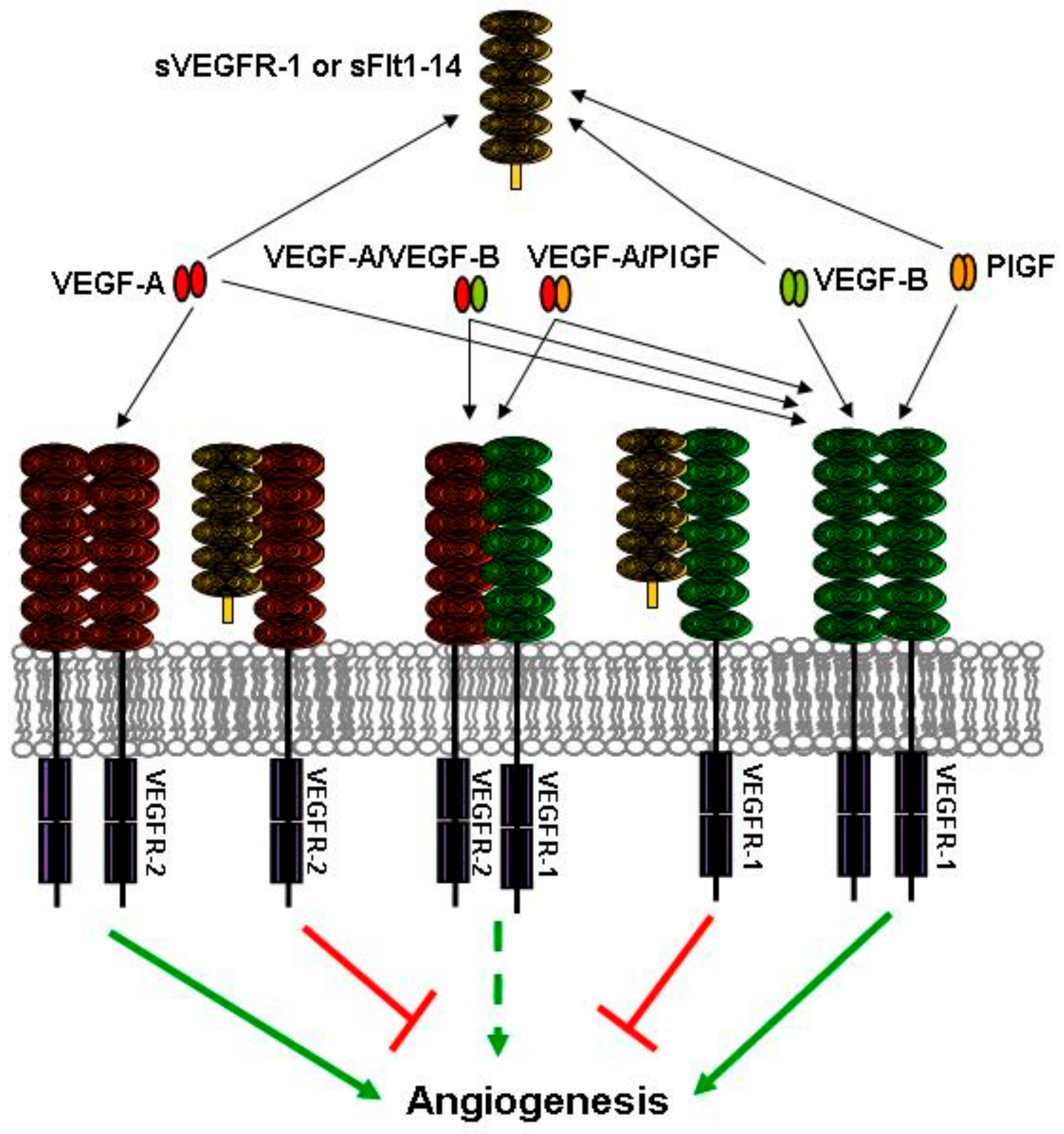
4. sVEGFR-1 and Vessel Sprouting
5. Soluble Forms of VEGFR-1 in Human Pathologies
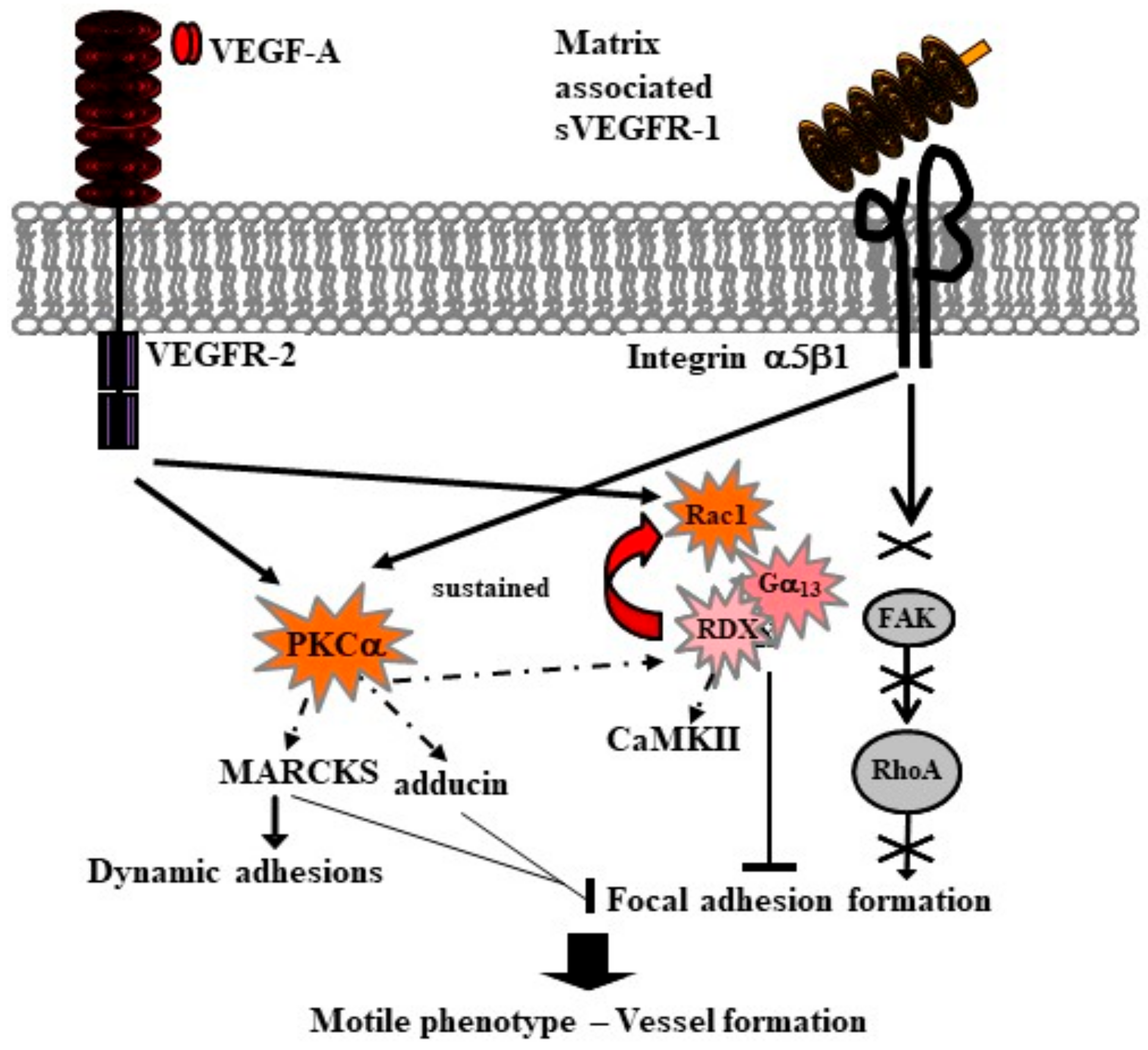
6. Soluble VEGFR-1 Isoforms in the Extracellular Matrix
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
| PlGF | placenta growth factor |
| s | soluble |
| Ig | immunoglubulin |
| HIF1α | hypoxia inducible factor 1α |
| FAK | focal adhesion kinase |
References
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Yamaguchi, S.; Yamane, A.; Ikeda, T.; Tojo, A.; Matsushime, H.; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase (Flt) closely related to the FMS family. Oncogene 1990, 8, 519–527. [Google Scholar]
- Cébe-Suarez, S.; Zehnder-Fjallman, A.; Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 2006, 63, 601–615. [Google Scholar] [CrossRef] [PubMed]
- De Vries, C.; Escobedo, J.; Ueno, H.; Houck, K.; Ferrara, N.; Williams, L. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.; Rossant, J.; Gertsenstein, M.; Breitman, M. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.; Zhang, L.; Bryce, D.; Peng, J. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in Flt-1 knock-out mice. Development 1999, 126, 3015–3025. [Google Scholar] [PubMed]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Wang, G.; Thomas, K.A. Identification of a natural soluble form of the vascular endothelial growth factor receptor, Flt-1, and its heterodimerization with KDR. Biochem. Biophys. Res. Commun. 1996, 226, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Kibata, K.; Suzuki, M.; Nakamura, S.; Motoda, R.; Orita, K. Identification of a vascular endothelial growth factor (VEGF) antagonist, sFlt-1, from a human hematopoietic cell line NALM-16. FEBS Lett. 2000, 469, 14–18. [Google Scholar] [CrossRef]
- Roberts, D.M.; Kearney, J.B.; Johnson, J.H.; Rosenberg, M.P.; Kumar, R.; Bautch, V.L. The VEGF receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am. J. Pathol. 2004, 164, 1531–1535. [Google Scholar] [CrossRef]
- Sela, S.; Itin, A.; Natanson-Yaron, S.; Greenfield, C.; Goldman-Wohl, D.; Yagel, S.; Keshet, E. A Novel Human-Specific Soluble Vascular Endothelial Growth Factor Receptor 1: Cell Type-Specific Splicing and Implications to Vascular Endothelial Growth Factor Homeostasis and Preeclampsia. Circ. Res. 2008, 102, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Andrews, J.I.; Liu, K.Z. Intronic polyadenylation signal sequences and alternate splicing generate human soluble Flt1 variants and regulate the abundance of soluble Flt1 in the placenta. FASEB J. 2007, 21, 3885–3895. [Google Scholar] [CrossRef] [PubMed]
- Raikwar, N.S.; Liu, K.Z.; Thomas, C.P. Protein kinase C regulates FLT1 abundance and stimulates its cleavage in vascular endothelial cells with the release of a soluble PlGF/VEGF antagonist. Exp. Cell Res. 2013, 319, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.J.; Hewett, P.W.; Ahmad, S.; Wang, K.-Q.; Cai, M.; Al-Ani, B.; Fujisawa, T.; Ma, B.; Sissaoui, S.; Ramma, W.; et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat. Commun. 2012, 3, 972. [Google Scholar] [CrossRef] [PubMed]
- Failla, C.M.; De Luca, N.; Avitabile, S.; Tatangelo, L.; Rossiter, H.; Tschachler, E.; Odorisio, T. Mice over-expressing placenta growth factor in the skin exhibit increased vascularization and vessel permeability independently of VEGF-A. J. Dermatol. Sci. 2018, 90, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tjwa, M.; Van Hove, I.; Enholm, B.; Neven, E.; Paavonen, K.; Jeltsch, M.; Diez Juan, T.; Sievers, R.E.; Chorianopoulos, E.; et al. Reevaluation of the role of VEGF-B suggests a restricted role in the revascularization of the ischemic myocardium. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Nakao, K.; Nakamura, K.; Katsuki, M.; Maru, Y.; Shibuya, M. Membrane fixation of vascular endothelial growth factor receptor 1 ligand-binding domain is important for vasculogenesis and angiogenesis in mice. Mol. Cell. Biol. 2005, 25, 346–354. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; de Vos, A.M. Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 1997, 91, 695–704. [Google Scholar] [CrossRef]
- Iyer, S.; Darley, P.I.; Acharya, K.R. Structural insights into the binding of vascular endothelial growth factor-B by VEGFR-1D2. J. Biol. Chem. 2010, 285, 23779–23789. [Google Scholar] [CrossRef] [PubMed]
- Christinger, H.W.; Fuh, G.; de Vos, A.M.; Wiesmann, C. The crystal structure of placental growth factor in complex with domain 2 of vascular endothelial growth factor receptor-1. J. Biol. Chem. 2004, 279, 10382–10388. [Google Scholar] [CrossRef] [PubMed]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the full-length VEGFR-1 extracellular domain in complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.; Minneci, F.; Nugent, T.C.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013, 41, W239–W357. [Google Scholar] [CrossRef] [PubMed]
- Dayringer, H.E.; Tramontano, A.; Sprang, R.S.; Fletterick, R.J. Interactive program for visualization and modelling of proteins, nucleic acids and small molecules. J. Mol. Graphics 1986, 4, 82–87. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Gough, J.; Karplus, K.; Hughey, R.; Clothia, C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 2001, 313, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- Yang, Y.; Xie, P.; Opatowsky, Y.; Schlessinger, J. Direct contacts between extracellular membrane-proxinal domains are required for VEGF receptor activation and cell signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 1906–1911. [Google Scholar] [CrossRef] [PubMed]
- Lacal, P.M.; Morea, V.; Ruffini, F.; Orecchia, A.; Dorio, A.S.; Failla, C.M.; Soro, S.; Tentori, L.; Zambruno, G.; Graziani, G.; et al. Inhibition of endothelial cell migration and angiogenesis by a vascular endothelial growth factor receptor-1 derived peptide. Eur. J. Cancer 2008, 44, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, S.; Marasco, D.; Tarallo, V.; Albuquerque, R.J.C.; Mitola, S.; Takeda, A.; Stassen, J.-M.; Presta, M.; Ambati, J.; Ruvo, M.; et al. Modulation of angiogenesis by a tetrameric tripeptide that antagonizes vascular endothelial growth factor receptor 1. J. Biol. Chem. 2008, 283, 34250–34259. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, L.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Broussy, S.; Zhang, T.; Ji, L.; Vidal, M.; Liu, W.-Q. Identification of peptidic antagonists of vascular endothelial growth factor receptor-1 by scanning the binding epitopes of its ligands. J. Med. Chem. 2017, 60, 6598–6606. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Raikwar, N.S.; Kelley, E.A.; Liu, K.Z. Alternate processing of Flt1 transcripts is directed by conserved cis-elements within an intronic region of FLT1 that reciprocally regulates splicing and polyadenylation. Nucleic Acid Res. 2010, 38, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.-P.; Condorelli, F.; Park, J.; Ferrara, N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef] [PubMed]
- Shima, D.T.; Deutsch, U.; D’Amore, P.A. Hypoxic induction of vascular endothelial growth factor (VEGF) in human epithelial cells is mediated by increases in mRNA stability. FEBS Lett. 1995, 370, 203–208. [Google Scholar] [CrossRef]
- Ikeda, T.; Sun, L.; Tsuruoka, N.; Ishigaki, Y.; Yoshitomi, Y.; Yoshitake, Y.; Yonekura, H. Hypoxia down-regulates sFlt-1 (sVEGFR-1) expression in human microvascular endothelial cells by a mechanism involving mRNA alternative processing. Biochem. J. 2011, 436, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Roda, J.M.; Liu, H.; O’Neil, T.; Marsh, C.B. Opposite roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 2011, 117, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liebermann, D.A.; Tront, J.S.; Holtzman, E.J.; Huang, Y.; Hoffman, B.; Geifman-Holtzman, O. Gadd45a stress signaling regulates sFlt-1 expression in preeclampsia. J. Cell. Physiol. 2009, 220, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Andrews, J.I.; Raikwar, N.S.; Kelley, E.A.; Herse, F.; Dechend, R.; Golos, T.G.; Liu, K.Z. A recently evolved novel trophoblast-enriched secreted form of fms-like tyrosine kinase-1 variant is up-regulated in hypoxia and preeclampsia. J. Clin. Endocrinol. Metab. 2009, 94, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Rajakumar, A.; Geahchan, C.; Salahuddin, S.; Cerdeira, A.S.; Burke, S.D.; George, E.M.; Granger, J.P.; Karumanchi, S.A. Ouabain inhibits placental sFlt1 production by repressing HSP27-dependent HIF-1α pathway. FASEB J. 2014, 28, 4324–4334. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Raikwar, N.S.; Santillan, M.K.; Santillan, D.A.; Thomas, C.P. Aspirin inhibits expression of sFlt1 from cytotrophoblasts induced by hypoxia, via cyclo-oxygenase 1. Placenta 2015, 36, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Touhey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1–356.e15. [Google Scholar] [CrossRef] [PubMed]
- Boeckel, J.-N.; Guarani, V.; Koyanagi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Sears, R.A.; Powers, L.S.; Nash, E.; Bair, T.; Gerke, A.K.; Hassan, I.; Thomas, C.P.; Gross, T.J.; Monick, M.M. Coordinated DNA methylation and gene expression changes in smoker alveolar macrophages: Specific effects on VEGF receptor 1 expression. J. Leukoc. Biol. 2012, 92, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Roberts, R.; Galloway, M.; Wang, Y.; Cohn, D.E.; Marsh, C.B. GM-CSF induces expression of soluble VEGF receptor-1 from human monocytes and inhibits angiogenesis in mice. Immunity 2004, 21, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-K.; Georgiadis, A.; Copland, D.A.; Liyanage, S.; Luhmann, U.F.O.; Robbie, S.J.; Liu, J.; Wu, J.; Bainbridge, J.W.; Bates, D.O.; et al. IL-4 regulates specific Arg-1+ macrophage sFlt-1-mediated inhibition of angiogenesis. Am. J. Pathol. 2015, 185, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Fellows, A.; Mierke, D.F.; Nichols, R.C. AUF1-RGG peptides up-regulate the VEGF antagonist, soluble VEGF receptor-1 (sFlt-1). Cytokine 2013, 64, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, R.; Houben, A.; Tohidnezhad, M.; Kweider, N.; Fragoulis, A.; Wruck, C.J.; Brandenburg, L.O.; Hermanns-Sachweh, B.; Goldring, M.B.; Pufe, T.; et al. Mechanical forces induce changes in VEGF and VEGFR-1/sFlt-1 expression in human chondrocytes. Int. J. Mol. Sci. 2014, 15, 15456–15474. [Google Scholar] [CrossRef] [PubMed]
- Sela, S.; Natanson-Yaron, S.; Zcharia, E.; Vlodavsky, I.; Yagel, S.; Keshet, E. Local retention versus systemic release of soluble VEGF receptor-1 are mediated by heparin-binding and regulated by heparanase. Circ. Res. 2011, 108, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Kearney, J.B.; Kappas, N.C.; Ellerstrom, C.; DiPaola, F.W.; Bautch, V.L. The VEGF receptor Flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 2004, 103, 4527–4535. [Google Scholar] [CrossRef] [PubMed]
- Brudno, Y.; Ennett-Shepard, A.B.; Chen, R.R.; Aizenberg, M.; Mooney, D.J. Enhancing microvascular formation and vessel maturation through temporal control over multiple pro-angiogenic and pro-maturation factors. Biomaterials 2013, 34, 9201–9209. [Google Scholar] [CrossRef] [PubMed]
- Kearney, J.B.; Ambler, C.; Monaco, K.A.; Johnson, N.; Rapoport, R.G.; Bautch, V.L. Vascular endothelial growth factor receptor Flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood 2002, 99, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Kappas, N.C.; Zeng, G.; Chappel, J.C.; Kearney, J.B.; Hazarika, S.; Kallianos, K.G.; Patterson, C.; Annex, B.H.; Bautch, V.L. The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J. Cell Biol. 2008, 181, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Taylor, S.M.; Ferrara, N.; Bautch, V.L. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev. Cell 2009, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Mouillesseaux, K.P.; Bautch, V.L. Flt-1 (vascular endothelial growth factor receptor-1) is essential for the vascular endothelial growth factor-Notch feedback loop during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; Failla, C.M.; Orecchia, A.; Bani, M.R.; Dorio, A.S.; Fortes, C.; Zambruno, G.; Graziani, G.; Giavazzi, R.; D’Atri, S.; et al. Expression of the soluble vascular endothelial growth factor receptor-1 in cutaneous melanoma: Role in tumour progression. Br. J. Dermatol. 2011, 164, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Koch, S.; Gualandi, L.; Li, X.; Claesson-Welsh, L. Vascular endothelial growth factors and receptors: Anti-angiogenic therapy in the treatment of cancer. Mol. Asp. Med. 2011, 32, 88–111. [Google Scholar] [CrossRef] [PubMed]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.C.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Uehara, H.; Zhang, X.; Das, S.K.; Olsen, T.; Holt, D.; Simonis, J.M.; Jackman, K.; Singh, N.; Miya, T.R.; et al. Photoreceptor avascular privilege is shielded by soluble VEGF receptor-1. eLife 2013, 2, e00324. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Mamalis, C.; McFadden, M.; Taggart, M.; Stagg, B.; Passi, S.; WEarle, P.; Chakravarthy, U.; Hogg, R.E.; Ambati, B.K. The reduction of serum soluble Flt-1 in patients with neovascular age-related macular degeneration. Am. J. Ophtalmol. 2015, 159, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Lauer, G.; Cole, M.; Jurk, S.; Christ, H.; Hornig, C.; Krieg, T.; Welch, H.A. Increased levels of the soluble variant of the vascular endothelial growth factor receptor VEGFR-1 are associated with a poor prognosis in wound healing. J. Investig. Dermatol. 2004, 123, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Banks, R.E.; Forbes, M.A.; Searles, J.; Pappin, D.; Canas, B.; Rahman, D.; Kaufmann, S.; Walters, C.E.; Jackson, A.; Eves, P.; et al. Evidence for the existence of a novel pregnancy-associated soluble variant of the vascular endothelial growth factor receptor, Flt-1. Mol. Hum. Reprod. 1998, 4, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- McKeeman, G.C.; Ardill, J.E.S.; Caldwell, C.M.; Humnter, A.J.; McClure, N. Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia develop. Am. J. Obstet. Gynecol. 2004, 191, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, O.; Bretschneider, A.; Zhong, E.; Webb, J.; Hopp, H.; Verlohren, S.; Fuhr, N.; Lanowska, M.; Nonnenmacher, A.; Vetter, R.; et al. Soluble vascular endothelial growth factor receptor-1 (sFlt-1) mediates downregulation of Flt-1 and prevents activated neutrophils from women with preeclampsia from additional migration by VEGF. Circ. Res. 2005, 97, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Placental specific sFLT-1: Role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol. Hum. Reprod. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennstrom, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive value of the sFlt-1:PlGF ratio in women with suspected preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Amraoui, F.; Spijkers, L.; Lahsinoui, H.H.; Vogt, L.; van der Post, J.; Peters, S.; Afink, G.; Ris-Stalpers, C.; van den Born, B.-J. sFlt-1 elevates blood pressure by augmenting endothelin-1-mediated vasoconstriction in mice. PLoS ONE 2014, 9, e91897. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Kentrup, D.; Reuter, S.; Mayer, A.B.; Golle, L.; Tiemann, K.; Fobker, M.; Engelbertz, C.; Breithardt, G.; Brand, E.; et al. Soluble Flt-1 links microvascular disease with heart failure in CKD. Basic Res. Cardiol. 2015, 110, 30. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, N.I.; Yano, K.; Okada, H.; Fischer, C.; Howell, M.; Spokes, K.C.; Ngo, L.; Angus, D.C.; Aird, W.C. A prospective, observational study of soluble Flt-1 and vascular endothelial growth factor in sepsis. Shock 2008, 29, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Jaroszewicz, J.; Januszkiewicz, M.; Flisiak, R.; Rogalska, M.; Kalinowska, A.; Wierzbicka, I. Circulating vascular endothelial growth factor and its soluble receptors in patients with liver cirrhosis: Possible association with epatic function impairment. Cytokine 2008, 44, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Dumnicka, P.; Kusnierz-Cabala, B.; Sporek, M.; Mazur-Laskowska, M.; Gil, K.; Kuzniewski, M.; Cernowicz, P.; Warzecha, Z.; Dembinski, A.; Bonior, J.; et al. Serum concentrations of angiopoietin-2 and soluble fms-like tyrosine kinase 1 (sFlt-1) are associated with coagulopathy among patients with acute pancreatitis. Int. J. Mol. Sci. 2017, 18, 753. [Google Scholar] [CrossRef] [PubMed]
- Justiniano, S.E.; Elavazhagan, S.; Fatehchand, K.; Shah, P.; Mehta, P.; Roda, J.M.; Mo, X.; Cheney, C.; Hertlein, E.; Eubank, T.; et al. Fcγ receptor-induced soluble vascular endothelial growth factor receptor-1 (VEGFR-1) production inhibits angiogenesis and enhances efficacy of anti-tumor antibodies. J. Biol. Chem. 2013, 288, 26800–26809. [Google Scholar] [CrossRef] [PubMed]
- Onoue, K.; Uemura, S.; Takeda, Y.; Somekawa, S.; Iwama, H.; Imagawa, K.; Nishida, T.; Morikawa, Y.; Takemoto, Y.; Asai, O.; et al. Reduction of circulating soluble fms-like tyrosine kinase-1 plays a significant role in renal dysfunction-associated aggravation of atherosclerosis. Circulation 2009, 120, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Lee, S.-H.; Park, S.H.; Kang, S.-M.; Chung, N.; Shim, W.-H.; Cho, S.-Y.; Manabe, I.; Jang, Y. Soluble fms-like tyrosine kinase-1 and the progression of carotid intima-media thickness. Circ. J. 2010, 74, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Lacal, P.M.; Schietroma, C.; Morea, V.; Zambruno, G.; Failla, C.M. Vascular endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the α5β1 integrin. J. Cell Sci. 2003, 116, 3479–3489. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Bell, K.; Mausa, S.A.; Varner, J.A. Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 2000, 156, 1345–1362. [Google Scholar] [CrossRef]
- Taverna, D.; Hynes, R.O. Reduced blood vessel formation and tumor growth in α5-integrin-negative teratocarcinomas and embryoid bodies. Cancer Res. 2001, 61, 5255–5261. [Google Scholar] [PubMed]
- Yang, J.T.; Rayburn, H.; Hynes, R.O. Embryonic mesodermal defects in α5 integrin-deficient mice. Development 1993, 119, 1093–1105. [Google Scholar] [PubMed]
- Soro, S.; Orecchia, A.; Morbidelli, L.; Lacal, P.M.; Morea, V.; Ballmer-Hofer, K.; Ruffini, F.; Ziche, M.; D’Atri, S.; Zambruno, G.; et al. A proangiogenic peptide derived from vascular endothelial growth factor receptor-1 acts through alpha5beta1 integrin. Blood 2008, 111, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Mettouchi, A.; Uva, P.; Simon, G.C.; Arcelli, D.; Avitabile, S.; Ragone, G.; Meneguzzi, G.; Pfenninger, K.H.; Zambruno, G.; et al. Endothelial cell adhesion to soluble vascular endothelial growth factor receptor-1 triggers a cell dynamic and angiogenic phenotype. FASEB J. 2014, 28, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Bernal, A.; Gatlin, J.C.; Sunpaweravong, S.; Pfenninger, K.H. Dynamic adhesions and MARCKS in melanoma cells. J. Cell Sci. 2009, 122, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Kalwa, H.; Michel, T. The MARCKS Protein Plays a Critical Role in Phosphatidylinositol 4,5-Bisphosphate Metabolism and Directed Cell Movement in Vascular Endothelial Cells. J. Biol. Chem. 2011, 286, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sison, K.; Li, C.; Tian, R.; Wnuk, M.; Sung, H.-K.; Jeansson, M.; Zhang, C.; Tucholska, M.; Jones, N.; et al. Soluble Flt1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell 2012, 151, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Bry, M.; Kivela, R.; Leppanen, V.-M.; Alitalo, K. Vascular endothelial growth factor-B in physiology and disease. Physiol. Rev. 2014, 94, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Dewerchin, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef] [PubMed]
- Parsons-Wingerter, P.; Kasman, I.M.; Norberg, S.; Magnussen, A.; Zanivan, S.; Rissone, A.; Baluk, P.; Favre, C.J.; Jeffrey, U.; Murray, R.; et al. Uniform overexpression and rapid accessibility of α5β1 integrin on blood vessels in tumors. Am. J. Pathol. 2005, 167, 193–211. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID: 1FLT | PDB ID: 2XAC | PDB ID: 1RV6 | |||||
| VEGFR-1 D2 | VEGF-A | VEGFR-1 D2 | VEGF-B | VEGFR-1 D2 | PlGF | ||
| 142 I | 17 F | 141 E | 16 S | 140 S | 25 F | ||
| 143 P | 18 M | 142 I | 17 W | 141 E | 26 Q | ||
| 145 I | 21 Y | 143 P | 21 Y | 142 I | 29 W | ||
| 147 H | 22 Q | 145 I | 22 T | 143 P | 30 G | ||
| 171 K | 25 Y | 172 F | 48 V | 145 I | 33 Y | ||
| 172 F | 46 I | 199 Y | 62 P | 171 K | 71 D | ||
| 173 P | 48 K | 202 I | 63 D | 172 F | 74 L | ||
| 199 Y | 63 D | 203 G | 66 L | 173 P | 89 L | ||
| 204 L | 65 G | 204 L | 79 Q | 199 Y | 91 I | ||
| 221 L | 66 L | 221 L | 81 L | 203 G | 97 P | ||
| 223 H | 81 M | 224 R | 88 S | 204 L | 99 Y | ||
| 224 R | 83 I | 89 Q | 219 N | 114 P | |||
| 225 Q | 86 H | 90 L | 221 L | ||||
| 89 Q | 105 P | 223 H | |||||
| 91 I | 224 R | ||||||
| 105 R | |||||||
| 106 P | |||||||
| PDB ID: 5T89 | PDB ID: 5T89 | PDB ID: 5T89 | Homology Model | ||||
| VEGFR-1 D3 | VEGF-A | VEGFR-1 D4 | VEGFR-1 D4 | VEGFR-1 D5 | VEGFR-1 D5 | VEGFR-1 D7 | VEGFR-1 D7 |
| 226 T | 34 D | 351 R | 351 R | 382 R | 675 S | 675 S | |
| 227 N | 36 F | 379 K | 379 K | 394 D | 676 S | 676 S | |
| 258 L | 40 P | 380 S | 380 S | 429 Q | 429 Q | 677 S | 677 S |
| 259 N | 41 D | 381 A | 381 A | 431 Y | 431 Y | 704 E | 704 E |
| 260 T | 43 I | 382 R | 382 R | 432 E | 432 E | 705 P | 705 P |
| 261 R | 44 E | 393 K | 393 K | 433 K | 433 K | 706 G | 706 G |
| 262 V | 45 Y | 394 D | 434 A | 434 A | 707 I | 707 I | |
| 263 Q | 46 I | 435 V | 435 V | 708 I | 708 I | ||
| 264 M | 63 D | 436 S | 436 S | 717 F | 717 F | ||
| 277 S | 64 E | 437 S | 437 S | 719 E | 719 E | ||
| 278 V | 65 G | 438 F | 438 F | 720 R | 720 R | ||
| 279 R | 84 K | 439 P | 439 P | 722 T | 722 T | ||
| 280 R | 85 P | 451 I | 451 I | 725 D | 725 D | ||
| 282 I | 86 H | 453 T | 453 T | ||||
| 284 Q | 107 K | 455 T | 455 T | ||||
| 287 S | 108 K | 508 R | 508 R | ||||
| 290 N | 510 A | 510 A | |||||
| 292 F | 512 I | 512 I | |||||
| 315 S | 513 E | 513 E | |||||
| 316 G | 517 K | 517 K | |||||
| 317 P | 519 A | 519 A | |||||
| 521 T | 521 T | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Failla, C.M.; Carbo, M.; Morea, V. Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. Int. J. Mol. Sci. 2018, 19, 1306. https://doi.org/10.3390/ijms19051306
Failla CM, Carbo M, Morea V. Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. International Journal of Molecular Sciences. 2018; 19(5):1306. https://doi.org/10.3390/ijms19051306
Chicago/Turabian StyleFailla, Cristina M., Miriam Carbo, and Veronica Morea. 2018. "Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1" International Journal of Molecular Sciences 19, no. 5: 1306. https://doi.org/10.3390/ijms19051306
APA StyleFailla, C. M., Carbo, M., & Morea, V. (2018). Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. International Journal of Molecular Sciences, 19(5), 1306. https://doi.org/10.3390/ijms19051306