Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2

 ,
, 
Abstract

1. Introduction
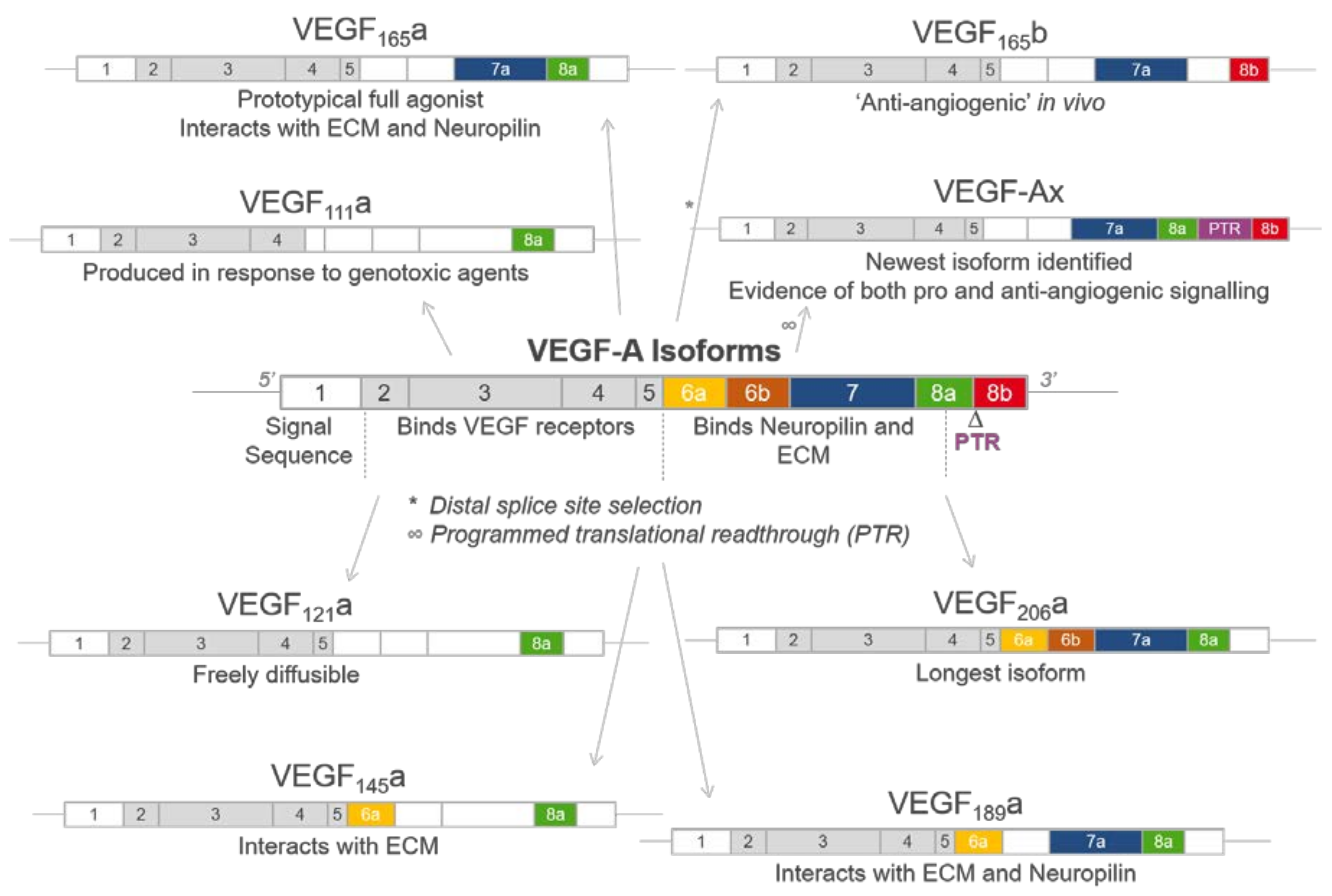
2. Generation of VEGF-A Isoforms by Alternative Splicing
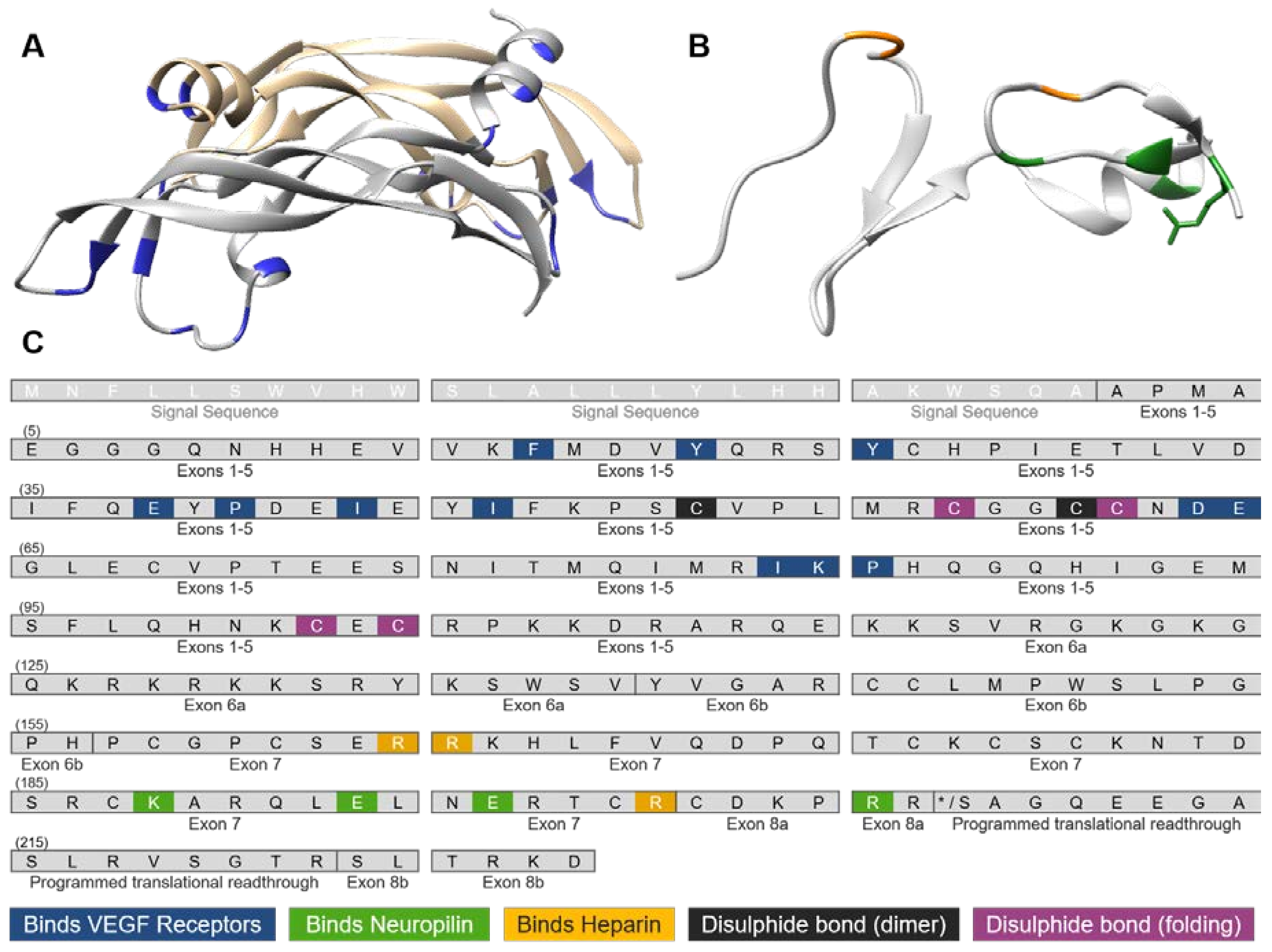
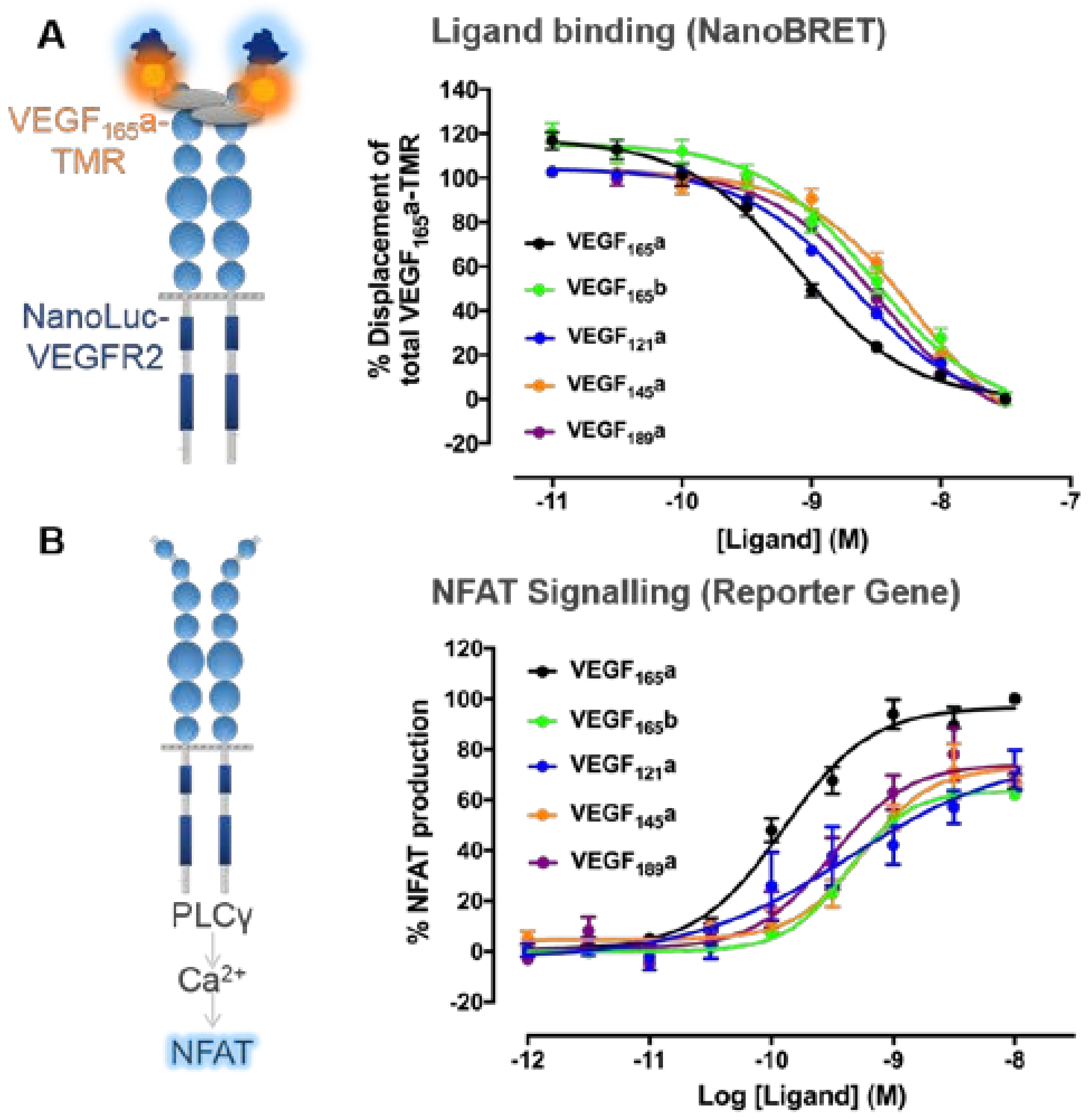
3. VEGF-A Ligand/Receptor Binding
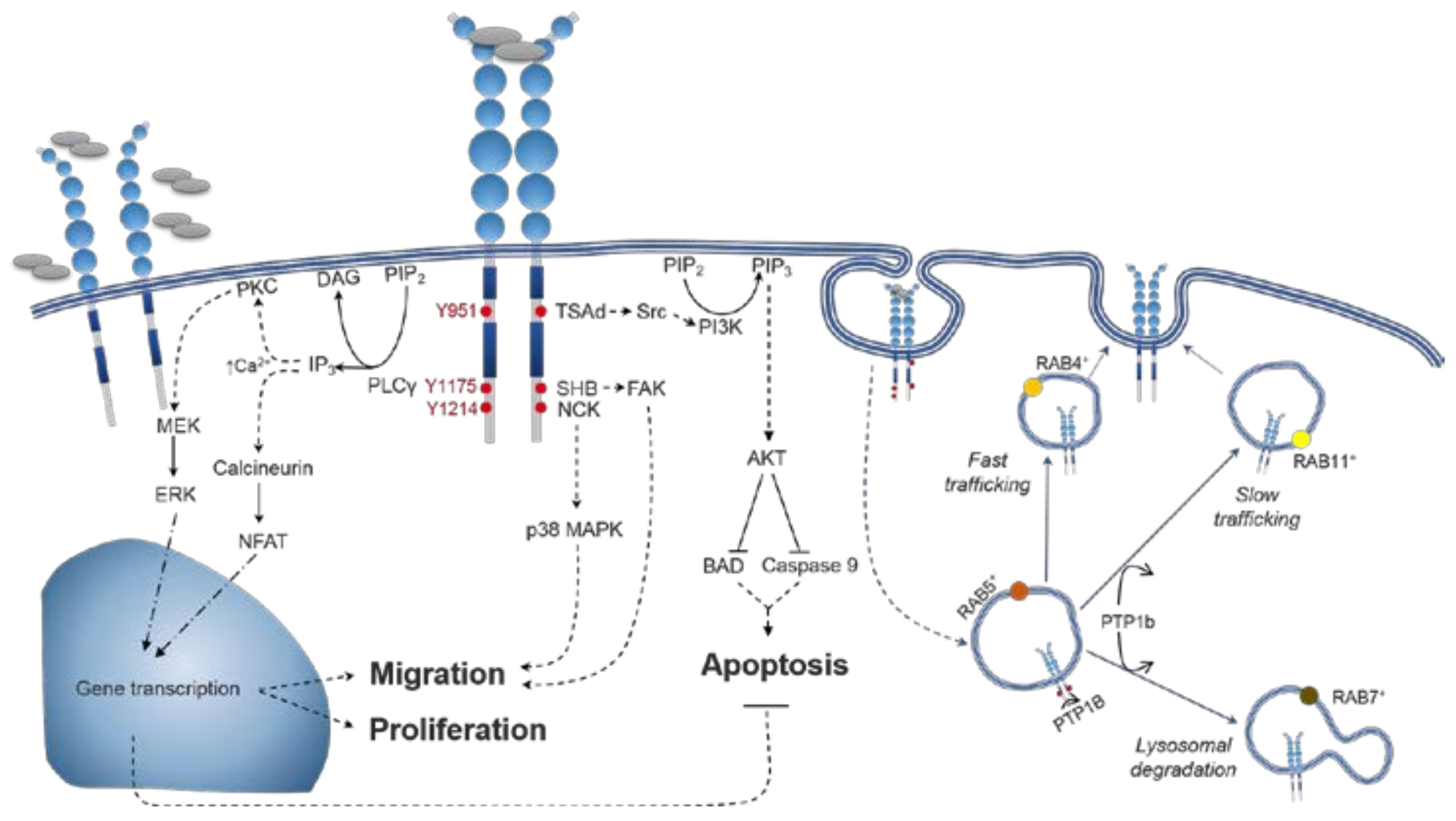
4. VEGFR2 Signalling
4.1. VEGFR2 Activation
4.2. Distinctions between VEGF-A Isoform Signalling
5. Molecular Mechanisms Distinguishing between VEGF-A Isoforms
5.1. Spatiotemporal Dynamics of VEGFR2 Trafficking
5.2. Spatiotemporal Dynamics of VEGF-A Bioavailability
5.3. Interactions with Co-Receptor Neuropilin-1 (NRP1)
5.4. Heterodimer Formation with VEGFR1
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| BRET | Bioluminescence Resonance Energy Transfer |
| eNOS | Endothelial Nitric Oxide Synthase |
| ERK | Extracellular Signal-Related Kinase |
| FAK | Focal Adhesion Kinase |
| GPCR | G Protein-Coupled Receptor |
| HEK | Human Embryonic Kidney cells |
| HMVECs | Human Microvascular Endothelial Cells |
| HPMECs | Human Pulmonary Microvascular Endothelial Cells |
| HUVECs | Human Umbilical Vein Endothelial Cells |
| MAPK | Mitogen-Activated Protein Kinases |
| NFAT | Nuclear Factor of Activated T-Cells |
| NRP1 | Neuropilin-1 |
| PAECs | Porcine Aortic Endothelial cells |
| PI3K | Phosphatidylinositol 3-Kinase |
| PLCγ | Phospholipase Cγ |
| RTK | Receptor Tyrosine Kinase |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kalka, C.; Masuda, H.; Chen, D.; Silver, M.; Kearney, M.; Magner, M.; Isner, J.; Asahara, T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med. 1999, 5, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Capla, J.M.; Galiano, R.D.; Ceradini, D.J.; Callaghan, M.J.; Kleinman, M.E.; Gurtner, G.C. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood 2005, 105, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.; Vlahos, N.; Shih, I.; Zhao, Y. Expression patterns of VEGF and Flk-1 in human endometrium at the various phases of the natural menstrual cycle. Hum. Reprod. 2014, 29, i195. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Boghozian, R.; Mirshafiey, A. The potential role of angiogenic factors in rheumatoid arthritis. Int. J. Rheum. Dis. 2014, 17, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in psoriasis: Therapeutic implications. J. Investig. Dermatol. 1972, 59, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Ved, N.; Hulse, R.P.; Bestall, S.M.; Donaldson, L.F.; Bainbridge, J.W.; Bates, D.O. Vascular endothelial growth factor-A 165 b ameliorates outer-retinal barrier and vascular dysfunction in the diabetic retina. Clin. Sci. 2017, 131, 1225–1243. [Google Scholar] [CrossRef] [PubMed]
- Alkim, C.; Alkim, H.; Koksal, A.R.; Boga, S.; Sen, I. Angiogenesis in inflammatory bowel disease. Int. J. Inflam. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Thervet, E.; Timsit, M.O. Angiogenic response following renal ischemia reperfusion injury: New players. Prog. Urol. 2014, 24, S20–S25. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Ema, M. Roles of VEGF-A signalling in development, regeneration, and tumours. J. Biochem. 2014, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation 2009, 16, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.; Galli, S.; Dvorak, A.; Perruzzi, C.; Harvey, V.; Dvorak, H. Tumor Cells Secrete a Vascular Permeability Factor That Promotes Accumulation of Ascites Fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.; Perruzzi, C.; Feder, J.; Dvorak, H. A Highly Conserved Vascular Permeability Factor Secreted by a Variety of Human and Rodent Tumor Cell Lines. Cancer Res. 1986, 46, 5629–5632. [Google Scholar] [PubMed]
- Ferrara, N.; Henzel, W. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Fearnley, G.W.; Odell, A.F.; Latham, A.M.; Mughal, N.A.; Bruns, A.F.; Burgoyne, N.J.; Homer-Vanniasinkam, S.; Zachary, I.C.; Hollstein, M.C.; Wheatcroft, S.B.; et al. VEGF-A isoforms differentially regulate ATF-2-dependent VCAM-1 gene expression and endothelial-leukocyte interactions. Mol. Biol. Cell 2014, 25, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Oku, A.; Sawano, A.; Yamaguchi, S.; Yazaki, Y.; Shibuya, M. A novel type of vascular endothelial growth factor, VEGF-E (NZ-7 VEGF). J. Biol. Chem. 1998, 273, 31273–31282. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Matsunaga, Y.; Tokunaga, Y.; Obayashi, S.; Saito, M.; Morita, T. Snake venom vascular endothelial growth factors (VEGF-Fs) exclusively vary their structures and functions among species. J. Biol. Chem. 2009, 284, 9885–9891. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Acharya, K.R. Tying the knot: The cystine signature and molecular-recognition processes of the vascular endothelial growth factor family of angiogenic cytokines. FEBS J. 2011, 278, 4304–4322. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.A.; Heiring, C.; Misselwitz, R.; Welfle, K.; Welfle, H. The cystine knot promotes folding and not thermodynamic stability in vascular endothelial growth factor. J. Biol. Chem. 2002, 277, 43410–43416. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Uchida, S.; Nitta, K.; Yumura, W.; Marumo, F.; Nihei, H. Glomerular endothelial cells in culture express and secrete vascular endothelial growth factor. Am. J. Physiol. 1994, 266, F81–F88. [Google Scholar] [CrossRef] [PubMed]
- Namiki, A.; Brogi, E.; Kearney, M.; Kim, E.A.; Wu, T.; Couffinhal, T.; Varticovski, L.; Isner, J.M. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J. Biol. Chem. 1995, 270, 31189–31195. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.N.; Polverini, P.J.; Koch, A.E.; Volin, M.V.; Gamelli, R.L.; DiPietro, L.A. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am. J. Pathol. 1998, 152, 1445–1452. [Google Scholar] [PubMed]
- Brogi, E.; Wu, T.; Namiki, A. Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation 1994, 90, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Banks, R.E.; Forbes, M.A.; Kinsey, S.E.; Stanley, A.; Ingham, E.; Walters, C.; Selby, P.J. Release of the angiogenic cytokine vascular endothelial growth factor (VEGF) from platelets: Significance for VEGF measurements and cancer biology. Br. J. Cancer 1998, 77, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Gaudry, M.; Brégerie, O.; Andrieu, V.; El Benna, J.; Pocidalo, M.-A.A.; Hakim, J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood 1997, 90, 4153–4161. [Google Scholar] [PubMed]
- Berse, B.; Brown, L.F.; Van De Water, L.; Dvorak, H.F.; Senger, D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 1992, 3, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-Asignaling and Bcl-w expression. Blood 2011, 118, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Fabbro, D.; Kelly, E.; Marrion, N.; Peters, J.A.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. The Concise Guide to pharmacology 2015/16: Catalytic receptors. Br. J. Pharmacol. 2015, 172, 5979–6023. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Fearnley, G.W.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. The cellular response to vascular endothelial growth factors requires co-ordinated signal transduction, trafficking and proteolysis. Biosci. Rep. 2015, 35, e00253. [Google Scholar] [CrossRef] [PubMed]
- Kabrun, N.; Bühring, H.J.; Choi, K.; Ullrich, A.; Risau, W.; Keller, G. Flk-1 expression defines a population of early embryonic hematopoietic precursors. Development 1997, 124, 2039–2048. [Google Scholar] [PubMed]
- Ishida, A.; Murray, J.; Saito, Y.; Kanthou, C.; Benzakour, O.; Shibuya, M.; Wijelath, E.S. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J. Cell. Physiol. 2001, 188, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Witmer, A.N.; Dai, J.; Weich, H.A.; Vrensen, G.F.; Schlingemann, R.O. Expression of vascular endothelial growth factor receptors 1, 2, and 3 in quiescent endothelia. J. Histochem. Cytochem. 2002, 50, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.D.; Mohammadi, M.; Rahimi, N. A single amino acid substitution in the activation loop defines the decoy characteristic of VEGFR-1/FLT-1. J. Biol. Chem. 2006, 281, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C. Different Signal-Transduction Properties of Kdr and Flt1, 2 Receptors for Vascular Endothelial Growth-Factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar] [PubMed]
- Li, Y.L.; Zhao, H.; Ren, X.-B.; Li, Y.L.; Zhao, H.; Ren, X.B. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Sawano, A.; Iwai, S.; Sakurai, Y.; Ito, M.; Shitara, K.; Nakahata, T.; Shibuya, M. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood 2001, 97, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Positive and Negative Modulation of Angiogenesis by VEGFR1 Ligands. Sci. Signal. 2009, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Hypoxia Regulates Vascular Endothelial Growth Factor Gene Expression in Endothelial Cells. Circ. Res. 1995, 77, 638–643. [Google Scholar]
- Forsythe, J.O.A.; Jiang, B.; Iyer, N.V.; Agani, F.; Leung, S.W. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor Activation of Vascular Endothelial Growth Factor Gene Transcription by Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 1996, 16, 4604–4612. [Google Scholar] [CrossRef] [PubMed]
- Mineur, P.; Colige, A.C.; Deroanne, C.F.; Dubail, J.; Kesteloot, F.; Habraken, Y.; Noël, A.; Vöö, S.; Waltenberger, J.; Lapière, C.M.; et al. Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. J. Cell Biol. 2007, 179, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. BioEssays 2006, 28, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J.A. The Human Gene for Vascular Endothelial Growth-Factor. Multiple Protein Forms Are Encoded Through Alternative Exon Splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar] [PubMed]
- Guyot, M.; Pages, G. VEGF Splicing and the Role of VEGF Splice Variants: From Physiological-Pathological Conditions to Specific Pre-mRNA Splicing. In Methods in Molecular Biology; Springer: Berlin, Germany, 2015; Volume 1332, pp. 3–24. ISBN 9781493929160. [Google Scholar]
- Gu, F.; Li, X.; Kong, J.; Pan, B.; Sun, M.; Zheng, L.; Yao, Y. VEGF111b, a new member of VEGFxxxb isoforms and induced by mitomycin C, inhibits angiogenesis. Biochem. Biophys. Res. Commun. 2013, 441, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Eswarappa, S.M.; Potdar, A.A.; Koch, W.J.; Fan, Y.; Vasu, K.; Lindner, D.; Willard, B.; Graham, L.M.; Dicorleto, P.E.; Fox, P.L. Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell 2014, 157, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Pritchard-Jones, R.O.; Dunn, D.B.A.; Qiu, Y.; Varey, A.H.R.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGFxxxb, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Mavrou, A.; Qiu, Y.; Carter, J.G.; Hamdollah-Zadeh, M.; Barratt, S.; Gammons, M.V.; Millar, A.B.; Salmon, A.H.J.; Oltean, S.; et al. Detection of VEGF-Axxxb Isoforms in Human Tissues. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Dehghanian, F.; Hojati, Z. Comparative insight into expression of recombinant human VEGF111b, a newly identified anti-angiogenic isoform, in eukaryotic cell lines. Gene 2014, 553, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Abou-Rayyah, Y.; Bischoff, J.; Ritchie, A.; Sebire, N.J.; Watts, P.; Churchill, A.J.; Bates, D.O. Altered ratios of pro- and anti-angiogenic VEGF-A variants and pericyte expression of DLL4 disrupt vascular maturation in infantile haemangioma. J. Pathol. 2016, 239, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Blythe, T.; Jarrett, C.; Ourradi, K.; Shelley-Fraser, G.; Day, M.J.; Qiu, Y.; Harper, S.; Maher, T.M.; Oltean, S.; et al. Differential Expression of VEGF-A xxx Isoforms Is Critical for Development of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.A.; Garbacki, N.; Colige, A.C. Chemotherapy induces alternative transcription and splicing: Facts and hopes for cancer treatment. Int. J. Biochem. Cell Biol. 2017, 91, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Gammons, M.; Hulse, R.; Hamdollah-Zadeh, M.; Mavrou, A.; Donaldson, L.; Salmon, A.H.; Harper, S.J.; Ladomery, M.R.; Bates, D. SRPK1 inhibition in vivo: Modulation of VEGF splicing and potential treatment for multiple diseases. Biochem. Soc. Trans. 2012, 40, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Oltean, S. Modulation of VEGF-A Alternative Splicing as a Novel Treatment in Chronic Kidney Disease. Genes (Basel) 2018, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Toop, H.D.; Redondo, C.; Babaei-Jadidi, R.; Chaikuad, A.; Wearmouth, S.F.; Gibbons, B.; Allen, C.; Tallant, C.; Zhang, J.; et al. Development of Potent, Selective SRPK1 Inhibitors as Potential Topical Therapeutics for Neovascular Eye Disease. ACS Chem. Biol. 2017, 12, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Keyt, B.A.; Berleau, L.T. The Carboxyl-terminal Domain(111–165) of Vascular Endothelial Growth Factor Is Critical for Its Mitogenic Potency. J. Biol. Chem. 1996, 271, 7788–7795. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.I.R.; Zachary, I.C. Vascular endothelial growth factor regulates Stanniocalcin-1 expression via Neuropilin-1-dependent regulation of KDR and synergism with fibroblast growth Factor-2. Cell Signal. 2008, 20, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, W.J.; Champe, M.A.; Christinger, H.W.; Keyt, B.A.; Starovasnik, M.A. Solution structure of the heparin-binding domain of vascular endothelial growth factor. Structure 1998, 6, 637–648. [Google Scholar] [CrossRef]
- Krilleke, D.; DeErkenez, A.; Schubert, W.; Giri, I.; Robinson, G.S.; Ng, Y.S.; Shima, D.T. Molecular mapping and functional characterization of the VEGF164 heparin-binding domain. J. Biol. Chem. 2007, 282, 28045–28056. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Folkman, J.; Javaherian, K. HSPG-Binding peptide corresponding to the exon 6a-encoded domain of VEGF inhibits tumor growth by blocking angiogenesis in Murine model. PLoS ONE 2010, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar] [PubMed]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The Vascular Endothelial Growth Factor Family: Identification of a Fourth Molecular Species and Characterization of Alternative Splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M.R.; Harper, S.J.; Bates, D.O. Alternative splicing in angiogenesis: The vascular endothelial growth factor paradigm. Cancer Lett. 2007, 249, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling–in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Wang, W.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF 165b, an Inhibitory Vascular Endothelial Growth Factor Splice Variant: Mechanism of Action, In vivo Effect On Angiogenesis and Endogenous Protein Expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef] [PubMed]
- Catena, R.; Larzabal, L.; Larrayoz, M.; Molina, E.; Hermida, J.; Agorreta, J.; Montes, R.; Pio, R.; Montuenga, L.M.; Calvo, A. VEGF121b and VEGF165b are weakly angiogenic isoforms of VEGF-A. Mol. Cancer 2010, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cébe Suarez, S.; Pieren, M.; Cariolato, L.; Arn, S.; Hoffman, U.; Bogucki, A.; Manlius, C.; Wood, J.; Ballmer-Hofer, K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell. Mol. Life Sci. 2006, 63, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.; Craze, M.; Newton, J.; Fisher, M.; Shima, D.T.; Tozer, G.M.; Kanthou, C. Do anti-angiogenic VEGF (VEGFxxxb) isoforms exist? a Cautionary Tale. PLoS ONE 2012, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bridgett, S.; Dellett, M.; Simpson, D.A. RNA-Sequencing data supports the existence of novel VEGFA splicing events but not of VEGFAxxxb isoforms. Sci. Rep. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Zhong, C.; Nudleman, E.; Ferrara, N. Evidence for Pro-angiogenic Functions of VEGF-Ax. Cell 2016, 167, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. VEGF receptor protein-tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 375, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Brozzo, M.S.; Bjelic, S.; Kisko, K.; Schleier, T.; Leppánen, V.-M.; Alitalo, K.; Winkler, F.K.; Ballmer-Hofer, K. Thermodynamic and structural description of allosterically regulated VEGF receptor 2 dimerization. Blood 2011, 119, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Leppanen, V.M.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Kalkkinen, N.; Strandin, T.; Lankinen, H.; Goldman, A.; Ballmer-Hofer, K.; Alitalo, K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc. Natl. Acad. Sci. USA 2010, 107, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; de Vos, A.M. Crystal Structure at 1.7 Å Resolution of VEGF in Complex with Domain 2 of the Flt-1 Receptor. Cell 1997, 91, 695–704. [Google Scholar] [CrossRef]
- Starovasnik, M.A.; Christinger, H.W.; Wiesmann, C.; Champe, M.A.; de Vos, A.M.; Skelton, N.J. Solution structure of the VEGF-binding domain of Flt-1: Comparison of its free and bound states. J. Mol. Biol. 1999, 293, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef] [PubMed]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Rockl, W.; Hornig, C.; Grone, E.; Theis, H.; Weich, H.; Fuchs, E.; Yayon, A.; Grone, H. Receptors of Vascular Endothelial Growth Factor/Vascular Permeability Factor (VEGF/VPF) in Fetal and Adult Human Kidney : Localization and [125I] VEGF Binding. J. Am. Soc. Nephrol. 1998, 9, 1032–1044. [Google Scholar] [PubMed]
- Whitaker, G.B.; Limberg, B.J.; Rosenbaum, J.S. Vascular Endothelial Growth Factor Receptor-2 and Neuropilin-1 Form a Receptor Complex that is Responsible for the Differential Signaling Potency of VEGF165 and VEGF121. J. Biol. Chem. 2001, 276, 25520–25531. [Google Scholar] [CrossRef] [PubMed]
- Gille, H.; Kowalski, J.; Li, B.; LeCouter, J.; Moffat, B.; Zioncheck, T.F.; Pelletier, N.; Ferrara, N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2): A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 2001, 276, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, T.; Toivanen, P.I.; Rintanen, N.; Heikura, T.; Jauhiainen, S.; Airenne, K.J.; Alitalo, K.; Marjomäki, V.; Ylä-Herttuala, S. The impact of the receptor binding profiles of the vascular endothelial growth factors on their angiogenic features. Biochim. Biophys. Acta 2014, 1840, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.; Johnstone, E.K.M.; Wheal, A.J.; Goulding, J.; Robers, M.B.; Machleidt, T.; Wood, K.V.; Hill, S.J.; Pfleger, K.D.G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Kilpatrick, L.E.; Hill, S.J. NanoBRET Approaches to Study Ligand Binding to GPCRs and RTKs. Trends Pharmacol. Sci. 2017, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Friedman-Ohana, R.; Alcobia, D.; Riching, K.; Peach, C.J.; Wheal, A.; Briddon, S.; Robers, M.; Zimmerman, K.; Machleidt, T.; et al. Real-time analysis of the binding of fluorescent VEGF165a to VEGFR2 in living cells: Effect of receptor tyrosine kinase inhibitors and fate of internalized agonist-receptor complexes. Biochem. Pharmacol. 2017, 136, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Clegg, L.E.; Mac Gabhann, F. A computational analysis of in vivo VEGFR activation by multiple co-expressed ligands. PLOS Comput. Biol. 2017, 13, e1005445. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Christopoulos, A.; Carmeliet, P. Allosteric targeting of receptor tyrosine kinases. Nat. Biotechnol. 2014, 32, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Ruch, C.; Skiniotis, G.; Steinmetz, M.O.; Walz, T.; Ballmer-Hofer, K. Structure of a VEGF–VEGF receptor complex determined by electron microscopy. Nat. Struct. Mol. Biol. 2007, 14, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Kisko, K.; Brozzo, M.S.; Missimer, J.; Schleier, T.; Menzel, A.; Leppänen, V.-M.; Alitalo, K.; Walzthoeni, T.; Aebersold, R.; Ballmer-Hofer, K. Structural analysis of vascular endothelial growth factor receptor-2/ligand complexes by small-angle X-ray solution scattering. FASEB J. 2011, 25, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xie, P.; Opatowsky, Y.; Schlessinger, J. Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 1906–1911. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.A.C.; Giese, A.; Stuttfeld, E.; Abram Saliba, J.; Villemagne, D.; Schleier, T.; Binz, H.K.; Ballmer-Hofer, K. Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Mol. Cell. Biol. 2012, 32, 3802–3813. [Google Scholar] [CrossRef] [PubMed]
- Thieltges, K.M.; Avramovic, D.; Piscitelli, C.L.; Markovic-Mueller, S.; Binz, H.K.; Ballmer-Hofer, K. Characterization of a drug-targetable allosteric site regulating vascular endothelial growth factor signaling. Angiogenesis 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dosch, D.D.; Ballmer-Hofer, K. Transmembrane domain-mediated orientation of receptor monomers in active VEGFR-2 dimers. FASEB J. 2010, 24, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Mineev, K.S.; Usmanova, D.; Lyukmanova, E.N.; Shulepko, M.A.; Kirpichnikov, M.P.; Winter, J.; Matkovic, M.; Deupi, X.; Arseniev, A.S.; et al. Structural and functional characterization of alternative transmembrane domain conformations in VEGF receptor 2 activation. Structure 2014, 22, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Ballmer-Hofer, K.; Hristova, K. VEGFR-2 conformational switch in response to ligand binding. Elife 2016, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast, C.V.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Kan, C.-C.; Villafranca, J.E.; et al. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef]
- Manni, S.; Kisko, K.; Schleier, T.; Missimer, J.; Ballmer-Hofer, K. Functional and structural characterization of the kinase insert and the carboxy terminal domain in VEGF receptor 2 activation. FASEB J. 2014, 28, 4914–4923. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-g and DNA synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2678–2778. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Ueno, H.; Shibuya, M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 1999, 18, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Abu-Ghazaleh, R.; Kabir, J.; Jia, H.; Lobo, M.; Zachary, I. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem. J. 2001, 360, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular Endothelial Growth Factor Regulates Endothelial Cell Survival through the Phosphatidylinositol 3’-Kinase/Akt Signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, K.; Cross, M.J.; Rolny, C.; Hägerkvist, R.; Rahimi, N.; Matsumoto, T.; Claesson-Welsh, L.; Welsh, M. The adaptor protein Shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J. Biol. Chem. 2004, 279, 22267–22275. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-Induced Vascular Permeability Is Mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- McMullen, M.E.; Bryant, P.W.; Glembotski, C.C.; Vincent, P.A.; Pumiglia, K.M. Activation of p38 has opposing effects on the proliferation and migration of endothelial cells. J. Biol. Chem. 2005, 280, 20995–21003. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Luciano, A.K.; Ackah, E.; Rodriguez-Vita, J.; Bancroft, T.; Eichmann, A.; Simons, M.; Kyriakides, T.R.; Morales-Ruiz, M.; Sessa, W.C. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc. Natl. Acad. Sci. USA 2014, 111, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Zhu, H.; Jiang, W.; Zhang, S. Protocatechuic Acid Induces Angiogenesis through PI3K-Akt-eNOS-VEGF Signalling Pathway. Basic Clin. Pharmacol. Toxicol. 2013, 113, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, J.; Zhuang, Z.W.; Eichmann, A.; Simons, M. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Haj, F.G.; Verveer, P.J.; Squire, A.; Neel, B.G.; Bastiaens, P.I.H. Imaging Sites of Receptor Dephosphorylation by PTP1B on the Surface of the Endoplasmic Reticulum. Science 2002, 295, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, R.P. A Modification of Receptor Theory. Br. J. Pharmacol. Chemother. 1956, 11, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. New concepts in pharmacological efficacy at 7TM receptors. Br. J. Pharmacol. 2013, 168, 554–575. [Google Scholar] [CrossRef] [PubMed]
- Galandrin, S.; Bouvier, M. Distinct Signaling Profiles of beta1 and beta2 Adrenergic Receptor Ligands toward Adenylyl Cyclase and Mitogen-Activated Protein Kinase Reveals the Pluridimensionality of Efficacy. Mol. Pharmacol. 2006, 70, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Li, X.; Harper, S.J.; Bates, D.; Claesson-Welsh, L. Vascular Endothelial Growth Factor (VEGF)-A165b Is A Weak In vitro Agonist for VEGF Receptor-2 Due to Lack of Coreceptor Binding and Deficient Regulation of Kinase Activity. Cancer Res. 2008, 68, 4683–4692. [Google Scholar] [CrossRef] [PubMed]
- Delcombel, R.; Janssen, L.; Vassy, R.; Gammons, M.; Haddad, O.; Richard, B.; Letourneur, D.; Bates, D.; Hendricks, C.; Waltenberger, J.; et al. New prospects in the roles of the C-terminal domains of VEGF-A and their cooperation for ligand binding, cellular signaling and vessels formation. Angiogenesis 2013, 16, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Hervé, M.A.; Buteau-Lozano, H.; Mourah, S.; Calvo, F.; Perrot-Applanat, M. VEGF189 stimulates endothelial cells proliferation and migration in vitro and up-regulates the expression of Flk-1/KDR mRNA. Exp. Cell Res. 2005, 309, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Ourradi, K.; Blythe, T.; Jarrett, C.; Barratt, S.L.; Welsh, G.I.; Millar, A.B. VEGF isoforms have differential effects on permeability of human pulmonary microvascular endothelial cells. Respir. Res. 2017, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pang, V.; Bates, D.O.; Leach, L. Regulation of human feto-placental endothelial barrier integrity by vascular endothelial growth factors: Competitive interplay between VEGF-A165a, VEGF-A165b, PIGF and VE-cadherin. Clin. Sci. 2017, 131, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Shiying, W.; Boyun, S.; Jianye, Y.; Wanjun, Z.; Ping, T.; Jiang, L.; Hongyi, H. The Different Effects of VEGFA121 and VEGFA165 on Regulating Angiogenesis Depend on Phosphorylation Sites of VEGFR2. Inflamm. Bowel Dis. 2017, 23, 603–616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fearnley, G.W.; Bruns, A.F.; Wheatcroft, S.B.; Ponnambalam, S. VEGF-A isoform-specific regulation of calcium ion flux, transcriptional activation and endothelial cell migration. Biol. Open 2015, 4, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, G.W.; Smith, G.A.; Abdul-Zani, I.; Yuldasheva, N.; Mughal, N.A.; Homer-Vanniasinkam, S.; Kearney, M.T.; Zachary, I.C.; Tomlinson, D.C.; Harrison, M.A.; et al. VEGF-A isoforms program differential VEGFR2 signal transduction, trafficking and proteolysis. Biol. Open 2016, 5, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Rennel, E.; Waine, E.; Guan, H.; Schüler, Y.; Leenders, W.; Woolard, J.; Sugiono, M.; Gillatt, D.; Kleinerman, E.; Bates, D.; et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br. J. Cancer 2008, 98, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Hueso, L.; Rios-Navarro, C.; Ruiz-Sauri, A.; Chorro, F.J.; Nunez, J.; Sanz, M.J.; Bodi, V.; Piqueras, L. Dynamics and implications of circulating anti-angiogenic VEGF-A165b isoform in patients with ST-elevation myocardial infarction. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Fuster, M.M.; Lawrence, R.; Esko, J.D. Heparan sulfate regulates VEGF165- and VEGF121- mediated vascular hyperpermeability. J. Biol. Chem. 2011, 286, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.M.; Waltenberger, J.; Yachechko, R.; Mirzapoiazova, T.; Sham, J.; Lee, C.; Elia, J.; Verin, A. Neuropilin-1 Regulates Vascular Endothelial Growth Factor-Mediated Endothelial Permeability. Circ. Res. 2005, 96, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Rundqvist, H.; Branco, C.; Johnson, R.S. Autocrine VEGF Isoforms Differentially Regulate Endothelial Cell Behavior. Front. Cell Dev. Biol. 2016, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Imoukhuede, P.I.; Popel, A.S. Quantification and cell-to-cell variation of vascular endothelial growth factor receptors. Exp. Cell Res. 2011, 317, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Baranska, P.; Jerczynska, H.; Pawlowska, Z.; Koziolkiewicz, W.; Cierniewski, C.S. Expression of Integrins and Adhesive Properties of Human Endothelial Cell Line EA.hy 926. Cancer Genom. Proteom. 2005, 270, 265–269. [Google Scholar] [CrossRef]
- Murphy, J.E.; Padilla, B.E.; Hasdemir, B.; Cottrell, G.S.; Bunnett, N.W. Endosomes: A legitimate platform for the signaling train. Proc. Natl. Acad. Sci. USA 2009, 106, 17615–17622. [Google Scholar] [CrossRef] [PubMed]
- Gourlaouen, M.; Welti, J.C.; Vasudev, N.S.; Reynolds, A.R. Essential Role for Endocytosis in the Growth Factor-stimulated Activation of ERK1/2 in Endothelial Cells. J. Biol. Chem. 2013, 288, 7467–7480. [Google Scholar] [CrossRef] [PubMed]
- Basagiannis, D.; Zografou, S.; Galanopoulou, K.; Christoforidis, S. Dynasore impairs VEGFR2 signalling in an endocytosis- independent manner. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jopling, H.M.; Howell, G.J.; Gamper, N.; Ponnambalam, S. The VEGFR2 receptor tyrosine kinase undergoes constitutive endosome-to-plasma membrane recycling. Biochem. Biophys. Res. Commun. 2011, 410, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Jopling, H.M.; Odell, A.F.; Pellet-Many, C.; Latham, A.M.; Frankel, P.; Sivaprasadarao, A.; Walker, J.H.; Zachary, I.C.; Ponnambalam, S. Endosome-to-Plasma Membrane Recycling of VEGFR2 Receptor Tyrosine Kinase Regulates Endothelial Function and Blood Vessel Formation. Cells 2014, 3, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 174, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Ewan, L.C.; Jopling, H.M.; Jia, H.; Mittar, S.; Bagherzadeh, A.; Howell, G.J.; Walker, J.H.; Zachary, I.C.; Ponnambalam, S. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic 2006, 7, 1270–1282. [Google Scholar] [CrossRef] [PubMed]
- Basagiannis, D.; Christoforidis, S. Constitutive endocytosis of VEGFR2 protects the receptor against shedding. J. Biol. Chem. 2016, 291, 16892–16903. [Google Scholar] [CrossRef] [PubMed]
- Basagiannis, D.; Zografou, S.; Murphy, C.; Fotsis, T.; Morbidelli, L.; Ziche, M.; Bleck, C.; Mercer, J.; Christoforidis, S. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation via macropinocytosis. J. Cell Sci. 2016, 129, 4091–4104. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Fearnley, G.W.; Abdul-Zani, I.; Wheatcroft, S.B.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. VEGFR2 Trafficking, Signaling and Proteolysis is Regulated by the Ubiquitin Isopeptidase USP8. Traffic 2016, 17, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Gampel, A.; Moss, L.; Jones, M.C.; Brunton, V.; Norman, J.C.; Mellor, H. VEGF regulates the mobilization of VEGFR2/KDR from an intracellular endothelial storage compartment. Blood 2006, 108, 2624–2631. [Google Scholar] [CrossRef] [PubMed]
- Bruns, A.F.; Herbert, S.P.; Odell, A.F.; Jopling, H.M.; Hooper, N.M.; Zachary, I.C.; Walker, J.H.; Ponnambalam, S. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 2010, 11, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Clegg, L.W.; Mac Gabhann, F. Site-Specific Phosphorylation of VEGFR2 Is Mediated by Receptor Trafficking: Insights from a Computational Model. PLOS Comput. Biol. 2015, 11, e1004158. [Google Scholar] [CrossRef] [PubMed]
- Ballmer-Hofer, K.; Andersson, A.E.; Ratcliffe, L.E.; Berger, P. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood 2011, 118, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Vempati, P.; Popel, A.S.; Mac Gabhann, F. Extracellular regulation of VEGF: Isoforms, proteolysis, and vascular patterning. Cytokine Growth Factor Rev. 2014, 25, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Melissa, E.S.; Skelton, N.J.; Fairbrother, W.J. Refinement of the solution structure of the heparin-binding domain of vascular endothelial growth factor using residual dipolar couplings. J. Biomol. NMR 2002, 23, 57–61. [Google Scholar] [CrossRef]
- Zhao, W.; McCallum, S.A.; Xiao, Z.; Zhang, F.; Linhardt, R.J. Binding affinities of vascular endothelial growth factor (VEGF) for heparin-derived oligosaccharides. Biosci. Rep. 2012, 32, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Gitay-Goren, H.; Soker, S.; Vlodavsky, I.; Neufeld, G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J. Biol. Chem. 1992, 267, 6093–6098. [Google Scholar] [PubMed]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 Is Expressed by Endothelial and Tumor Cells as an Isoform-Specific Receptor for Vascular Endothelial Growth Factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Teran, M.; Nugent, M.A. Synergistic binding of vascular endothelial growth factor-a and its receptors to heparin selectively modulates complex affinity. J. Biol. Chem. 2015, 290, 16451–16462. [Google Scholar] [CrossRef] [PubMed]
- Ashikari-Hada, S.; Habuchi, H.; Kariya, Y.; Kimata, K. Heparin regulates vascular endothelial growth factor165-dependent mitogenic activity, tube formation, and its receptor phosphorylation of human endothelial cells. Comparison of the effects of heparin and modified heparins. J. Biol. Chem. 2005, 280, 31508–31515. [Google Scholar] [CrossRef] [PubMed]
- Mamluk, R.; Gechtman, Z.; Kutcher, M.E.; Gasiunas, N.; Gallagher, J.; Klagsbrun, M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002, 277, 24818–24825. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Limberg, B.J.; Brian Whitaker, G.; Perman, B.; Leahy, D.J.; Rosenbaum, J.S.; Ginty, D.D.; Kolodkin, A.L. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 18069–18076. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, S.; Driscoll, P.C. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov. Today 2013, 18, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.F.; Vander Kooi, C.W. Neuropilin Functions as an Essential Cell Surface Receptor. J. Biol. Chem. 2015, 290, 29120–29126. [Google Scholar] [CrossRef] [PubMed]
- Appleton, B.; Wu, P.; Maloney, J.; Yin, J.P.; Liang, W.-C.; Stawicki, S.; Mortara, K.; Bowman, K.K.; Elliott, J.M.; Desmarais, W.; et al. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. EMBO J. 2007, 26, 4902–4912. [Google Scholar] [CrossRef] [PubMed]
- Kitsukawa, T.; Shimizu, M.; Sanbo, M.; Hirata, T.; Taniguchi, M.; Bekku, Y.; Yagi, T.; Fujisawa, H. Neuropilin–Semaphorin III/D-Mediated Chemorepulsive Signals Play a Crucial Role in Peripheral Nerve Projection in Mice. Neuron 1997, 19, 995–1005. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kitsukawa, T.; Bekku, Y.; Matsuda, Y.; Sanbo, M.; Yagi, T.; Fujisawa, H. A requirement for neuropilin-1 in embryonic vessel formation. Development 1999, 126, 4895–4902. [Google Scholar] [PubMed]
- Gu, C.; Rodriguez, E.R.; Reimert, D.V.; Shu, T.; Fritzsch, B.; Richards, L.J.; Kolodkin, A.L.; Ginty, D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 2003, 5, 45–57. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Strickland, L.A.; Liu, S.D.; Mak, J.; Schmidt, M.; Koeppen, H. Neuropilin-1 expression in cancer and development. J. Pathol. 2012, 226, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Lee, J.E.; Yoo, C.Y.; Ko, M.S.; Park, C.S.; Yang, S.H. NRP-1 expression is strongly associated with the progression of pituitary adenomas. Oncol. Rep. 2014, 32, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Chittenden, T.W.; Claes, F.; Lanahan, A.A.; Autiero, M.; Palac, R.T.; Tkachenko, E.V.; Elfenbein, A.; Ruiz de Almodovar, C.; Dedkov, E.; Tomanek, R.; et al. Selective Regulation of Arterial Branching Morphogenesis by Synectin. Dev. Cell 2006, 10, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Schwarz, Q.; Davidson, K.; Normando, E.M.; Denti, L.; Ruhrberg, C. The cytoplasmic domain of neuropilin 1 is dispensable for angiogenesis, but promotes the spatial separation of retinal arteries and veins. Development 2011, 138, 4185–4191. [Google Scholar] [CrossRef] [PubMed]
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Mota, F.; Fotinou, C.; Rhana, R.; Edith Chan, A.W.; Yelland, T.; Arooz, M.T.; O’Leary, A.P.; Hutton, J.; Frankel, P.; Zachary, I.; et al. Architecture and Hydration of the Arginine Binding Site of Neuropilin-1. FEBS J. 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol. Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Von Wronski, M.A.; Raju, N.; Pillai, R.; Bogdan, N.J.; Marinelli, E.R.; Nanjappan, P.; Ramalingam, K.; Arunachalam, T.; Eaton, S.; Linder, K.E.; et al. Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor. J. Biol. Chem. 2006, 281, 5702–5710. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Ladam, P.; Vassy, R.; Badache, S.; Bouchemal, N.; Navaza, A.; du Penhoat, C.H.; Perret, G.Y. Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides 2007, 28, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Vassy, R.; Martin, A.; Lecouvey, M.; Di Benedetto, M.; Crépin, M.; Perret, G.Y. Antiangiogenic and antitumor activities of peptide inhibiting the vascular endothelial growth factor binding to neuropilin-1. Life Sci. 2006, 79, 2370–2381. [Google Scholar] [CrossRef] [PubMed]
- Fuh, G.; Garcia, K.C.; de Vos, A.M. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol.Chem. 2000, 275, 26690–26695. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Guo, H.F.; Li, X.; Linkugel, A.D.; Vander Kooi, C.W. Function of members of the neuropilin family as essential pleiotropic cell surface receptors. Biochemistry 2012, 51, 9437–9446. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Van Meeteren, L.A.; Morin, E.; Testini, C.; Weström, S.; Björkelund, H.; Le Jan, S.; Adler, J.; Berger, P.; Claesson-Welsh, L. NRP1 Presented in trans to the endothelium arrests VEGFR2 endocytosis, preventing angiogenic signaling and tumor initiation. Dev. Cell 2014, 28, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Reed, R.R. Cloning and characterization of neuropilin-1-interacting protein: A PSD-95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J. Neurosci. 1999, 19, 6519–6527. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mukhopadhyay, D.; Xu, X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB J. 2006, 20, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Prahst, C.; Héroult, M.; Lanahan, A.A.; Uziel, N.; Kessler, O.; Shraga-Heled, N.; Simons, M.; Neufeld, G.; Augustin, H.G. Neuropilin-1-VEGFR-2 complexing requires the PDZ-binding domain of neuropilin-1. J. Biol. Chem. 2008, 283, 25110–25114. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.L.; Lin, A.W.; Chen, L.Q.; Safer, D.; Cain, S.M.; Hasson, T.; Carragher, B.O.; Milligan, R.A.; Sweeney, H.L. Myosin VI is an actin-based motor that moves backwards. Nature 1999, 401, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Brautigam, C.A.; Chen, R.; Lu, D.; Torres-vázquez, J. Structure analyses reveal a regulated oligomerization mechanism of the PlexinD1/GIPC/myosin VI complex. Elife 2017, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.; Hasson, T.; Horowitz, A. Binding of internalized receptors to the PDZ domain of GIPC/synectin recruits myosin VI to endocytic vesicles. PNAS 2006, 103, 12735–12740. [Google Scholar] [CrossRef] [PubMed]
- Reed, B.; Cefalu, C.; Bellaire, B.; Cardelli, J.; Louis, T.; Salamon, J.; Bloecher, M.; Bunn, R. GLUT1CBP(TIP2/GIPC1) Interactions with GLUT1 and Myosin VI: Evidence Supporting an Adapter Function for GLUT1CBP. Mol. Biol. Cell 2005, 16, 4183–4201. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Xu, P.; Guo, H.F.; Vander Kooi, C.W. Mechanism of Selective VEGF-A Binding by Neuropilin-1 Reveals a Basis for Specific Ligand Inhibition. PLoS ONE 2012, 7, e49177. [Google Scholar] [CrossRef] [PubMed]
- Tillo, M.; Erskine, L.; Cariboni, A.; Fantin, A.; Joyce, A.; Denti, L.; Ruhrberg, C. VEGF189 binds NRP1 and is sufficient for VEGF/NRP1-dependent neuronal patterning in the developing brain. Development 2015, 142, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Mac Gabhann, F. VEGF-A121a binding to Neuropilins–A concept revisited. Cell Adh. Migr. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N.; Dayanir, V.; Lashkari, K. Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J. Biol. Chem. 2000, 275, 16986–16992. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.J.; Hewett, P.W.; Ahmad, S.; Wang, K.-Q.; Cai, M.; Al-Ani, B.; Fujisawa, T.; Ma, B.; Sissaoui, S.; Ramma, W.; et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat. Commun. 2012, 3, 972. [Google Scholar] [CrossRef] [PubMed]
- Dixelius, J.; Mäkinen, T.; Wirzenius, M.; Karkkainen, M.J.; Wernstedt, C.; Alitalo, K.; Claesson-Welsh, L. Ligand-induced Vascular Endothelial Growth Factor Receptor-3 (VEGFR-3) Heterodimerization with VEGFR-2 in Primary Lymphatic Endothelial Cells Regulates Tyrosine Phosphorylation Sites. J. Biol. Chem. 2003, 278, 40973–40979. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.; Bahram, F.; Li, X.; Gualandi, L.; Koch, S.; Jarvius, M.; Söderberg, O.; Anisimov, A.; Kholová, I.; Pytowski, B.; et al. VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 2010, 29, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Coon, B.G.; Baeyens, N.; Han, J.; Budatha, M.; Ross, T.D.; Fang, J.S.; Yun, S.; Thomas, J.-L.; Schwartz, M.A. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 2015, 208, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Dunk, C.; Grohman, M.; Kontos, C.D.; Mason, J.; Ahmed, A. Vascular Endothelial Growth Factor Receptor-1 Modulates Vascular Endothelial Growth Factor-Mediated Angiogenesis via Nitric Oxide. Am. J. Pathol. 2001, 159, 993–1008. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Popel, A.S. Dimerization of VEGF receptors and implications for signal transduction: A computational study. Biophys. Chem. 2007, 128, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Neagoe, P.E.; Lemieux, C.; Sirois, M.G. Vascular endothelial growth factor (VEGF)-A165-induced prostacyclin synthesis requires the activation of VEGF receptor-1 and -2 heterodimer. J. Biol. Chem. 2005, 280, 9904–9912. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Andersson, C.; Roomans, G.M.; Ito, N.; Claesson-Welsh, L. Signaling properties of VEGF receptor-1 and -2 homo- and heterodimers. Int. J. Biochem. Cell Biol. 2001, 33, 315–324. [Google Scholar] [CrossRef]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; de Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Danastas, K.; Miller, E.J.; Hey-Cunningham, A.J.; Murphy, C.R.; Lindsay, L.A. Expression of vascular endothelial growth factor A isoforms is dysregulated in women with endometriosis. Reprod. Fertil. Dev. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Advani, A. VEGF and the diabetic kidney: More than too much of a good thing. J. Diabetes Complic. 2016, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hulse, R.P.; Beazley-Long, N.; Ved, N.; Bestall, S.M.; Riaz, H.; Singhal, P.; Ballmer-Hofer, K.; Harper, S.J.; Bates, D.; Donaldson, L.F. Vascular endothelial growth factor-A 165b prevents diabetic neuropathic pain and sensory neuronal degeneration. Clin. Sci. 2015, 129, 741–756. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.G.; Adamis, A.P. Ocular Angiogenesis: Vascular Endothelial Growth Factor and Other Factors. Dev. Ophthalmol. 2016, 55, 28–37. [Google Scholar] [PubMed]
- Shibata, Y.; Kikuchi, R.; Ishii, H.; Suzuki, S.; Harada, K.; Hirayama, K.; Suzuki, A.; Tatami, Y.; Kondo, K.; Murohara, T. Balance between angiogenic and anti-angiogenic isoforms of VEGF-A is associated with the complexity and severity of coronary artery disease. Clin. Chim. Acta 2018, 478, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Ganta, V.C.; Choi, M.; Kutateladze, A.; Annex, B.H. VEGF165b Modulates Endothelial VEGFR1-STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Pediatr. Neurol. 2016, 52, 566–584. [Google Scholar] [CrossRef]
- Kikuchi, R.; Nakamura, K.; Maclauchlan, S.; Ngo, D.T. An anti-angiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2015, 20, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Clara, A.; Newell, P.; Villanueva, A. VEGF signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Melillo, G. Role of the VEGF/VEGFR Axis in Cancer Biology and Therapy. Adv. Cancer Res. 2012, 114, 237–267. [Google Scholar] [PubMed]
- Guyot, M.; Hilmi, C.; Ambrosetti, D.; Merlano, M.; Lo Nigro, C.; Durivault, J.; Grépin, R.; Pagès, G. Targeting the pro-angiogenic forms of VEGF or inhibiting their expression as anti-cancer strategies. Oncotarget 2016, 8, 9174–9188. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new?: A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Comunanza, V.; Bussolino, F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front. Cell Dev. Biol. 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Li, B.; Houck, K.; Winer, J.; Ferrara, N. The Vascular Endothelial Growth Factor Proteins: Identification of Biologically Relevant Regions by Neutralizing Monoclonal Antibodies. Growth Factors 1992, 7, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Varey, A.H.R.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF165b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: Balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Gagliano, T.; Giamas, G. Direct Effects of Anti-Angiogenic Therapies on Tumor Cells: VEGF Signaling. Trends Mol. Med. 2017, 23, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration : A comprehensive review. Drug Des. Dev. Ther. 2016, 1857–1867. [Google Scholar] [CrossRef]
- Bahrami, B.; Zhu, M.; Hong, T.; Chang, A. Diabetic macular oedema: Pathophysiology, management challenges and treatment resistance. Diabetologia 2016, 59, 1594–1608. [Google Scholar] [CrossRef] [PubMed]
- Schmidinger, M. Understanding and managing toxicities of vascular endothelial growth factor (VEGF) inhibitors. Eur. J. Cancer Suppl. 2013, 11, 172–191. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Fretwell, L.V.; Woolard, J. Effects of 4 multitargeted receptor tyrosine kinase inhibitors on regional hemodynamics in conscious, freely moving rats. FASEB J. 2017, 31, 1193–1203. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
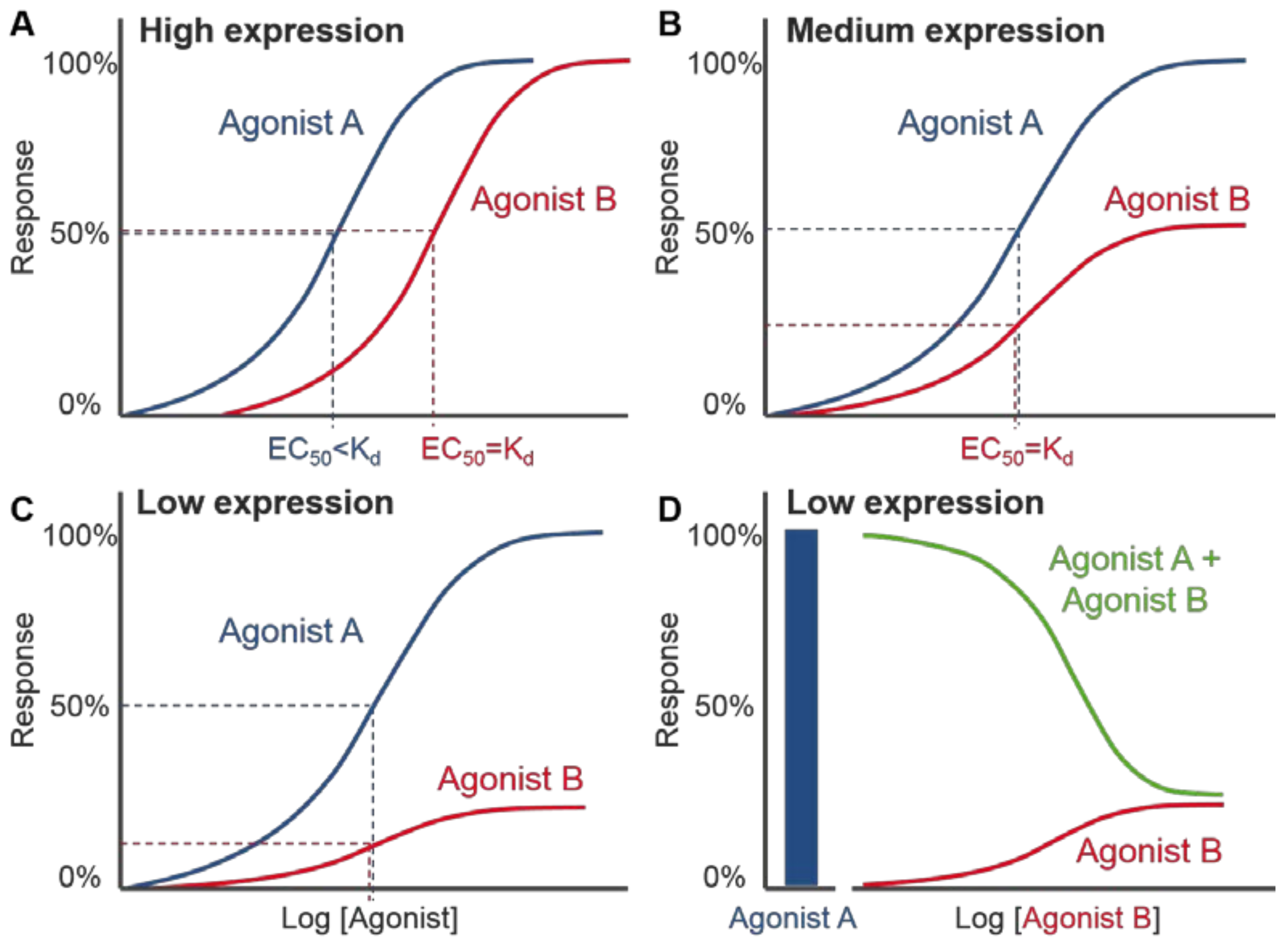{kind=link}
| Isoform | Technique | Expression System | Binding Affinity * | Ref. |
|---|---|---|---|---|
| VEGF165a | Radioligand binding | Human kidney tissue in situ | 0.01–0.04 nM | [91] |
| HUVECs | 0.17 nM | [92] | ||
| Balb/c expressing VEGFR2 | 0.29 nM | [92] | ||
| COS-1 cells expressing VEGFR2 | 0.34 nM | [92] | ||
| PAE cells expressing VEGFR2 | 0.76 nM | [44] | ||
| PAE cells expressing VEGFR2 | 0.097 nM | [93] | ||
| SPR | VEGFR2 ligand binding domains (D2/D3) | 36.7 nM | [85] | |
| ITC | VEGFR2 ligand binding domains (D2/D3) | 18 nM | [85] | |
| VEGFR2 ligand binding domains (D2/D3) | 170 nM | [84] | ||
| VEGFR2 extracellular domain (D1–D7) | 2670 nM | [84] | ||
| NanoBRET | HEK293 cells expressing NanoLuc-VEGFR2 | 0.15 nM | [97] | |
| VEGF165b | NanoBRET | HEK293 cells expressing NanoLuc-VEGFR2 | 0.39 nM | [97] |
| VEGF121a | ITC | VEGFR2 extracellular domain (D1–D7) | 1120 nM | [84] |
| VEGFR2 ligand binding domains (D2/D3) | 93 nM | [84] | ||
| NanoBRET | HEK293 cells expressing NanoLuc-VEGFR2 | 0.34 nM | [97] | |
| VEGF145a | NanoBRET | HEK293 cells expressing NanoLuc-VEGFR2 | 1.82 nM | [97] |
| VEGF189a | NanoBRET | HEK293 cells expressing NanoLuc-VEGFR2 | 1.02 nM | [97] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. https://doi.org/10.3390/ijms19041264
Peach CJ, Mignone VW, Arruda MA, Alcobia DC, Hill SJ, Kilpatrick LE, Woolard J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. International Journal of Molecular Sciences. 2018; 19(4):1264. https://doi.org/10.3390/ijms19041264
Chicago/Turabian StylePeach, Chloe J., Viviane W. Mignone, Maria Augusta Arruda, Diana C. Alcobia, Stephen J. Hill, Laura E. Kilpatrick, and Jeanette Woolard. 2018. "Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2" International Journal of Molecular Sciences 19, no. 4: 1264. https://doi.org/10.3390/ijms19041264
APA StylePeach, C. J., Mignone, V. W., Arruda, M. A., Alcobia, D. C., Hill, S. J., Kilpatrick, L. E., & Woolard, J. (2018). Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. International Journal of Molecular Sciences, 19(4), 1264. https://doi.org/10.3390/ijms19041264