miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis

 ,
,  ,
, 
Abstract
1. Introduction
2. miRNAs
Exosomal miRNAs
3. miRNA Signature in NAFLD
3.1. miR-122
3.2. miR-192
3.3. miR-375
4. miRNA Signature in Progressive NAFLD
4.1. miR-33a and miR-33b
4.2. miR-34a
4.3. miR-451
4.4. miR-155
5. Role of miRNAs in Adipose Tissue and Gut Homeostasis
6. miRNAs in Liver Fibrosis and HCC
6.1. miR-15 and miR-16
6.2. miR-34
6.3. miR-21
6.4. miR-221/222
7. miRNA Hereditability in NAFLD
8. Epigenetic Therapies Which Target miRNAs
9. Single Nucleotide Polymorphism Can Modify miRNA Target Sites
10. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAFLD | Nonalcoholic Fatty Liver Disease |
| NASH | Nonalcoholic Steatohepatitis |
| HCC | Hepatocellular Carcinoma |
| HBV | Viral Hepatitis B |
| HCV | Viral Hepatitis C |
| T2DM | Type 2 Diabetes Mellitus |
| VAT | Visceral Adipose Tissue |
| ALT | Alanine Aminotransferase |
| AST | Aspartate Aminotransferase |
| BMI | Body Mass Index |
| NAS | NAFLD Activity Score |
| FIB 4 | Fibrosis 4 |
| TNM | Tumor, Node and Metastasis |
| SNP | Single Nucleotide Polymorphism |
| PNPLA3 | Patatin-like phospholipase domain containing-3 |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| MBOAT7/TMC4 | Membrane bound O-acyltransferase domain containing 7-Transmembrane channel-like 4 |
| HFD | High Fat Diet |
| MCD | Methionine Choline-deficient Diet |
| TAA | Thioacetamide |
| VLDL | Very Low-Density Lipoprotein |
| HDL | High Density Lipoprotein |
| DSS | Dextran Sulfate Sodium |
| CCl4 | Carbon Tetrachloride |
| UDCA | Ursodeoxycholic Acid |
| OCA | Obeticholic Acid |
| FFA | Free Fatty Acid |
| KCs | Kupffer cells |
| CDCA | Chenodeoxycholic Acid |
| HSCs | Hepatic Stellate Cells |
| ATM | Adipose Tissue Macrophages |
| IEC | Intestinal Epithelial Cell |
| ECM | Extracellular Matrix |
| SREBP | Sterol Regulatory Element-binding Protein |
| miRNAs | microRNAs |
| exomiRs | Exosomal miRNAs |
| LPS | Lipopolysaccharides |
| TLRs | Toll-like receptors |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| PPARα | Peroxisome Proliferator Activated Receptor-alpha |
| PPARγ | Peroxisome Proliferator Activated Receptor-gamma |
| PTEN | Phosphatase and Tensin Homolog |
| ACLS-1 | Acyl-CoA Synthetase 1 Long-chain Family Member 1 |
| TGF-β/Smad | Transforming Growth Factor-Beta/Suppressor of Mothers Against Decapentaplegic Miscellaneous |
| AMPK | AMP-activated Protein Kinase |
| AdipoR2 | Adiponectin Receptor 2 |
| ATG7 | Autophagy-related Gene 7 |
| ATG12-5 | Autophagy-related Gene 12-5 |
| AEG-1 | Astrocyte Elevated Gene-1 |
| YAP1 | Yes-associated Protein |
| SIRT1 | Sirtuin 1 |
| HNF4α | Hepatocyte Nuclear Factor 4 Alpha |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IL-10 | Interleukin 10 |
| IL-12p40 | p40 Subunit of Interleukin 12 |
| ZO-1 | Zonula Occludens-1 |
| UTR | Untranslated region |
| IL-6/STAT3 | Interleukin-6/Signal Transducer and Activator of Transcription 3 |
| HNF6 | Hepatocyte Nuclear Factor 6 |
| CK-18 | Cytokeratin-18 |
| TNF-α | Tumor Necrosis Factor-alpha |
| CCL2 | C-C Motif Chemokine Ligand 2 (CCL2) |
| AGO2 | Argonaute-2 |
| HIF1-α | Hypoxia-Inducible Factor 1-α |
| MAPK1 | Mitogen-Activated Protein Kinase 1 |
| ABCA1 | ATP-binding Cassette Transporter sub-family A member 1 |
| APOA1 | Apolipoprotein A1 |
| Col1A1 | Type I Collagen A1 |
| αSMA | alpha-smooth muscle actin |
| PI3k/Akt | Phosphatidylinositol-3-Kinase/ Protein Kinase B |
| HMGCR | Hydroxymethylglutaryl-CoA Reductase |
| FGF19 | Fibroblast Growth Factor 19 |
| LXRα | Liver X Receptor alpha |
| FXR | Farnesoid-X Activated Receptor |
| iNOS | Inducible Nitric oxide synthases |
| Gα12 | Guanine Nucleotide-binding α-subunit 12 |
| GPCRs | G-protein Coupled Receptors |
| Bcl2 | B-cell lymphoma-2 |
| CASP2 | Caspase 2 |
| MMP2 | Metalloproteinase 2 |
| MMP9 | Metalloproteinase 9 |
| HDAC1 | Histone deacetylase 1 |
| ERK1 | Extracellular Signal-Regulated Kinase 1 |
| SPRY2 | Sprouty 2 |
| HBP1 | HMG-Box Transcription Factor 1 |
| CCN | Central Communication Network |
| DDIT4 | DNA Damage-Inducible Transcript 4 |
| AlncRNA | Artificial Long Non-coding RNA |
| DGCR8 | DiGeorge critical region 8 |
| GGT | γ Glutamyl Transferase |
References
- Rinella, M.E. Nonalcoholic fatty liver disease a systematic review. J. Am. Med. Assoc. 2015, 22, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 3, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. A nutrigenomic approach to non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2017, 7, 1534. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. BioMed Res. Int. 2015, 2015, 460190. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Diehl, A.M. Nonalcoholic Steatohepatitis. Annu. Rev. Med. 2017, 68, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, V.; Lammert, F. Genetics and epigenetics in the fibrogenic evolution of chronic liver diseases. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.-W. Epigenetics in non-alcoholic fatty liver disease. Mol. Aspects Med. 2016, 54, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. MicroRNAs in Nonalcoholic Fatty Liver Disease. J. Clin. Med. 2015, 4, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; El Andaloussi, S.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed]
- Enache, L.S.; Enache, E.L.; Ramière, C.; Diaz, O.; Bancu, L.; Sin, A.; André, P. Circulating RNA molecules as biomarkers in liver disease. Int. J. Mol. Sci. 2014, 15, 17644–17666. [Google Scholar] [CrossRef]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef]
- Goldie, B.J.; Dun, M.D.; Lin, M.; Smith, N.D.; Verrills, N.M.; Dayas, C.V.; Cairns, M.J. Activity-associated miRNA are packaged in Map1b-enriched exosomes released from depolarized neurons. Nucleic Acids Res. 2014, 42, 9195–9208. [Google Scholar] [CrossRef] [PubMed]
- Guduric-Fuchs, J.; O’Connor, A.; Camp, B.; O’Neill, C.L.; Medina, R.J.; Simpson, D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genom. 2012, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.I.; Yamaguchi, K.; et al. Let-7 microRNA family Is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 2010, 5, E13247. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Rekker, K.; Saare, M.; Roost, A.M.; Kubo, A.L.; Zarovni, N.; Chiesi, A.; Salumets, A.; Peters, M. Comparison of serum exosome isolation methods for microRNA profiling. Clin. Biochem. 2014, 47, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microRNA: Trafficking, sorting, and function. Genom. Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. MicroRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 6, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Panera, N.; Gnani, D.; Crudele, A.; Ceccarelli, S.; Nobili, V.; Alisi, A. MicroRNAs as controlled systems and controllers in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15079–15086. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Castaño, G.O.; Mallardi, P.; Martino, J.S.; Ledesma, M.M.G.L.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef]
- Feng, Y.Y.; Xu, X.Q.; Ji, C.B.; Shi, C.M.; Guo, X.R.; Fu, J.F. Aberrant hepatic MicroRNA expression in nonalcoholic fatty liver disease. Cell. Physiol. Biochem. 2014, 34, 1983–1997. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and epigenetic regulation in nonalcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Ampuero, J.; Gil-Gomez, A.; Montero-Vallejo, R.; Rojas, A.; Munoz-Hernandez, R.; Gallego-Duran, R.; Romero-Gomez, M. miRNAs in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2018, 69, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Deiters, A. MicroRNA miR-122 as a therapeutic target for oligonucleotides and small molecules. Curr. Med. Chem. 2013, 20, 36293–36640. [Google Scholar] [CrossRef]
- Laudadio, I.; Manfroid, I.; Achouri, Y.; Schmidt, D.; Wilson, M.D.; Cordi, S.; Thorrez, L.; Knoops, L.; Jacquemin, P.; Schuit, F.; et al. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology 2012, 142, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Sharma, A.D.; Roll, G.R.; Ng, R.; Lee, A.Y.; Blelloch, R.H.; Frandsen, N.M.; Willenbring, H. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology 2010, 51, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.P.; Rau, M.; Schmitt, J.; Malsch, C.; Hammer, C.; Bantel, H.; Müllhaupt, B.; Geier, A. Performance of Serum microRNAs -122, -192 and -21 as Biomarkers in Patients with Non-Alcoholic Steatohepatitis. PLoS ONE 2015, 10, e0142661. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Sharapova, T.; Lake, A.D.; Blomme, E.; Maher, J.; Cherrington, N.J. Circulating microRNA 122 in the methionine and choline-deficient mouse model of non-alcoholic steatohepatitis. J. Appl. Toxicol. 2014, 34, 726–732. [Google Scholar] [CrossRef]
- Yamada, H.; Suzuki, K.; Ichino, N.; Ando, Y.; Sawada, A.; Osakabe, K.; Sugimoto, K.; Ohashi, K.; Teradaira, R.; Inoue, T.; et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin. Chim. Acta 2013, 424, 99–103. [Google Scholar] [CrossRef]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; Perle, K.L.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Rayner, K.J.; Suárez, Y.; Fernández-Hernando, C. The Role of MicroRNAs in Cholesterol Efflux and Hepatic Lipid Metabolism. Annu. Rev. Nutr. 2011, 31, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Sacco, J.; Adeli, K. MicroRNAs: Emerging roles in lipid and lipoprotein metabolism. Curr. Opin. Lipidol. 2012, 23, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Luna, J.M.; Michailidis, E.; Rice, C.M. Mopping up miRNA: An integrated HBV transcript disrupts liver homeostasis by sequestering miR-122. J. Hepatol. 2016, 64, 257–259. [Google Scholar] [CrossRef]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and MIR-122-mediated HIF-1α activation. Sci. Rep. 2016, 6, 21340. [Google Scholar] [CrossRef]
- McGill, M.; Jaeschke, H. MicroRNAs as Signaling Mediators and Biomarkers of Drug- and Chemical-Induced Liver Injury. J. Clin. Med. 2015, 4, 1063–1078. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G1-modulated P53 activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef]
- Roberts, A.P.E.; Lewis, A.P.; Jopling, C.L. MiR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011, 39, 7716–7729. [Google Scholar] [CrossRef]
- Conrad, K.D.; Giering, F.; Erfurth, C.; Neumann, A.; Fehr, C.; Meister, G.; Niepmann, M. microRNA-122 Dependent Binding of Ago2 Protein to Hepatitis C Virus RNA Is Associated with Enhanced RNA Stability and Translation Stimulation. PLoS ONE 2013, 8, e56272. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Bukong, T.N.; Kodys, K.; Szabo, G. Alcohol Facilitates HCV RNA Replication Via Up-Regulation of miR-122 Expression and Inhibition of Cyclin G1 in Human Hepatoma Cells. Alcohol. Clin. Exp. Res. 2013, 37, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Hou, W.; Kodys, K.; Szabo, G. Ethanol facilitates hepatitis C virus replication via up-regulation of GW182 and heat shock protein 90 in human hepatoma cells. Hepatology 2013, 57, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; El Taghdouini, A.; Perea, L.; Mannaerts, I.; Vila-Casadesús, M.; Blaya, D.; Rodrigo-Torres, D.; Affò, S.; Morales-Ibanez, O.; Graupera, I.; et al. Integrative miRNA and Gene Expression Profiling Analysis of Human Quiescent Hepatic Stellate Cells. Sci. Rep. 2015, 5, 11549. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Zhou, C.; Yang, X.; Li, L. Down-regulation of microRNA-375 regulates adipokines and inhibits inflammatory cytokines by targeting AdipoR2 in non-alcoholic fatty liver disease. Clin. Exp. Pharmacol. Physiol. 2018, 45, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Yan, W.; He, X.; Zhang, L.; Li, C.; Huang, H.; Nace, G.; Geller, D.A.; Lin, J.; Tsung, A. MiR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology 2012, 143, 177–187.e8. [Google Scholar] [CrossRef] [PubMed]
- He, X.-X.; Chang, Y.; Meng, F.-Y.; Wang, M.-Y.; Xie, Q.-H.; Tang, F.; Li, P.-Y.; Song, Y.-H.; Lin, J.-S. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene 2012, 31, 3357–3369. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.M.; Poon, R.T.P.; Luk, J.M. MicroRNA-375 targets Hippo-signaling effector YAP in liver cancer and inhibits tumor properties. Biochem. Biophys. Res. Commun. 2010, 394, 623–627. [Google Scholar] [CrossRef]
- Tan, Y.; Ge, G.; Pan, T.; Wen, D.; Gan, J. A pilot study of serum micrornas panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e105192. [Google Scholar] [CrossRef]
- Guo, Y.; Xiong, Y.; Sheng, Q.; Zhao, S.; Wattacheril, J.; Flynn, C.R. A micro-RNA expression signature for human NAFLD progression. J. Gastroenterol. 2016, 51, 1022–1030. [Google Scholar] [CrossRef]
- Willeit, P.; Skroblin, P.; Kiechl, S.; Fernández-Hernando, C.; Mayr, M. Liver microRNAs: Potential mediators and biomarkers for metabolic and cardiovascular disease? Eur. Heart J. 2016, 37, 3260–3266. [Google Scholar] [CrossRef] [PubMed]
- Blaya, D.; Aguilar-Bravo, B.; Hao, F.; Casacuberta-Serra, S.; Coll, M.; Perea, L.; Vallverdú, J.; Graupera, I.; Pose, E.; Llovet, L.; et al. Expression of microRNA-155 in inflammatory cells modulates liver injury. Hepatology 2018, 68, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, Q.; Zhou, W.; Liu, T.; Yang, L.; Zheng, P.; Zhang, L.; Ji, G. Integrated analysis of hepatic mRNA and miRNA profiles identified molecular networks and potential biomarkers of NAFLD. Sci. Rep. 2018, 8, 7628. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V.; Najafi-Shoushtari, S.H.; Kristo, F.; Gurumurthy, S.; Zhong, L.; Li, Y.; Cohen, D.E.; Gerszten, R.E.; Bardeesy, N.; Mostoslavsky, R.; et al. Micrornas in metabolism and metabolic diseases. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; Van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Gori, M.; Arciello, M.; Balsano, C. MicroRNAs in Nonalcoholic Fatty Liver Disease : Novel Biomarkers and Prognostic Tools during the Transition from Steatosis to Hepatocarcinoma. BioMed Res. Int. 2014, 2014, 741465. [Google Scholar] [CrossRef]
- Li, Z.J.; Ou-Yang, P.H.; Han, X.P. Profibrotic effect of miR-33a with Akt activation in hepatic stellate cells. Cell. Signal. 2014, 26, 141–148. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Zarfeshani, A.; Ngo, S.; Sheppard, A. MicroRNA expression relating to dietary-induced liver steatosis and NASH. J. Clin. Med. 2015, 4, 1938–1950. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.E.; Ferreira, D.M.S.; Afonso, M.B.; Borralho, P.M.; MacHado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.P. MiR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, K.; Li, P.-F. MicroRNA-34 Family and Its Role in Cardiovascular Disease. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of MIR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Gao, L.; Zeng, W.; Hu, Y.; Wang, G.; Li, M.; Zhou, J.; Ma, X.; Tian, X.; Yao, J. Activation of the SIRT1/p66shc antiapoptosis pathway via carnosic acid-induced inhibition of MIR-34a protects rats against nonalcoholic fatty liver disease. Cell Death Dis. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Choi, S.-E.; Kim, D.-H.; Seok, S.; Suino-Powell, K.M.; Xu, H.E.; Kemper, J.K. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor—Klotho. Proc. Natl. Acad. Sci. USA 2012, 109, 16137–16142. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Nguyen, D.T.; Lu, L.F. Progress and challenge of microRNA research in immunity. Front. Genet. 2014, 5, 178. [Google Scholar] [CrossRef] [PubMed]
- Hur, W.; Lee, J.H.; Kim, S.W.; Kim, J.H.; Bae, S.H.; Kim, M.; Hwang, D.; Kim, Y.S.; Park, T.; Um, S.J.; et al. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/AKT pathway. Int. J. Biochem. Cell Biol. 2015, 64, 265–276. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-regulation of microRNA-155 in macrophages contributes to increased Tumor Necrosis Factor α (TNFα) production via increased mRNA half-life in alcoholic liver disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef]
- Csak, T.; Bala, S.; Lippai, D.; Kodys, K.; Catalano, D.; Iracheta-Vellve, A.; Szabo, G. MicroRNA-155 deficiency attenuates liver steatosis and fibrosis without reducing inflammation in a mouse model of steatohepatitis. PLoS ONE 2015, 10, e0129251. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J. MicroRNA-155 in the Heart. Circulation 2015, 131, 1533–1535. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Broering, R.; Trippler, M.; Wu, J.; Zhang, E.; Zhang, X.; Gerken, G.; Lu, M.; Schlaak, J.F. MicroRNA-155 controls Toll-like receptor 3- and hepatitis C virus-induced immune responses in the liver. J. Viral Hepat. 2014, 21, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Wang, Z.; Qi, G.; Yuan, S.; Yu, S.; Li, B.; Wei, Y.; Huang, Q.; Zhai, R.; He, S. MicroRNA-155 deficiency attenuates ischemia-reperfusion injury after liver transplantation in mice. Transpl. Int. 2015, 28, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Abente, E.J.; Subramanian, M.; Ramachandran, V.; Najafi-Shoushtari, S.H. MicroRNAs in obesity-associated disorders. Arch. Biochem. Biophys. 2016, 589, 108–119. [Google Scholar] [CrossRef]
- Estep, M.; Armistead, D.; Hossain, N.; Elarainy, H.; Goodman, Z.; Baranova, A.; Chandhoke, V.; Younossi, Z.M. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 32, 487–497. [Google Scholar] [CrossRef]
- Sharma, H.; Estep, M.; Birerdinc, A.; Afendy, A.; Moazzez, A.; Elariny, H.; Goodman, Z.; Chandhoke, V.; Baranova, A.; Younossi, Z.M. Expression of genes for microRNA-processing enzymes is altered in advanced non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2013, 28, 1410–1415. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate in Vivo and in Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef]
- Chai, C.; Rivkin, M.; Berkovits, L.; Simerzin, A.; Zorde-Khvalevsky, E.; Rosenberg, N.; Klein, S.; Yaish, D.; Durst, R.; Shpitzen, S.; et al. Metabolic Circuit Involving Free Fatty Acids, microRNA 122, and Triglyceride Synthesis in Liver and Muscle Tissues. Gastroenterology 2017, 153, 1404–1415. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Ye, D.; Guo, S.; Alsadi, R.; Ma, T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 2011, 141, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Baque, V.; Coupé, B.; Fabre, A.; Handgraaf, S.; Gourdy, P.; Arnal, J.F.; Courtney, M.; Schuster-Klein, C.; Guardiola, B.; Tercé, F.; et al. Associations between hepatic miRNA expression, liver triacylglycerols and gut microbiota during metabolic adaptation to high-fat diet in mice. Diabetologia 2017, 60, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 Knockout Improve the Survival Rate in DSS Induced Fatal Colitis through Protecting against Inflammation and Tissue Injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e17. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Limaye, A.; Cabrera, R. Hepatocellular carcinoma: Current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepatic Med. Evid. Res. 2012, 4, 19–37. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Halász, T.; Horváth, G.; Pár, G.; Werling, K.; Kiss, A.; Schaff, Z.; Lendvai, G. miR-122 negatively correlates with liver fibrosis as detected by histology and fibroscan. World J. Gastroenterol. 2015, 21, 7814–7823. [Google Scholar] [CrossRef]
- Celikbilek, M.; Baskol, M.; Taheri, S.; Deniz, K.; Dogan, S.; Zararsiz, G.; Gursoy, S.; Guven, K.; Ozbakir, O.; Dundar, M.; et al. Circulating microRNAs in patients with non-alcoholic fatty liver disease. World J. Hepatol. 2014, 6, 613–620. [Google Scholar] [CrossRef]
- Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Ohno, M.; Takata, A.; Shibata, C.; Koike, K. The role of microRNAs in hepatocarcinogenesis: Current knowledge and future prospects. J. Gastroenterol. 2014, 49, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Tijsen, A.J.; Van Der Made, I.; Van Den Hoogenhof, M.M.; Wijnen, W.J.; Van Deel, E.D.; De Groot, N.E.; Alekseev, S.; Fluiter, K.; Schroen, B.; Goumans, M.J.; et al. The microRNA-15 family inhibits the TGFβ-pathway in the heart. Cardiovasc. Res. 2014, 104, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Han, C.Y.; Kim, J.Y.; Cho, S.S.; Kim, Y.S.; Koo, J.H.; Lee, J.M.; Lim, S.C.; Kang, K.W.; Kim, J.S.; et al. Gα12overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J. Hepatol. 2018, 68, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Hernndezgea, V.; Ghiassinejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.J.; Pan, Q.; Li, D.G.; Sun, H.; Liu, B.W. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. J. Hepatol. 2009, 50, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.J.; Pan, Q.; Cheng, T.; Jiang, B.; Chen, G.Y.; Li, D.G. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J. 2009, 276, 5163–5176. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I.; Calin, G.A.; Croce, C.M. MiR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2010, 17, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Sun, T.; Wu, G.; Shang-Guan, H.; Jiang, Z.J.; Zhang, J.R.; Zheng, Y.F. MiR-15a suppresses hepatocarcinoma cell migration and invasion by directly targeting cMyb. Am. J. Transl. Res. 2017, 9, 520–532. [Google Scholar]
- Liu, K.; Liu, S.; Zhang, W.; Jia, B.; Tan, L.; Jin, Z.; Liu, Y. miR-494 promotes cell proliferation, migration and invasion, and increased sorafenib resistance in hepatocellular carcinoma by targeting PTEN. Oncol. Rep. 2015, 34, 1003–1010. [Google Scholar] [CrossRef]
- Wu, W.-L.; Wang, W.-Y.; Yao, W.-Q.; Li, G.-D. Suppressive effects of microRNA-16 on the proliferation, invasion and metastasis of hepatocellular carcinoma cells. Int. J. Mol. Med. 2015, 36, 1713–1719. [Google Scholar] [CrossRef]
- Meng, F.; Glaser, S.S.; Francis, H.; Yang, F.; Han, Y.; Stokes, A.; Staloch, D.; McCarra, J.; Liu, J.; Venter, J.; et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am. J. Pathol. 2012, 181, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Parkes, H.A.; Preston, E.; Wilks, D.; Ballesteros, M.; Carpenter, L.; Wood, L.; Kraegen, E.W.; Furler, S.M.; Cooney, G.J. Overexpression of acyl-CoA synthetase-1 increases lipid deposition in hepatic (HepG2) cells and rodent liver in vivo. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E737–E744. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. MicroRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor γ. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Li, B.; Xin, X.; Xu, M.; Ji, G.; Yu, H. MicroRNA-34a Promotes Hepatic Stellate Cell Activation via Targeting ACSL1. Med. Sci. Monit. 2015, 21, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Shen, H.; Li, X.; Li, Z.; Liu, Z.; Xu, J.; Ma, S.; Zhao, X.; Bai, X.; Li, M.; et al. MiR-125a-5p decreases after long non-coding RNA HOTAIR knockdown to promote cancer cell apoptosis by releasing caspase 2. Cell Death Dis. 2016, 7, e2137. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef]
- Misso, G.; Teresa, M.; Martino, D.; Rosa, G.D.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gullà, A.; Tagliaferri, P.; et al. Mir-34 : A New Weapon Against Cancer ? Mol. Ther. 2014, 3, e194. [Google Scholar] [CrossRef]
- Sun, T.; Xie, H.; Li, Z.; Kong, L.; Gou, X.; Li, D.; Shi, Y.; Ding, Y. miR-34a regulates HDAC1 expression to affect the proliferation and apoptosis of hepatocellular carcinoma. Am. J. Transl. Res. 2017, 9, 103–114. [Google Scholar]
- Xia, C.; Shui, L.; Lou, G.; Ye, B.; Zhu, W.; Wang, J.; Wu, S. 0404 inhibits hepatocellular carcinoma through a p53/miR-34a/SIRT1 positive feedback loop. Sci. Rep. 2017, 7, 43–96. [Google Scholar] [CrossRef]
- Bonci, D. MicroRNA-21 as therapeutic target in cancer and cardiovascular disease. Recent Pat. Cardiovasc. Drug Discov. 2010, 5, 156–161. [Google Scholar] [CrossRef]
- Zhao, J.; Tang, N.; Wu, K.; Dai, W.; Ye, C.; Shi, J.; Zhang, J.; Ning, B.; Zeng, X.; Lin, Y. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PLoS ONE 2014, 9, e108005. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Afonso, M.B.; Simaõ, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; MacHado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.; et al. MiR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Feng, L.; Li, Z.; Xu, G.; Fan, X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed. Pharmacother. 2013, 67, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ. Res. 2007, 100, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Enomoto, M.; Fujii, H.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; Kawada, N. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut 2012, 61, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, C.; Jiao, X.; Zhao, S.; Liu, X.; Wang, Y.; Zhang, J. miR-221 promotes growth and invasion of hepatocellular carcinoma cells by constitutive activation of NFκB. Am. J. Transl. Res. 2016, 8, 4764–4777. [Google Scholar]
- Gramantieri, L.; Fornari, F.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Croce, C.M.; Bolondi, L.; Negrini, M. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin. Cancer Res. 2009, 15, 5073–5081. [Google Scholar] [CrossRef]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of Nonalcoholic Fatty Liver Disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. Genetics of nonalcoholic fatty liver disease. Metabolism 2016, 65, 1026–1037. [Google Scholar] [CrossRef]
- Loomba, R.; Rao, F.; Zhang, L.; Khandrika, S.; Ziegler, M.G.; Brenner, D.A.; O’Connor, D.T. Genetic covariance between gamma-glutamyl transpeptidase and fatty liver risk factors: Role of beta2-adrenergic receptor genetic variation in twins. Gastroenterology 2010, 139, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. From The Cover: Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Gupta, S.; Maurya, M.R.; Subramaniam, S.; Loomba, R. Serum microRNAs explain discordance of non-alcoholic fatty liver disease in monozygotic and dizygotic twins: a prospective study. Gut 2016, 65, 1546–1554. [Google Scholar] [CrossRef]
- Fornari, F.; Pollutri, D.; Patrizi, C.; Bella, T.L.; Marinelli, S.; Gardini, A.C.; Marisi, G.; Toaldo, M.B.; Baglioni, M.; Callegari, E.; et al. In hepatocellular carcinoma miR-221 modulates Sorafenib resistance through inhibition of caspase-3 mediated apoptosis. Clin. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Gottwein, J.M.; Scheel, T.K.; Jensen, T.B.; Bukh, J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5’ UTR. Proc. Natl. Acad. Sci. USA 2011, 108, 4991–4996. [Google Scholar] [CrossRef]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Rum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, L.; Liu, J.; Zhang, Y.; Zhu, J.; Chen, J.; Liu, S.; Liu, Z.; Shi, H.; Shen, H.; et al. A potentially functional polymorphism in the promoter region of miR-34b/c is associated with an increased risk for primary hepatocellular carcinoma. Int. J. Cancer 2011, 128, 412–417. [Google Scholar] [CrossRef]
- Yoon, E.L.; Eun Yeon, J.; Ko, E.; Jung Lee, H.; Hye Je, J.; Jae Yoo, Y.; Hee Kang, S.; Jun Suh, S.; Hoon Kim, J.; Seok Seo, Y.; et al. An Explorative Analysis for the Role of Serum miR-10b-3p Levels in Predicting Response to Sorafenib in Patients with Advanced Hepatocellular Carcinoma. J. Korean Med. Sci. 2017, 32, 212–220. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Dong, X.; Zhai, B.; Jiang, X.; Dong, D.; Li, B.; Jiang, H.; Xu, S.; Sun, X. MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget 2015, 6, 28867–28881. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tan, G.; Jiang, X.; Han, P.; Zhai, B.; Dong, X.; Qiao, H.; Jiang, H.; Sun, X. An artificial lncRNA targeting multiple miRNAs overcomes sorafenib resistance in hepatocellular carcinoma cells. Oncotarget 2016, 7, 73257–73269. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from MIR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Yang, Q.; Wang, T.; Cao, Y.; Jiang, Q.-Y.; Ma, H.-D.; Sun, H.-W.; Hou, M.-X.; Yang, Y.-P.; Feng, F. Rhamnetin induces sensitization of hepatocellular carcinoma cells to a small molecular kinase inhibitor or chemotherapeutic agents. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Yamagami, M.; Ohno, M.; Takata, A.; Shibata, C.; Ishibashi, R.; Koike, K. MicroRNAs and liver disease. J. Hum. Genet. 2017, 62, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Krattinger, R.; Boström, A.; Lee, S.M.L.; Thasler, W.E.; Schiöth, H.B.; Kullak-Ublick, G.A.; Mwinyi, J. Chenodeoxycholic acid significantly impacts the expression of miRNAs and genes involved in lipid, bile acid and drug metabolism in human hepatocytes. Life Sci. 2016, 156, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Katsura, A.; Morishita, A.; Iwama, H.; Tani, J.; Sakamoto, T.; Tatsuta, M.; Toyota, Y.; Fujita, K.; Kato, K.; Maeda, E.; et al. MicroRNA profiles following metformin treatment in a mouse model of non-alcoholic steatohepatitis. Int. J. Mol. Med. 2015, 35, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Hepatocellular carcinoma in nonalcoholic fatty liver: Role of environmental and genetic factors. World J. Gastroenterol. 2014, 20, 12945–12955. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. PNPLA3, the triacylglycerol synthesis/hydrolysis/storage dilemma, and nonalcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 6018–6026. [Google Scholar] [CrossRef]
- Suresh, P.S.; Venkatesh, T.; Tsutsumi, R. In silico analysis of polymorphisms in microRNAs that target genes affecting aerobic glycolysis. Ann. Transl. Med. 2016, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Pirooz, H.J.; Jafari, N.; Rastegari, M.; Fathi-Roudsari, M.; Tasharrofi, N.; Shokri, G.; Tamadon, M.; Sazegar, H.; Kouhkan, F. Functional SNP in microRNA-491-5p binding site of MMP9 3’-UTR affects cancer susceptibility. J. Cell. Biochem. 2018, 119, 5126–5134. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
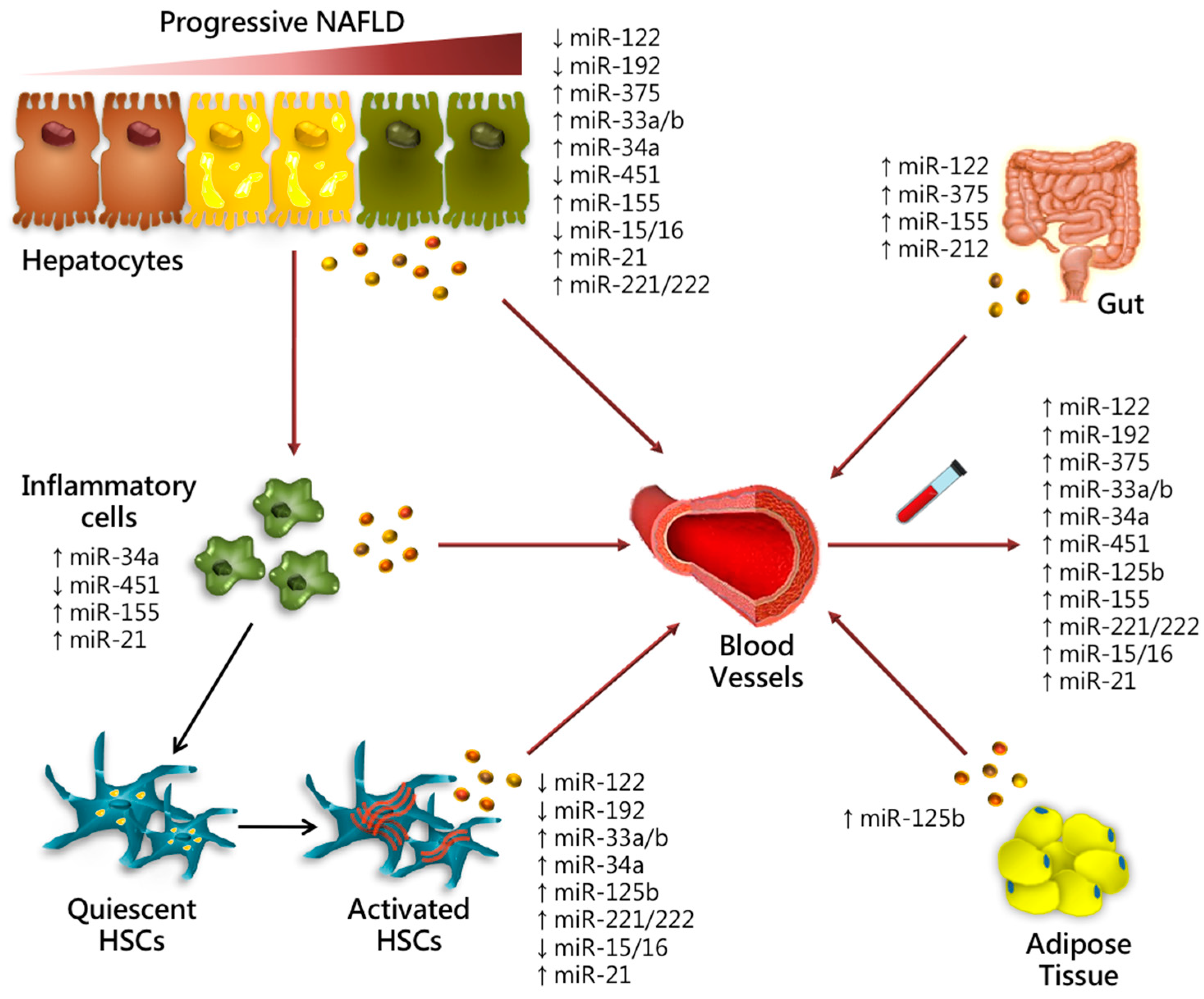
| Candidate Biomarkers | Serum | Liver | Function | Experimental Model | NAFLD Severity | Ref. |
|---|---|---|---|---|---|---|
| miR-122 | Up-regulated | Down-regulated | Lipid metabolism Intestinal permeability modulation Inflammation Fibrogenesis Proliferation | Human MCD mice In vitro | Steatosis NASH Fibrosis HCC | [29,38,39,40,71] |
| miR-192 | Up-regulated | Down-regulated | HSCs activation | Human | Steatosis NASH Fibrosis | [29,54] |
| miR-375 | Up-regulated | Up-regulated | Glucose homeostasis Intestinal permeability modulation Inflammation | Human HFD mice In vitro | Steatosis NASH HCC | [29,55,56] |
| miR-125b | Up-regulated | Up-regulated | Lipid and glucose homeostasis Adipocytes differentiation HSCs activation Fibrogenesis | Human | Steatosis NASH Fibrosis | [29,87] |
| miR-33a/b | Up-regulated | Up-regulated | Lipid and cholesterol metabolism Glucose homeostasis HSCs activation | Human Non-human primates HFD mice MCD mice In vitro | Steatosis NASH Fibrosis | [66,69,70] |
| miR-34a | Up-regulated | Up-regulated | Lipid metabolism Oxidative stress Apoptosis Proliferation | Human HFD mice In vitro | Steatosis NASH Fibrosis HCC | [71,72,75,76] |
| miR-451 | Up-regulated | Down-regulated | Inflammation | Human HFD mice In vitro | NASH | [39,79] |
| miR-155 | Up-regulated | Up-regulated | Lipid metabolism Intestinal permeability modulation Inflammation | Human MCD mice In vitro | NASH | [81] |
| miR-221/222 | Up-regulated | Up-regulated | HSCs activation Fibrogenesis Sorafenib resistance | Human TAA mice MCD mice In vitro | NASH Fibrosis HCC | [116,127,137] |
| miR-15/16 | Up-regulated | Down-regulated | HSCs activation Proliferation and Metastasis | Human CCl4 rats In vitro | Fibrosis HCC | [45,108,109] |
| miR-21 | Up-regulated | Up-regulated | Gut microbiota modulation Inflammation EMT Proliferation | Human HFD mice In vitro | NASH Fibrosis HCC | [92,93,122,125] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. https://doi.org/10.3390/ijms19123966
Dongiovanni P, Meroni M, Longo M, Fargion S, Fracanzani AL. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. International Journal of Molecular Sciences. 2018; 19(12):3966. https://doi.org/10.3390/ijms19123966
Chicago/Turabian StyleDongiovanni, Paola, Marica Meroni, Miriam Longo, Silvia Fargion, and Anna Ludovica Fracanzani. 2018. "miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis" International Journal of Molecular Sciences 19, no. 12: 3966. https://doi.org/10.3390/ijms19123966
APA StyleDongiovanni, P., Meroni, M., Longo, M., Fargion, S., & Fracanzani, A. L. (2018). miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. International Journal of Molecular Sciences, 19(12), 3966. https://doi.org/10.3390/ijms19123966