Werner Syndrome Protein and DNA Replication

Abstract
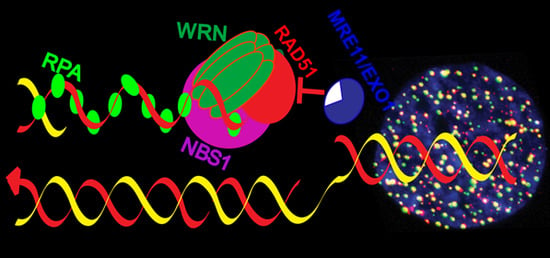
{kind=link}
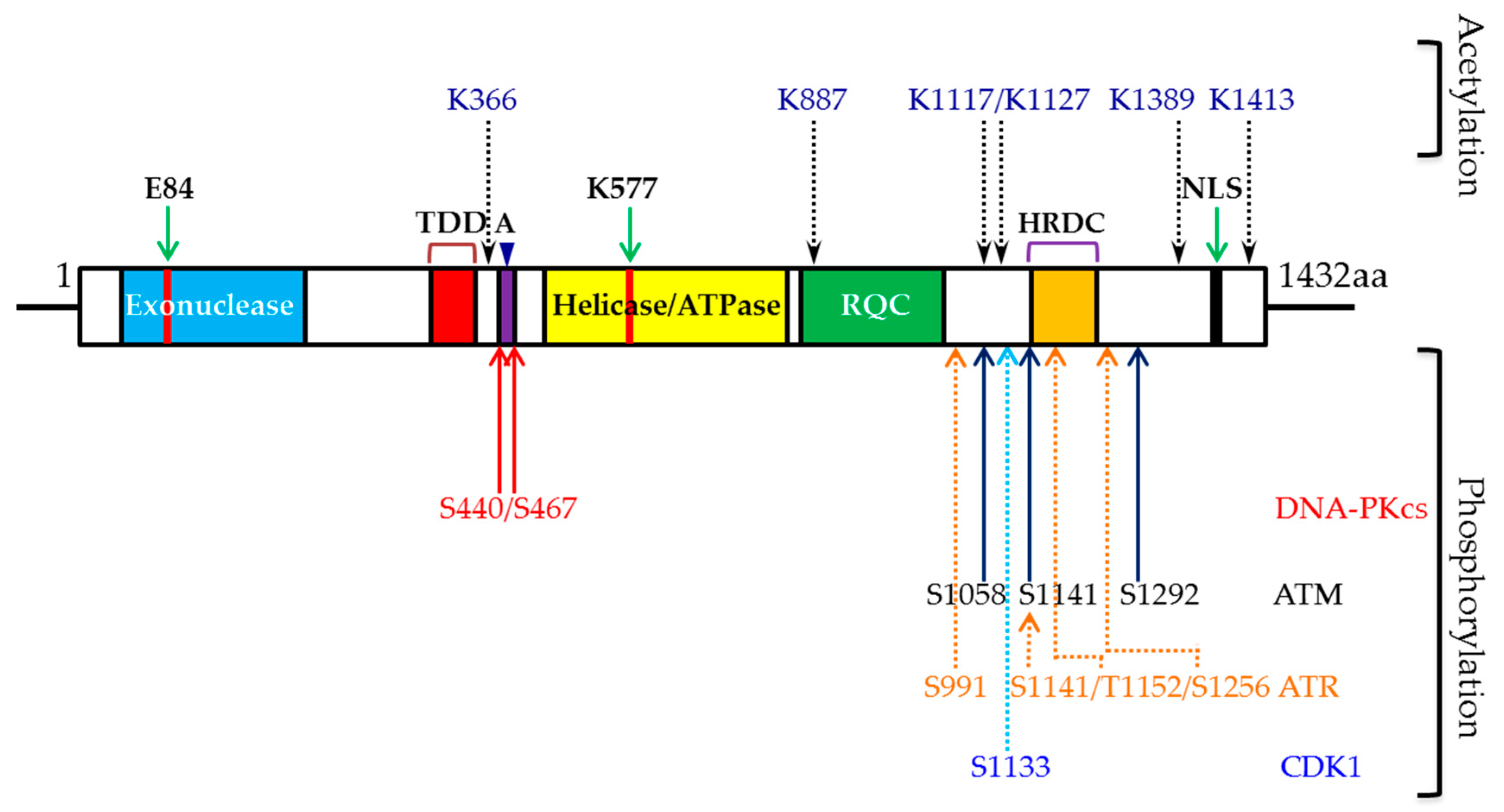{kind=link}
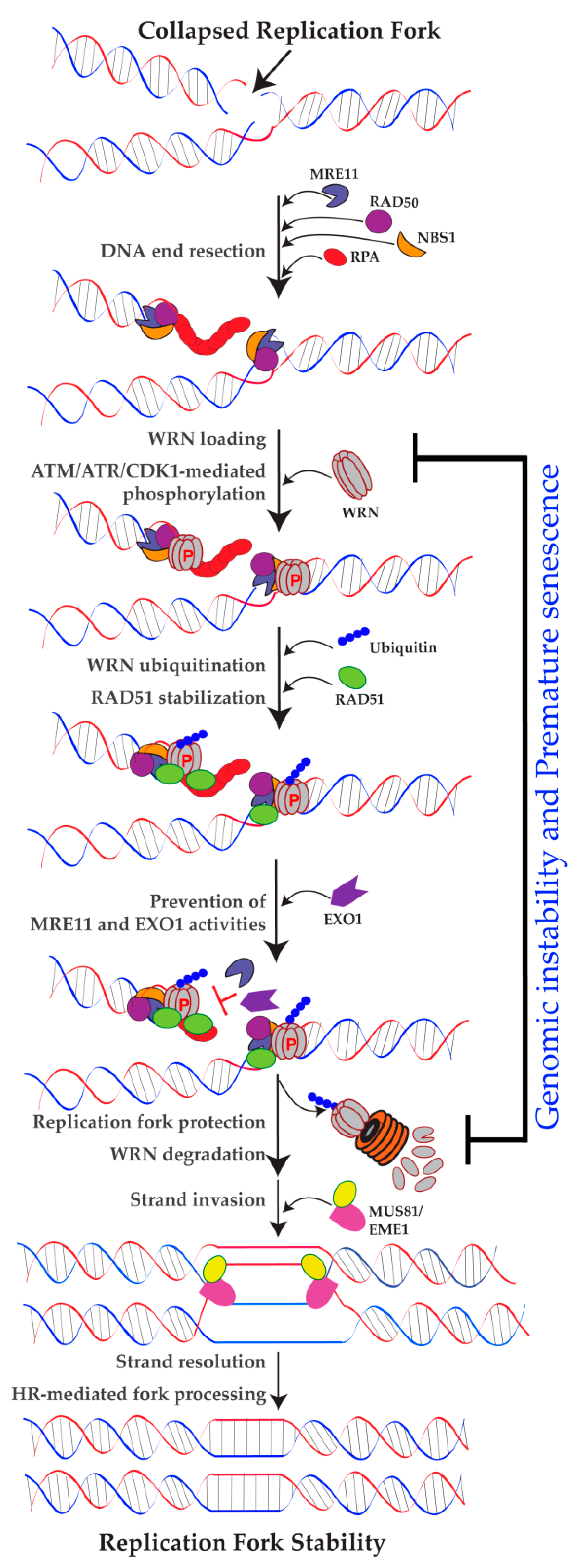{kind=link}
{kind=link}
1. Introduction
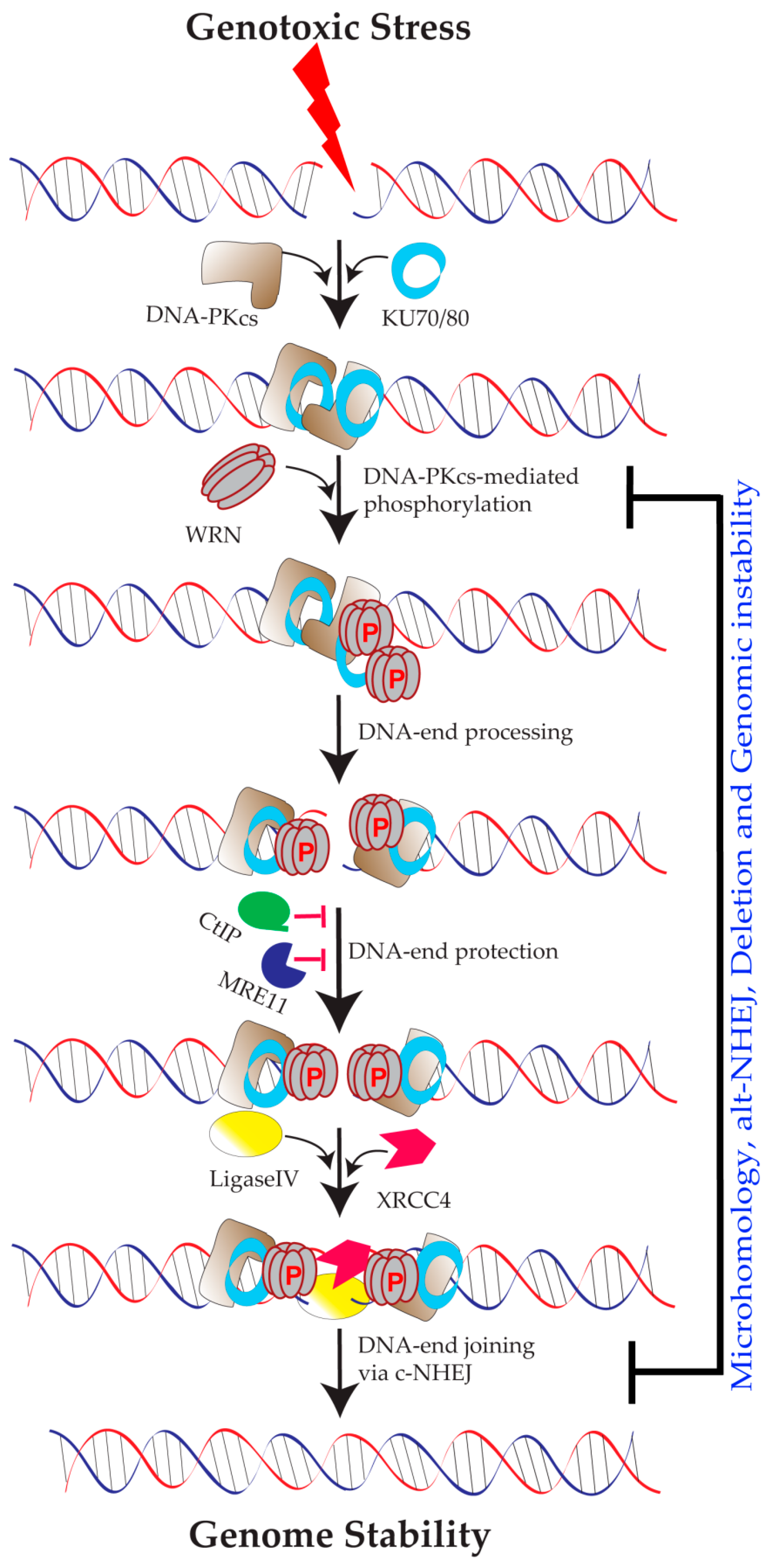
2. WRN in DNA Replication
2.1. Role of WRN in Replication Fork Progression
2.2. Role of WRN in Replication Fork Arrest Recovery
2.3. Role of WRN in Replication Fork Protection
3. WRN and Phosphorylation
3.1. Role of DNA-PKCS-Mediated WRN Phosphorylation
3.2. Role of ATR-Mediated WRN Phosphorylation
3.3. Role of ATM-Mediated WRN Phosphorylation
3.4. Role of CDK1-Mediated WRN Phosphorylation
4. WRN Stability and Degradation
4.1. Role of WRN Ubiquitination
4.2. Role of WRN Acetylation
5. WRN and DSB Repair Pathway Choice
5.1. Role of WRN in Classical NHEJ
5.2. Role of WRN in Alternative NHEJ Pathway
5.3. Role of WRN in HR
6. Consequences of WRN Deficiency and Replication Stress
6.1. Role of WRN in Maintaining Genome Stability
6.2. Role of WRN in Suppressing Premature Aging
7. Conclusions and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Friedrich, K.; Lee, L.; Leistritz, D.F.; Nurnberg, G.; Saha, B.; Hisama, F.M.; Eyman, D.K.; Lessel, D.; Nurnberg, P.; Li, C.; et al. WRN mutations in Werner syndrome patients: Genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum. Genet. 2010, 128, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Goto, M. Hierarchical deterioration of body systems in Werner’s syndrome: Implications for normal ageing. Mech. Ageing Dev. 1997, 98, 239–254. [Google Scholar] [CrossRef]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Salk, D. Werner’s syndrome: A review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Hum. Genet. 1982, 62, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.J.; Motulsky, A.G. Werner syndrome: Entering the helicase era. Bioessays 1996, 18, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Biomark. Prev. 1996, 5, 239–246. [Google Scholar]
- Salk, D.; Au, K.; Hoehn, H.; Martin, G.M. Cytogenetics of Werner’s syndrome cultured skin fibroblasts: Variegated translocation mosaicism. Cytogenet. Genome Res. 1981, 30, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, K.; Martin, G.M.; Monnat, R.J., Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl. Acad. Sci. USA 1989, 86, 5893–5897. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.M.; Sprague, C.A.; Epstein, C.J. Replicative life-span of cultivated human cells. Effects of donor’s age, tissue, and genotype. Lab. Investig. 1970, 23, 86–92. [Google Scholar] [PubMed]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.J.; Yannone, S.M.; Holden, L.G.; Hitomi, C.; Asaithamby, A.; Han, S.; Cooper, P.K.; Chen, D.J.; Tainer, J.A. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol. 2006, 13, 414. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Beresten, S.; Li, B.; Oshima, J.; Ellis, N.A.; Campisi, J. Characterization of the human and mouse WRN 3′→5′ exonuclease. Nucleic Acids Res. 2000, 28, 2396–2405. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ratcliff, G.C.; Wang, H.; Davis-Searles, P.R.; Gray, M.D.; Erie, D.A.; Redinbo, M.R. A minimal exonuclease domain of WRN forms a hexamer on DNA and possesses both 3′–5′ exonuclease and 5′-protruding strand endonuclease activities. Biochemistry 2002, 41, 2901–2912. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Kang, S.Y.; Bae, W.J.; Jin, K.S.; Ree, M.; Cho, Y. Probing the roles of active site residues in the 3’-5’ exonuclease of the Werner syndrome protein. J. Biol. Chem. 2007, 282, 9941–9951. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.J.; Asaithamby, A.; Barnebey, A.; Kiamanesch, F.; Chen, D.J.; Han, S.; Tainer, J.A.; Yannone, S.M. Identification of a coiled coil in werner syndrome protein that facilitates multimerization and promotes exonuclease processivity. J. Biol. Chem. 2010, 285, 25699–25707. [Google Scholar] [CrossRef] [PubMed]
- Kitano, K.; Yoshihara, N.; Hakoshima, T. Crystal structure of the HRDC domain of human Werner syndrome protein, WRN. J. Biol. Chem. 2007, 282, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Von Kobbe, C.; Thoma, N.H.; Czyzewski, B.K.; Pavletich, N.P.; Bohr, V.A. Werner syndrome protein contains three structure-specific DNA binding domains. J. Biol. Chem. 2003, 278, 52997–53006. [Google Scholar] [CrossRef] [PubMed]
- Compton, S.A.; Tolun, G.; Kamath-Loeb, A.S.; Loeb, L.A.; Griffith, J.D. The Werner syndrome protein binds replication fork and holliday junction DNAs as an oligomer. J. Biol. Chem. 2008, 283, 24478–24483. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.D.; Shen, J.C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997, 17, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, S.; Lee, L.; Davalos, A.; Schiestl, R.H.; Campisi, J.; Oshima, J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2003, 2, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kamath-Loeb, A.; Loeb, L.A.; Fry, M. The Werner syndrome protein is distinguished from the Bloom syndrome protein by its capacity to tightly bind diverse DNA structures. PLoS ONE 2012, 7, e30189. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Mukherjee, S.; Yang, Y.; Mori, E.; Bhattacharya, S.; Kobayashi, J.; Yannone, S.M.; Chen, D.J.; Asaithamby, A. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 2014, 9, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Okui, M.; Asaithamby, A.; Burma, S.; Chen, B.P.; Tanimoto, K.; Matsuura, S.; Komatsu, K.; Chen, D.J. WRN participates in translesion synthesis pathway through interaction with NBS1. Mech. Ageing Dev. 2010, 131, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; von Kobbe, C.; Opresko, P.L.; Arthur, L.M.; Komatsu, K.; Seidman, M.M.; Carney, J.P.; Bohr, V.A. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 2004, 279, 21169–21176. [Google Scholar] [CrossRef] [PubMed]
- Otterlei, M.; Bruheim, P.; Ahn, B.; Bussen, W.; Karmakar, P.; Baynton, K.; Bohr, V.A. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell Sci. 2006, 119, 5137–5146. [Google Scholar] [CrossRef] [PubMed]
- Trego, K.S.; Chernikova, S.B.; Davalos, A.R.; Perry, J.J.; Finger, L.D.; Ng, C.; Tsai, M.S.; Yannone, S.M.; Tainer, J.A.; Campisi, J.; et al. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle 2011, 10, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Lozada, E.; Wold, M.S.; Li, G.M.; Orren, D.K. Molecular cooperation between the Werner syndrome protein and replication protein A in relation to replication fork blockage. J. Biol. Chem. 2011, 286, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Orren, D.K. TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNA. Oncogene 2004, 23, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Popuri, V.; Huang, J.; Ramamoorthy, M.; Tadokoro, T.; Croteau, D.L.; Bohr, V.A. RECQL5 plays co-operative and complementary roles with WRN syndrome helicase. Nucleic Acids Res. 2013, 41, 881–899. [Google Scholar] [CrossRef] [PubMed]
- Yannone, S.M.; Roy, S.; Chan, D.W.; Murphy, M.B.; Huang, S.; Campisi, J.; Chen, D.J. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J. Biol. Chem. 2001, 276, 38242–38248. [Google Scholar] [PubMed]
- Karmakar, P.; Piotrowski, J.; Brosh, R.M., Jr.; Sommers, J.A.; Miller, S.P.; Cheng, W.H.; Snowden, C.M.; Ramsden, D.A.; Bohr, V.A. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J. Biol. Chem. 2002, 277, 18291–18302. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Kehrli, K.; Mao, F.; Monnat, R., Jr. Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair. 2013, 12, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Li, N.; Folch, A.; Monnat, R.J., Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 2008, 7, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, A.M.; Jackson, D.A.; Iborra, F.; Cox, L.S. Asymmetry of DNA replication fork progression in Werner’s syndrome. Aging Cell 2002, 1, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Baynton, K.; Otterlei, M.; Bjoras, M.; von Kobbe, C.; Bohr, V.A.; Seeberg, E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 2003, 278, 36476–36486. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M. Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair. 2008, 7, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Nicolai, S.; Cignolo, L.; Bignami, M.; Franchitto, A. The RAD9-RAD1-HUS1 (9.1.1) complex interacts with WRN and is crucial to regulate its response to replication fork stalling. Oncogene 2012, 31, 2809–2823. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Pirzio, L.M.; Prosperi, E.; Sapora, O.; Bignami, M.; Pichierri, P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008, 183, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Murfuni, I.; De Santis, A.; Federico, M.; Bignami, M.; Pichierri, P.; Franchitto, A. Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis 2012, 33, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Iannascoli, C.; Palermo, V.; Murfuni, I.; Franchitto, A.; Pichierri, P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015, 43, 9788–9803. [Google Scholar] [CrossRef] [PubMed]
- Kehrli, K.; Phelps, M.; Lazarchuk, P.; Chen, E.; Monnat, R., Jr.; Sidorova, J.M. Class I Histone Deacetylase HDAC1 and WRN RECQ Helicase Contribute Additively to Protect Replication Forks upon Hydroxyurea-induced Arrest. J. Biol. Chem. 2016, 291, 24487–24503. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, R.; Muftuoglu, M.; Bohr, V.A. The role of WRN in DNA repair is affected by post-translational modifications. Mech. Ageing Dev. 2007, 128, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Rosselli, F.; Franchitto, A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene 2003, 22, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto-Matsuo, R.; Ghosh, D.; Karmakar, P.; May, A.; Ramsden, D.; Bohr, V.A. Serines 440 and 467 in the Werner syndrome protein are phosphorylated by DNA-PK and affects its dynamics in response to DNA double strand breaks. Aging 2014, 6, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; von Kobbe, C.; Opresko, P.L.; Fields, K.M.; Ren, J.; Kufe, D.; Bohr, V.A. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol. Cell. Biol. 2003, 23, 6385–6395. [Google Scholar] [CrossRef] [PubMed]
- Palermo, V.; Rinalducci, S.; Sanchez, M.; Grillini, F.; Sommers, J.A.; Brosh, R.M., Jr.; Zolla, L.; Franchitto, A.; Pichierri, P. CDK1 phosphorylates WRN at collapsed replication forks. Nat. Commun. 2016, 7, 12880. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Bhattacharya, S.; Abdisalaam, S.; Mukherjee, S.; Yajima, H.; Yang, Y.; Mishra, R.; Srinivasan, K.; Ghose, S.; Chen, D.J.; et al. Replication stress induced site-specific phosphorylation targets WRN to the ubiquitin-proteasome pathway. Oncotarget 2016, 7, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Signer, R.A.; Morrison, S.J. Cellular differences in protein synthesis regulate tissue homeostasis. Cell 2014, 159, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Shamanna, R.A.; Lu, H.; Croteau, D.L.; Arora, A.; Agarwal, D.; Ball, G.; Aleskandarany, M.A.; Ellis, I.O.; Pommier, Y.; Madhusudan, S.; et al. Camptothecin targets WRN protein: Mechanism and relevance in clinical breast cancer. Oncotarget 2016, 7, 13269–13284. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, R.; Lozada, E.; Fan, W.; Orren, D.K.; Luo, J. Acetylation of WRN protein regulates its stability by inhibiting ubiquitination. PLoS ONE 2010, 5, e10341. [Google Scholar] [CrossRef] [PubMed]
- Lozada, E.; Yi, J.; Luo, J.; Orren, D.K. Acetylation of Werner syndrome protein (WRN): Relationships with DNA damage, DNA replication and DNA metabolic activities. Biogerontology 2014, 15, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.P.; Machwe, A.; Orren, D.K.; Brosh, R.M.; Ramsden, D.; Bohr, V.A. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000, 14, 907–912. [Google Scholar] [PubMed]
- Oshima, J.; Huang, S.; Pae, C.; Campisi, J.; Schiestl, R.H. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 2002, 62, 547–551. [Google Scholar] [PubMed]
- Lee-Theilen, M.; Matthews, A.J.; Kelly, D.; Zheng, S.; Chaudhuri, J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol. 2011, 18, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Zheng, H.; He, H.; Luo, Y.; Multani, A.; Carpenter, P.B.; Chang, S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010, 29, 2598–2610. [Google Scholar] [CrossRef] [PubMed]
- Badie, S.; Carlos, A.R.; Folio, C.; Okamoto, K.; Bouwman, P.; Jonkers, J.; Tarsounas, M. BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. EMBO J. 2015, 34, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Shamanna, R.A.; Lu, H.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [PubMed]
- Shamanna, R.A.; Croteau, D.L.; Lee, J.H.; Bohr, V.A. Recent Advances in Understanding Werner Syndrome. F1000Research 2017, 6, 1779. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, K.K.; Sidorova, J.; Saintigny, Y.; Poot, M.; Gollahon, K.; Rabinovitch, P.S.; Monnat, R.J., Jr. Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell 2007, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Poot, M.; Hoehn, H.; Runger, T.M.; Martin, G.M. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp. Cell Res. 1992, 202, 267–273. [Google Scholar] [CrossRef]
- Pichierri, P.; Franchitto, A.; Mosesso, P.; Palitti, F. Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol. Biol. Cell 2001, 12, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, A.M.; Whitby, M.C.; Borer, C.M.; Bachler, M.A.; Cox, L.S. Correction of proliferation and drug sensitivity defects in the progeroid Werner’s Syndrome by Holliday junction resolution. Rejuvenation Res. 2007, 10, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Salk, D.; Bryant, E.; Hoehn, H.; Johnston, P.; Martin, G.M. Growth characteristics of Werner syndrome cells in vitro. Adv. Exp. Med. Biol. 1985, 190, 305–311. [Google Scholar] [PubMed]
- Ogburn, C.E.; Oshima, J.; Poot, M.; Chen, R.; Hunt, K.E.; Gollahon, K.A.; Rabinovitch, P.S.; Martin, G.M. An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum. Genet. 1997, 101, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Poot, M.; Gollahon, K.A.; Emond, M.J.; Silber, J.R.; Rabinovitch, P.S. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: Implications for the disease phenotype. FASEB J. 2002, 16, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Poot, M.; Yom, J.S.; Whang, S.H.; Kato, J.T.; Gollahon, K.A.; Rabinovitch, P.S. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J. 2001, 15, 1224–1226. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Groden, J.; Orren, D.K. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 2006, 45, 13939–13946. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Xiao, L.; Groden, J.; Matson, S.W.; Orren, D.K. RecQ family members combine strand pairing and unwinding activities to catalyze strand exchange. J. Biol. Chem. 2005, 280, 23397–23407. [Google Scholar] [CrossRef] [PubMed]
- Orren, D.K.; Theodore, S.; Machwe, A. The Werner syndrome helicase/exonuclease (WRN) disrupts and degrades D-loops in vitro. Biochemistry 2002, 41, 13483–13488. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar] [CrossRef] [PubMed]
- Machwe, A.; Karale, R.; Xu, X.; Liu, Y.; Orren, D.K. The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) holliday junctions to functional replication forks. Biochemistry 2011, 50, 6774–6788. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.N.; Machwe, A.; Chen, L.; Bohr, V.A.; Orren, D.K. The DNA structure and sequence preferences of WRN underlie its function in telomeric recombination events. Nat. Commun. 2015, 6, 8331. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.; Tarsounas, M.; Karow, J.K.; Brosh, R.M.; Bohr, V.A.; Hickson, I.D.; West, S.C. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000, 1, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Kusumoto, R.; Opresko, P.L.; Sui, X.; Huang, S.; Nicolette, M.L.; Paull, T.T.; Campisi, J.; Seidman, M.; Bohr, V.A. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006, 34, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Nishikawa, K.; Heo, S.J.; Goto, M.; Furuichi, Y.; Shimamoto, A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells 2001, 6, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Agrelo, R. A new molecular model of cellular aging based on Werner syndrome. Med. Hypotheses 2007, 68, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Anitei, M.G.; Zeitoun, G.; Mlecnik, B.; Marliot, F.; Haicheur, N.; Todosi, A.M.; Kirilovsky, A.; Lagorce, C.; Bindea, G.; Ferariu, D.; et al. Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin. Cancer Res. 2014, 20, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J., Jr. Spectrum and risk of neoplasia in Werner syndrome: A systematic review. PLoS ONE 2013, 8, e59709. [Google Scholar] [CrossRef] [PubMed]
- Yamaga, M.; Takemoto, M.; Takada-Watanabe, A.; Koizumi, N.; Kitamoto, T.; Sakamoto, K.; Ishikawa, T.; Koshizaka, M.; Maezawa, Y.; Yokote, K. Recent Trends in WRN Gene Mutation Patterns in Individuals with Werner Syndrome. J. Am. Geriatr. Soc. 2017, 65, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Yokote, K.; Chanprasert, S.; Lee, L.; Eirich, K.; Takemoto, M.; Watanabe, A.; Koizumi, N.; Lessel, D.; Mori, T.; Hisama, F.M.; et al. WRN Mutation Update: Mutation Spectrum, Patient Registries, and Translational Prospects. Hum. Mutat. 2017, 38, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, H.; Bryant, E.M.; Au, K.; Norwood, T.H.; Boman, H.; Martin, G.M. Variegated translocation mosaicism in human skin fibroblast cultures. Cytogenet. Genome Res. 1975, 15, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Monnat, R.J., Jr. Werner syndrome (WRN) gene variants and their association with altered function and age-associated diseases. Ageing Res. Rev. 2018, 41, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A Cell-Intrinsic Interferon-like Response Links Replication Stress to Cellular Aging Caused by Progerin. Cell Rep. 2018, 22, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.T.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Turaga, R.V.; Paquet, E.R.; Sild, M.; Vignard, J.; Garand, C.; Johnson, F.B.; Masson, J.Y.; Lebel, M. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle 2009, 8, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Massip, L.; Garand, C.; Paquet, E.R.; Cogger, V.C.; O’Reilly, J.N.; Tworek, L.; Hatherell, A.; Taylor, C.G.; Thorin, E.; Zahradka, P.; et al. Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J. 2010, 24, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Sugimoto, K.; Hayashi, S.; Ogino, T.; Sugimoto, M.; Furuichi, Y.; Matsuura, M.; Ishikawa, Y.; Iwaki-Egawa, S.; Watanabe, Y. Aging-associated inflammation in healthy Japanese individuals and patients with Werner syndrome. Exp. Gerontol. 2012, 47, 936–939. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Sinha, D.; Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Asaithamby, A. Werner Syndrome Protein and DNA Replication. Int. J. Mol. Sci. 2018, 19, 3442. https://doi.org/10.3390/ijms19113442
Mukherjee S, Sinha D, Bhattacharya S, Srinivasan K, Abdisalaam S, Asaithamby A. Werner Syndrome Protein and DNA Replication. International Journal of Molecular Sciences. 2018; 19(11):3442. https://doi.org/10.3390/ijms19113442
Chicago/Turabian StyleMukherjee, Shibani, Debapriya Sinha, Souparno Bhattacharya, Kalayarasan Srinivasan, Salim Abdisalaam, and Aroumougame Asaithamby. 2018. "Werner Syndrome Protein and DNA Replication" International Journal of Molecular Sciences 19, no. 11: 3442. https://doi.org/10.3390/ijms19113442
APA StyleMukherjee, S., Sinha, D., Bhattacharya, S., Srinivasan, K., Abdisalaam, S., & Asaithamby, A. (2018). Werner Syndrome Protein and DNA Replication. International Journal of Molecular Sciences, 19(11), 3442. https://doi.org/10.3390/ijms19113442