Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism


Abstract

1. Introduction
2. Roles for AMPK and Glycogen in Metabolism
2.1. AMPK Activation and Signaling
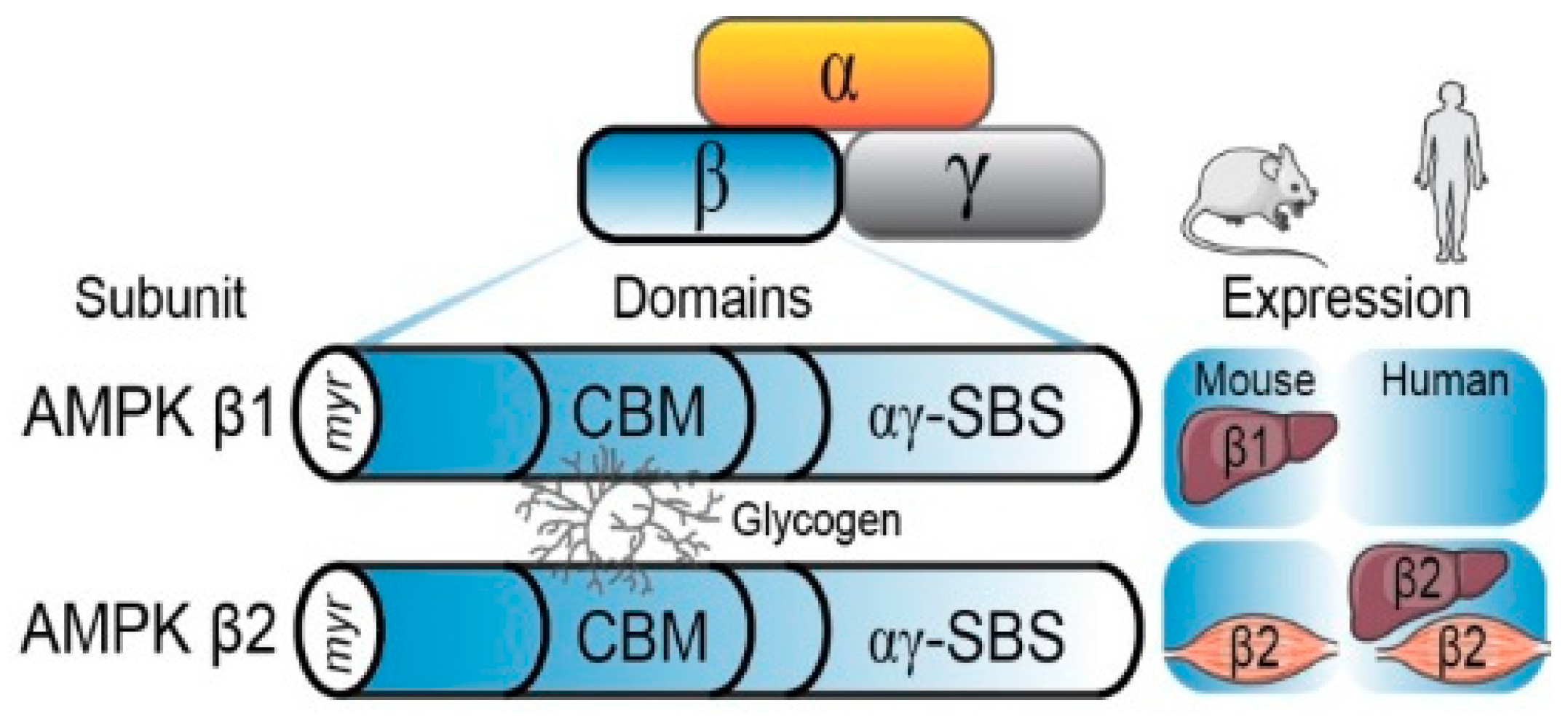
2.2. The AMPK β Subunit and Carbohydrate-Binding Module
2.3. Glycogen Dynamics
2.4. Glycogen Localization
3. Molecular Evidence of AMPK-Glycogen Binding
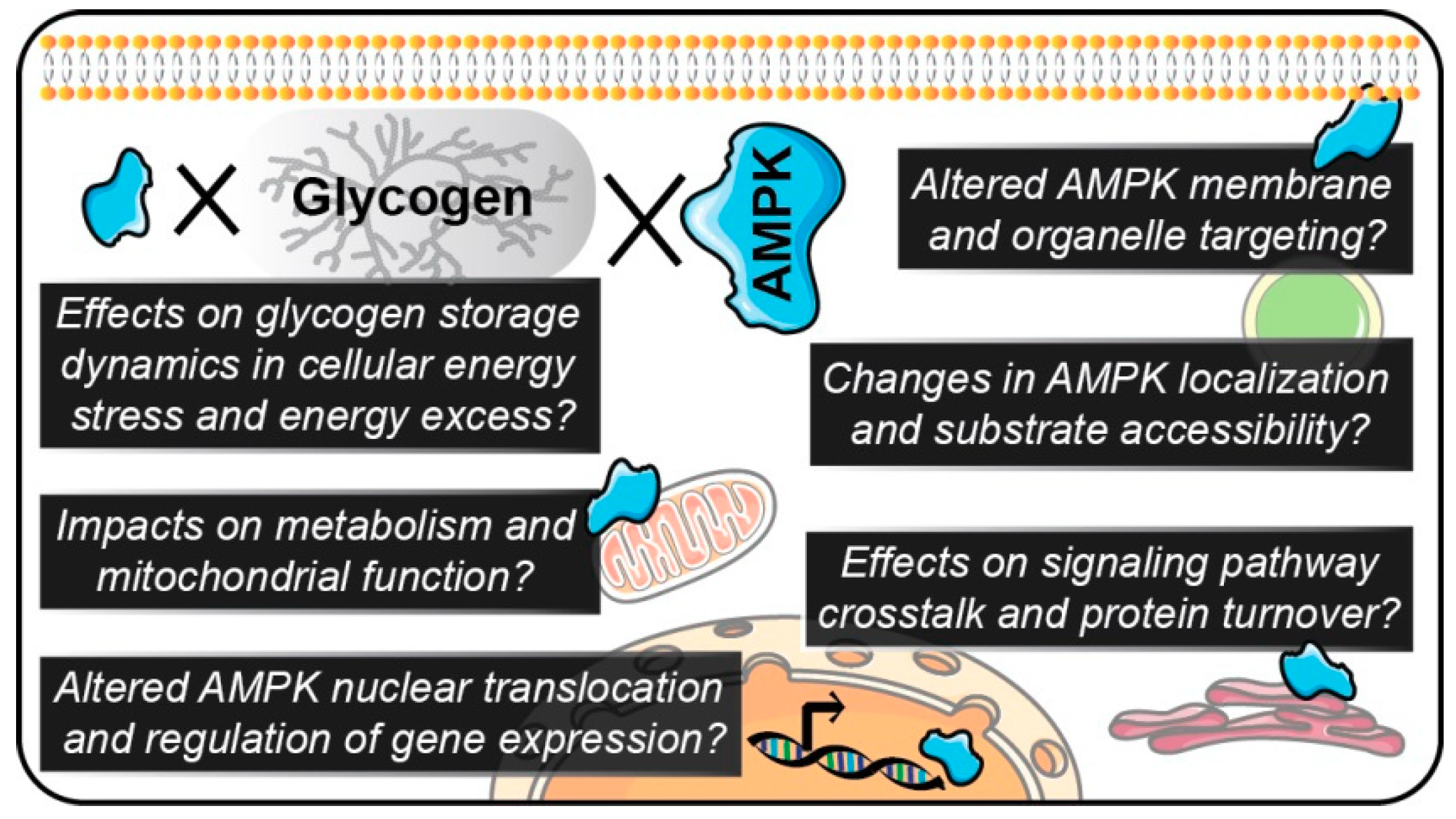
4. Regulation of Cellular Energy Sensing by AMPK-Glycogen Binding
5. Linking AMPK and Glycogen to Exercise Metabolism in Physiological Settings
5.1. Regulation of Glycogen Storage by AMPK
5.2. Roles for Glycogen Availability in the Regulation of AMPK Activity
5.3. Metabolic and Glycogen Storage Diseases as Models to Investigate AMPK-Glycogen Binding
6. Multidisciplinary Techniques and Models to Interrogate Roles for AMPK-Glycogen Interactions
7. Potential Therapeutic Relevance of Targeting AMPK-Glycogen Binding
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ADP | Adenosine diphosphate |
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| CaMKKβ | Calcium/calmodulin-dependent protein kinase β |
| cAMP | Cyclic AMP |
| CBM | Carbohydrate-binding module |
| CBS | Cystathionine-β-synthase domains |
| EDL | Extensor digitorum longus |
| FA | Fatty acid |
| FRET | Fluorescence resonance energy transfer |
| G6P | Glucose-6-phosphate |
| GBE | Glycogen branching enzyme |
| GDE | Glycogen debranching enzyme |
| GLUT4 | Glucose transporter 4 |
| GP | Glycogen phosphorylase |
| GS | Glycogen synthase |
| interMF | Intermyofibrillar |
| intraMF | Intramyofibrillar |
| KO | Knock-out |
| LKB1 | Liver kinase B1 |
| mTOR | Mechanistic target of rapamycin |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-α |
| Raptor | Regulatory associated protein of mechanistic target of rapamycin |
| SS | Subsarcolemmal |
| TBCID1 | Tre-2, BUB2, CDC16, 1 domain family, member 1 |
| TBC1D4 | Tre-2, BUB2, CDC16, 1 domain family, member 4 |
| TSC2 | Tuberous sclerosis complex 2 |
| T2D | Type 2 diabetes |
| UGP2 | UDP-glucose pyrophosphorylase 2 |
| WT | Wild type |
References
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Chen, Z.P.; Scott, J.W.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Guo, H.; Zhang, C.S.; Lin, S.Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Haro, L.; Garcia-Gimeno, M.A.; Neumann, D.; Beullens, M.; Bollen, M.; Sanz, P. The PP1-R6 protein phosphatase holoenzyme is involved in the glucose-induced dephosphorylation and inactivation of AMP-activated protein kinase, a key regulator of insulin secretion, in MIN6 β cells. FASEB J. 2010, 24, 5080–5091. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.K.; Liu, H.Y.; Francisco, J.; Pandya, D.; Donigan, M.; Gallo-Ebert, C.; Giordano, C.; Bata, A.; Nickels, J.T., Jr. Inhibition of AMP kinase by the protein phosphatase 2A heterotrimer, PP2APpp2r2d. J. Biol. Chem. 2015, 290, 10588–10598. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Ling, N.; Issa, S.M.; Dite, T.A.; O’Brien, M.T.; Chen, Z.P.; Galic, S.; Langendorf, C.G.; Steinberg, G.R.; Kemp, B.E.; et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem. Biol. 2014, 21, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Kjobsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.N.; Pehmoller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.E.; Rose, A.J.; Jorgensen, S.B.; Brandt, N.; Schjerling, P.; Wojtaszewski, J.F.; Richter, E.A. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1308–E1317. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kjobsted, R.; Munk-Hansen, N.; Birk, J.B.; Foretz, M.; Viollet, B.; Bjornholm, M.; Zierath, J.R.; Treebak, J.T.; Wojtaszewski, J.F. Enhanced muscle insulin sensitivity after contraction/exercise is mediated by AMPK. Diabetes 2017, 66, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Pehmoller, C.; Treebak, J.T.; Birk, J.B.; Chen, S.; Mackintosh, C.; Hardie, D.G.; Richter, E.A.; Wojtaszewski, J.F. Genetic disruption of AMPK signaling abolishes both contraction-and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E665–E675. [Google Scholar] [CrossRef] [PubMed]
- Vichaiwong, K.; Purohit, S.; An, D.; Toyoda, T.; Jessen, N.; Hirshman, M.F.; Goodyear, L.J. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem. J. 2010, 431, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.; Paglialunga, S.; Smith, B.K.; Miotto, P.M.; Simnett, G.; Robson, H.L.; Jain, S.S.; Herbst, E.A.F.; Desjardins, E.M.; Dyck, D.J.; et al. Ablating the protein TBC1D1 impairs contraction-induced sarcolemmal glucose transporter 4 redistribution but not insulin-mediated responses in rats. J. Biol. Chem. 2017, 292, 16653–16664. [Google Scholar] [CrossRef] [PubMed]
- Stockli, J.; Meoli, C.C.; Hoffman, N.J.; Fazakerley, D.J.; Pant, H.; Cleasby, M.E.; Ma, X.; Kleinert, M.; Brandon, A.E.; Lopez, J.A.; et al. The RabGAP TBC1D1 plays a central role in exercise-regulated glucose metabolism in skeletal muscle. Diabetes 2015, 64, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Hargreaves, M.; Joyner, M.J.; Zierath, J.R. Integrative biology of exercise. Cell 2014, 159, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. Tsc2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Yan, Y.; Novick, S.J.; Kovach, A.; Goswami, D.; Ke, J.; Tan, M.H.E.; Wang, L.; Li, X.; de Waal, P.W.; et al. Deconvoluting AMP-activated protein kinase (AMPK) adenine nucleotide binding and sensing. J. Biol. Chem. 2017, 292, 12653–12666. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [PubMed]
- Olivier, S.; Foretz, M.; Viollet, B. Promise and challenges for direct small molecule AMPK activators. Biochem. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stephenne, X.; Foretz, M.; Taleux, N.; van der Zon, G.C.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Puppala, D.; Feng, X.; Monetti, M.; Lapworth, A.L.; Geoghegan, K.F. Chemoproteomic analysis of intertissue and interspecies isoform diversity of AMP-activated protein kinase (AMPK). J. Biol. Chem. 2013, 288, 35904–35912. [Google Scholar] [CrossRef] [PubMed]
- Polekhina, G.; Gupta, A.; Michell, B.J.; van Denderen, B.; Murthy, S.; Feil, S.C.; Jennings, I.G.; Campbell, D.J.; Witters, L.A.; Parker, M.W.; et al. AMPK β subunit targets metabolic stress sensing to glycogen. Curr. Biol. 2003, 13, 867–871. [Google Scholar] [CrossRef]
- Polekhina, G.; Gupta, A.; van Denderen, B.J.; Feil, S.C.; Kemp, B.E.; Stapleton, D.; Parker, M.W. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 2005, 13, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Koay, A.; Woodcroft, B.; Petrie, E.J.; Yue, H.; Emanuelle, S.; Bieri, M.; Bailey, M.F.; Hargreaves, M.; Park, J.T.; Park, K.H.; et al. AMPK β subunits display isoform specific affinities for carbohydrates. FEBS Lett. 2010, 584, 3499–3503. [Google Scholar] [PubMed]
- Mobbs, J.I.; Di Paolo, A.; Metcalfe, R.D.; Selig, E.; Stapleton, D.I.; Griffin, M.D.W.; Gooley, P.R. Unravelling the carbohydrate-binding preferences of the carbohydrate-binding modules of AMP-activated protein kinase. Chembiochem 2017. [Google Scholar] [CrossRef] [PubMed]
- Mobbs, J.I.; Koay, A.; Di Paolo, A.; Bieri, M.; Petrie, E.J.; Gorman, M.A.; Doughty, L.; Parker, M.W.; Stapleton, D.I.; Griffin, M.D.; et al. Determinants of oligosaccharide specificity of the carbohydrate-binding modules of AMP-activated protein kinase. Biochem. J. 2015, 468, 245–257. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M. AMPK and exercise: Glucose uptake and insulin sensitivity. Diabetes Metab. J. 2013, 37, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.D.; Lomako, J.; Lomako, W.M.; Whelan, W.J. A new look at the biogenesis of glycogen. FASEB J. 1995, 9, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Treebak, J.T.; Wojtaszewski, J.F.; Sakamoto, K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 2011, 60, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [PubMed]
- Chasiotis, D.; Sahlin, K.; Hultman, E. Regulation of glycogenolysis in human muscle at rest and during exercise. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1982, 53, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Ruderman, N.B.; Gavras, H.; Belur, E.R.; Galbo, H. Muscle glycogenolysis during exercise: Dual control by epinephrine and contractions. Am. J. Physiol. 1982, 242, E25–E32. [Google Scholar] [CrossRef] [PubMed]
- Shearer, J.; Graham, T.E. Novel aspects of skeletal muscle glycogen and its regulation during rest and exercise. Exerc. Sport Sci. Rev. 2004, 32, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Prats, C.; Graham, T.E.; Shearer, J. The dynamic life of the glycogen granule. J. Biol. Chem. 2018, 293, 7089–7098. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.E.; Yuan, Z.; Hill, A.K.; Wilson, R.J. The regulation of muscle glycogen: The granule and its proteins. Acta Physiol. 2010, 199, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Ortenblad, N. Physiological aspects of the subcellular localization of glycogen in skeletal muscle. Appl. Physiol. Nutr. Metab. 2013, 38, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ortenblad, N.; Nielsen, J.; Saltin, B.; Holmberg, H.C. Role of glycogen availability in sarcoplasmic reticulum Ca2+ kinetics in human skeletal muscle. J. Physiol. 2011, 589, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Ortenblad, N.; Westerblad, H.; Nielsen, J. Muscle glycogen stores and fatigue. J. Physiol. 2013, 591, 4405–4413. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.; Hargreaves, M.; Baar, K. More than a store: Regulatory roles for glycogen in skeletal muscle adaptation to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1343–E1351. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Holmberg, H.C.; Schroder, H.D.; Saltin, B.; Ortenblad, N. Human skeletal muscle glycogen utilization in exhaustive exercise: Role of subcellular localization and fibre type. J. Physiol. 2011, 589, 2871–2885. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.K.; Paton, C.D.; Garnham, A.P.; Burke, L.M.; Carey, A.L.; Hawley, J.A. Skeletal muscle adaptation and performance responses to once a day versus twice every second day endurance training regimens. J. Appl. Physiol. 2008, 105, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Cheng, A.J.; Ortenblad, N.; Westerblad, H. Subcellular distribution of glycogen and decreased tetanic Ca2+ in fatigued single intact mouse muscle fibres. J. Physiol. 2014, 592, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Romijn, J.A.; Coyle, E.F.; Sidossis, L.S.; Gastaldelli, A.; Horowitz, J.F.; Endert, E.; Wolfe, R.R. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am. J. Physiol. 1993, 265, E380–E391. [Google Scholar] [CrossRef] [PubMed]
- van Loon, L.J.; Greenhaff, P.L.; Constantin-Teodosiu, D.; Saris, W.H.; Wagenmakers, A.J. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J. Physiol. 2001, 536, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hughey, C.C.; James, F.D.; Bracy, D.P.; Donahue, E.P.; Young, J.D.; Viollet, B.; Foretz, M.; Wasserman, D.H. Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jorgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.; Ghilagaber, S.; Nikolaev, A.; Hardie, D.G. The glycogen-binding domain on the AMPK β subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009, 9, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hudson, E.R.; Pan, D.A.; James, J.; Lucocq, J.M.; Hawley, S.A.; Green, K.A.; Baba, O.; Terashima, T.; Hardie, D.G. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr. Biol. 2003, 13, 861–866. [Google Scholar] [CrossRef]
- Bendayan, M.; Londono, I.; Kemp, B.E.; Hardie, G.D.; Ruderman, N.; Prentki, M. Association of AMP-activated protein kinase subunits with glycogen particles as revealed in situ by immunoelectron microscopy. J. Histochem. Cytochem. 2009, 57, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; van Denderen, B.J.; Jorgensen, S.B.; Honeyman, J.E.; Steinberg, G.R.; Oakhill, J.S.; Iseli, T.J.; Koay, A.; Gooley, P.R.; Stapleton, D.; et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase β1-containing complexes. Chem. Biol. 2008, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Oligschlaeger, Y.; Miglianico, M.; Chanda, D.; Scholz, R.; Thali, R.F.; Tuerk, R.; Stapleton, D.I.; Gooley, P.R.; Neumann, D. The recruitment of AMP-activated protein kinase to glycogen is regulated by autophosphorylation. J. Biol. Chem. 2015, 290, 11715–11728. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Frankenberg, N.T.; Lamb, G.D.; Gooley, P.R.; Stapleton, D.I.; Murphy, R.M. When phosphorylated at Thr148, the β2-subunit of AMP-activated kinase does not associate with glycogen in skeletal muscle. Am. J. Physiol. Cell Physiol. 2016, 311, C35–C42. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Hardie, D.G. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim. Biophys. Acta 1989, 1012, 81–86. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Nielsen, J.N.; Birk, J.B.; Olsen, G.S.; Viollet, B.; Andreelli, F.; Schjerling, P.; Vaulont, S.; Hardie, D.G.; Hansen, B.F.; et al. The α2-5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 2004, 53, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjobsted, R.; Jorgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Fritzen, A.M.; Lundsgaard, A.M.; Jeppesen, J.; Christiansen, M.L.; Bienso, R.; Dyck, J.R.; Pilegaard, H.; Kiens, B. 5′-AMP activated protein kinase α2 controls substrate metabolism during post-exercise recovery via regulation of pyruvate dehydrogenase kinase 4. J. Physiol. 2015, 593, 4765–4780. [Google Scholar] [CrossRef] [PubMed]
- Prats, C.; Helge, J.W.; Nordby, P.; Qvortrup, K.; Ploug, T.; Dela, F.; Wojtaszewski, J.F. Dual regulation of muscle glycogen synthase during exercise by activation and compartmentalization. J. Biol. Chem. 2009, 284, 15692–15700. [Google Scholar] [CrossRef] [PubMed]
- Barre, L.; Richardson, C.; Hirshman, M.F.; Brozinick, J.; Fiering, S.; Kemp, B.E.; Goodyear, L.J.; Witters, L.A. Genetic model for the chronic activation of skeletal muscle AMP-activated protein kinase leads to glycogen accumulation. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E802–E811. [Google Scholar] [CrossRef] [PubMed]
- Prats, C.; Gomez-Cabello, A.; Hansen, A.V. Intracellular compartmentalization of skeletal muscle glycogen metabolism and insulin signalling. Exp. Physiol. 2011, 96, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Leighton, B.; Radda, G.K. Glycogen phosphorylase may be activated by AMP-kinase in skeletal muscle. Biochem. Soc. Trans. 1996, 24, 268S. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Radda, G.K.; Leighton, B. Activation of glycogen phosphorylase and glycogenolysis in rat skeletal muscle by AICAR—An activator of AMP-activated protein kinase. FEBS Lett. 1996, 382, 43–47. [Google Scholar] [CrossRef]
- Daignan-Fornier, B.; Pinson, B. 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranosyl 5′-monophosphate (AICAR), a highly conserved purine intermediate with multiple effects. Metabolites 2012, 2, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, H.; Fujishiro, M.; Fujio, J.; Shojima, N.; Ogihara, T.; Kushiyama, A.; Fukushima, Y.; Anai, M.; Ono, H.; Kikuchi, M.; et al. Glycogen debranching enzyme association with β-Subunit regulates AMP-activated protein kinase activity. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E474–E481. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, D.; Nelson, C.; Parsawar, K.; McClain, D.; Gilbert-Wilson, R.; Barker, E.; Rudd, B.; Brown, K.; Hendrix, W.; O’Donnell, P.; et al. Analysis of hepatic glycogen-associated proteins. Proteomics 2010, 10, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, D.; Nelson, C.; Parsawar, K.; Flores-Opazo, M.; McClain, D.; Parker, G. The 3T3-L1 adipocyte glycogen proteome. Proteome Sci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; O’Neill, H.M.; Dzamko, N.L.; Galic, S.; Naim, T.; Koopman, R.; Jorgensen, S.B.; Honeyman, J.; Hewitt, K.; Chen, Z.P.; et al. Whole body deletion of AMP-activated protein kinase β2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem. 2010, 285, 37198–37209. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Ju, J.S.; Sasaki, Y.; Liu, X.; Jung, S.R.; Higashida, K.; Lindquist, D.; Milbrandt, J. The AMPK β2 subunit is required for energy homeostasis during metabolic stress. Mol. Cell. Biol. 2012, 32, 2837–2848. [Google Scholar] [CrossRef] [PubMed]
- Hasenour, C.M.; Ridley, D.E.; James, F.D.; Hughey, C.C.; Donahue, E.P.; Viollet, B.; Foretz, M.; Young, J.D.; Wasserman, D.H. Liver AMP-activated protein kinase is unnecessary for gluconeogenesis but protects energy state during nutrient deprivation. PLoS ONE 2017, 12, e0170382. [Google Scholar]
- Hingst, J.R.; Bruhn, L.; Hansen, M.B.; Rosschou, M.F.; Birk, J.B.; Fentz, J.; Foretz, M.; Viollet, B.; Sakamoto, K.; Faergeman, N.J.; et al. Exercise-induced molecular mechanisms promoting glycogen supercompensation in human skeletal muscle. Mol. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszewski, J.F.; Jorgensen, S.B.; Hellsten, Y.; Hardie, D.G.; Richter, E.A. Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes 2002, 51, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszewski, J.F.; MacDonald, C.; Nielsen, J.N.; Hellsten, Y.; Hardie, D.G.; Kemp, B.E.; Kiens, B.; Richter, E.A. Regulation of 5′AMP-activated protein kinase activity and substrate utilization in exercising human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E813–E822. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Watt, M.J.; McGee, S.L.; Chan, S.; Hargreaves, M.; Febbraio, M.A.; Stapleton, D.; Kemp, B.E. Reduced glycogen availability is associated with increased AMPKα2 activity, nuclear AMPKα2 protein abundance, and GLUT4 mRNA expression in contracting human skeletal muscle. Appl. Physiol. Nutr. Metab. 2006, 31, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Steinberg, G.R.; Chan, S.; Garnham, A.; Kemp, B.E.; Febbraio, M.A. β-Adrenergic stimulation of skeletal muscle HSL can be overridden by AMPK signaling. FASEB J. 2004, 18, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.K.; Lessard, S.J.; Chen, Z.P.; Garnham, A.P.; Burke, L.M.; Rivas, D.A.; Kemp, B.E.; Hawley, J.A. Fat adaptation followed by carbohydrate restoration increases AMPK activity in skeletal muscle from trained humans. J. Appl. Physiol. 2008, 105, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.K.; McGee, S.L.; Carey, A.L.; Paton, C.D.; Garnham, A.P.; Hargreaves, M.; Hawley, J.A. Acute signalling responses to intense endurance training commenced with low or normal muscle glycogen. Exp. Physiol. 2010, 95, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.C.; Camera, D.M.; Lassiter, D.G.; Areta, J.L.; Bird, S.R.; Yeo, W.K.; Jeacocke, N.A.; Krook, A.; Zierath, J.R.; Burke, L.M.; et al. Effects of sleeping with reduced carbohydrate availability on acute training responses. J. Appl. Physiol. 2015, 119, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.; MacKenzie, M.G.; Belew, M.Y.; Towler, M.C.; Corstorphine, A.; Papalamprou, A.; Hardie, D.G.; Baar, K. Glycogen content regulates peroxisome proliferator activated receptor-∂ (PPAR-∂) activity in rat skeletal muscle. PLoS ONE 2013, 8, e77200. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.D.; Louhelainen, J.; Iqbal, Z.; Cochran, A.J.; Gibala, M.J.; Gregson, W.; Close, G.L.; Drust, B.; Morton, J.P. Reduced carbohydrate availability enhances exercise-induced p53 signaling in human skeletal muscle: Implications for mitochondrial biogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R450–R458. [Google Scholar] [CrossRef] [PubMed]
- Psilander, N.; Frank, P.; Flockhart, M.; Sahlin, K. Exercise with low glycogen increases PGC-1α gene expression in human skeletal muscle. Eur. J. Appl. Physiol. 2013, 113, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Sriwijitkamol, A.; Coletta, D.K.; Wajcberg, E.; Balbontin, G.B.; Reyna, S.M.; Barrientes, J.; Eagan, P.A.; Jenkinson, C.P.; Cersosimo, E.; DeFronzo, R.A.; et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: A time-course and dose-response study. Diabetes 2007, 56, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Witters, L.A.; Kemp, B.E. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5′-AMP-activated protein kinase. J. Biol. Chem. 1992, 267, 2864–2867. [Google Scholar] [PubMed]
- Musi, N.; Fujii, N.; Hirshman, M.F.; Ekberg, I.; Froberg, S.; Ljungqvist, O.; Thorell, A.; Goodyear, L.J. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 2001, 50, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Hojlund, K.; Staehr, P.; Hansen, B.F.; Green, K.A.; Hardie, D.G.; Richter, E.A.; Beck-Nielsen, H.; Wojtaszewski, J.F. Increased phosphorylation of skeletal muscle glycogen synthase at NH2-terminal sites during physiological hyperinsulinemia in type 2 diabetes. Diabetes 2003, 52, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.N.; Wojtaszewski, J.F.; Haller, R.G.; Hardie, D.G.; Kemp, B.E.; Richter, E.A.; Vissing, J. Role of 5′AMP-activated protein kinase in glycogen synthase activity and glucose utilization: Insights from patients with mcardle’s disease. J. Physiol. 2002, 541, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Pinos, T.; Nielsen, T.L.; Duran, J.; Garcia-Rocha, M.; Andreu, A.L.; Vissing, J. Differential glucose metabolism in mice and humans affected by mcardle disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R307–R314. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.L.; Pinos, T.; Brull, A.; Vissing, J.; Krag, T.O. Exercising with blocked muscle glycogenolysis: Adaptation in the mcardle mouse. Mol. Genet. Metab. 2018, 123, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kollberg, G.; Tulinius, M.; Gilljam, T.; Ostman-Smith, I.; Forsander, G.; Jotorp, P.; Oldfors, A.; Holme, E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N. Engl. J. Med. 2007, 357, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Pederson, B.A.; Schroeder, J.M.; Parker, G.E.; Smith, M.W.; DePaoli-Roach, A.A.; Roach, P.J. Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes 2005, 54, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Pederson, B.A.; Chen, H.; Schroeder, J.M.; Shou, W.; DePaoli-Roach, A.A.; Roach, P.J. Abnormal cardiac development in the absence of heart glycogen. Mol. Cell. Biol. 2004, 24, 7179–7187. [Google Scholar] [CrossRef] [PubMed]
- Xirouchaki, C.E.; Mangiafico, S.P.; Bate, K.; Ruan, Z.; Huang, A.M.; Tedjosiswoyo, B.W.; Lamont, B.; Pong, W.; Favaloro, J.; Blair, A.R.; et al. Impaired glucose metabolism and exercise capacity with muscle-specific glycogen synthase 1 (gys1) deletion in adult mice. Mol. Metab. 2016, 5, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, B.E.; Levin, R.S.; Hertz, N.T.; Maures, T.J.; Schoof, M.L.; Hollstein, P.E.; Benayoun, B.A.; Banko, M.R.; Shaw, R.J.; Shokat, K.M.; et al. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 2015, 22, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Nunez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J. Omics and exercise: Global approaches for mapping exercise biological networks. Cold Spring Harb. Perspect. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Zheng, B.; Hsu, C.H.; Sasaki, A.T.; Cantley, L.C. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011, 13, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Chennell, G.; Willows, R.J.; Warren, S.C.; Carling, D.; French, P.M.; Dunsby, C.; Sardini, A. Imaging of metabolic status in 3D cultures with an improved AMPK fret biosensor for flim. Sensors 2016, 16, 1312. [Google Scholar] [CrossRef] [PubMed]
- Konagaya, Y.; Terai, K.; Hirao, Y.; Takakura, K.; Imajo, M.; Kamioka, Y.; Sasaoka, N.; Kakizuka, A.; Sumiyama, K.; Asano, T.; et al. A highly sensitive fret biosensor for AMPK exhibits heterogeneous AMPK responses among cells and organs. Cell Rep. 2017, 21, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Depry, C.; Mehta, S.; Li, R.; Zhang, J. Visualization of compartmentalized kinase activity dynamics using adaptable BimKARs. Chem. Biol. 2015, 22, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Gu, X.; Xu, H.E.; Melcher, K. A highly sensitive non-radioactive activity assay for AMP-activated protein kinase (AMPK). Methods Protoc. 2018, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Benson, D.W.; Perez-Atayde, A.R.; McKenna, W.J.; Sparks, E.A.; Kanter, R.J.; McGarry, K.; Seidman, J.G.; Seidman, C.E. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J. Clin. Investig. 2002, 109, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Milan, D.; Jeon, J.T.; Looft, C.; Amarger, V.; Robic, A.; Thelander, M.; Rogel-Gaillard, C.; Paul, S.; Iannuccelli, N.; Rask, L.; et al. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 2000, 288, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Roves, P.M.; Osler, M.E.; Holmstrom, M.H.; Zierath, J.R. Gain-of-function R225Q mutation in AMP-activated protein kinase γ3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J. Biol. Chem. 2008, 283, 35724–35734. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Galic, S.; Graham, K.L.; Foitzik, R.; Ling, N.X.; Dite, T.A.; Issa, S.M.; Langendorf, C.G.; Weng, Q.P.; Thomas, H.E.; et al. Inhibition of AMP-activated protein kinase at the allosteric drug-binding site promotes islet insulin release. Chem. Biol. 2015, 22, 705–711. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janzen, N.R.; Whitfield, J.; Hoffman, N.J. Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism. Int. J. Mol. Sci. 2018, 19, 3344. https://doi.org/10.3390/ijms19113344
Janzen NR, Whitfield J, Hoffman NJ. Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism. International Journal of Molecular Sciences. 2018; 19(11):3344. https://doi.org/10.3390/ijms19113344
Chicago/Turabian StyleJanzen, Natalie R., Jamie Whitfield, and Nolan J. Hoffman. 2018. "Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism" International Journal of Molecular Sciences 19, no. 11: 3344. https://doi.org/10.3390/ijms19113344
APA StyleJanzen, N. R., Whitfield, J., & Hoffman, N. J. (2018). Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism. International Journal of Molecular Sciences, 19(11), 3344. https://doi.org/10.3390/ijms19113344