Evolution and Virulence of Influenza A Virus Protein PB1-F2

Abstract
1. Introduction
2. Structure
3. Expression
4. Molecular Mechanisms of Pathogenesis
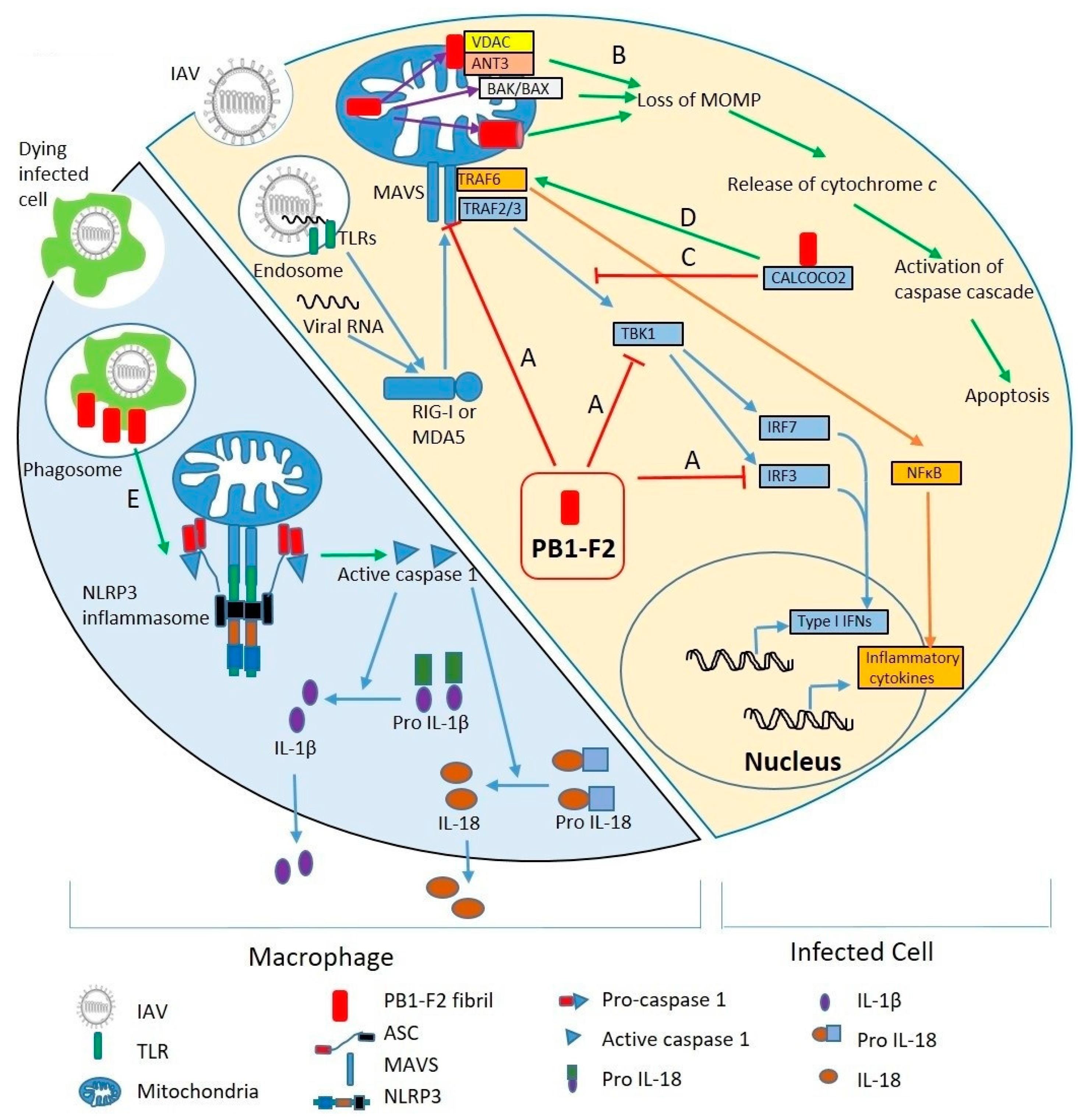
4.1. Regulation of Antiviral Innate Immunity
4.2. Enhancement of Viral Polymerase Activity
4.3. Induction of Cell Death
5. Pathogenesis in Animals
5.1. PB1-F2 Deletion
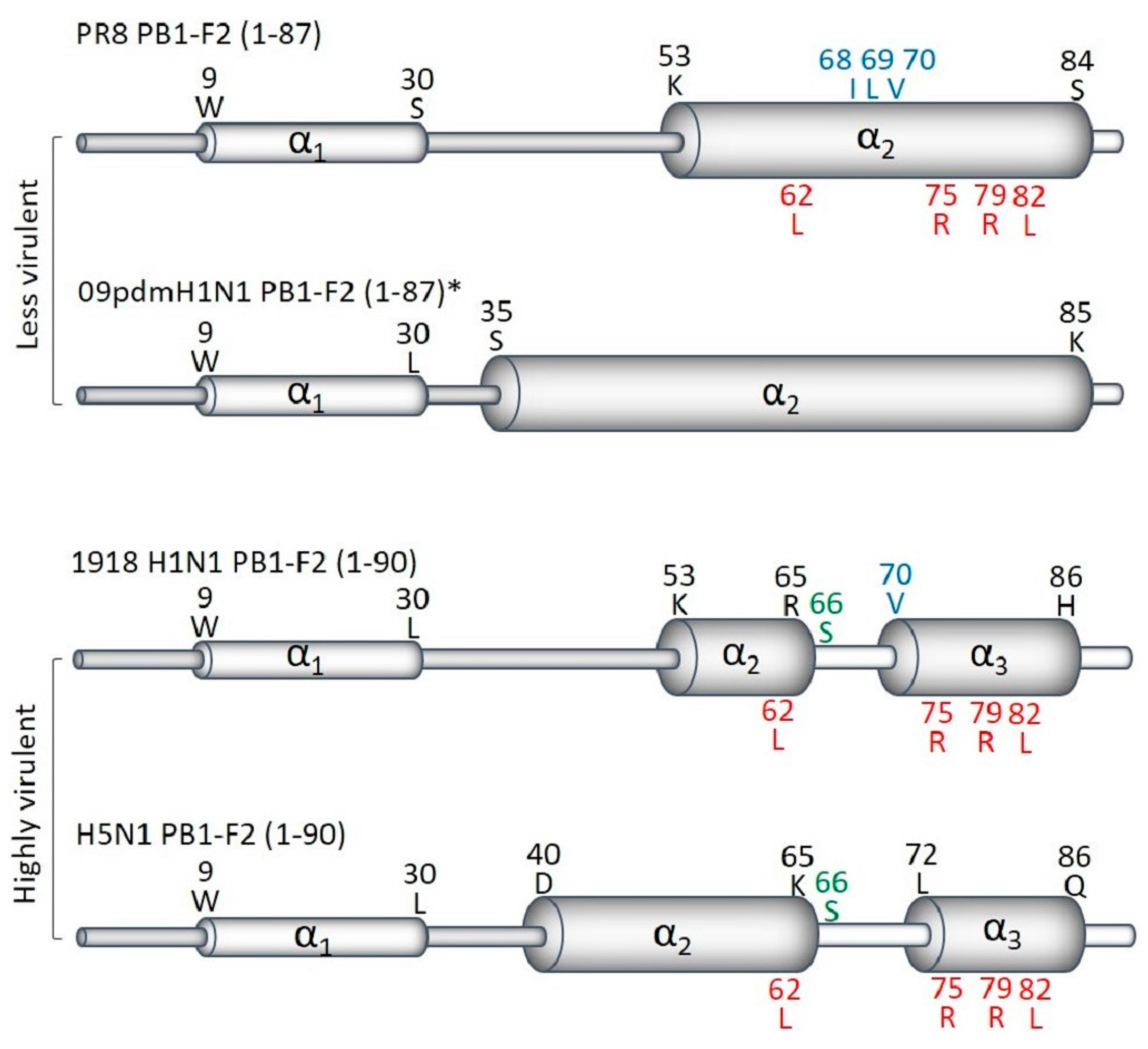
5.2. PB1-F2 Sequence Variants
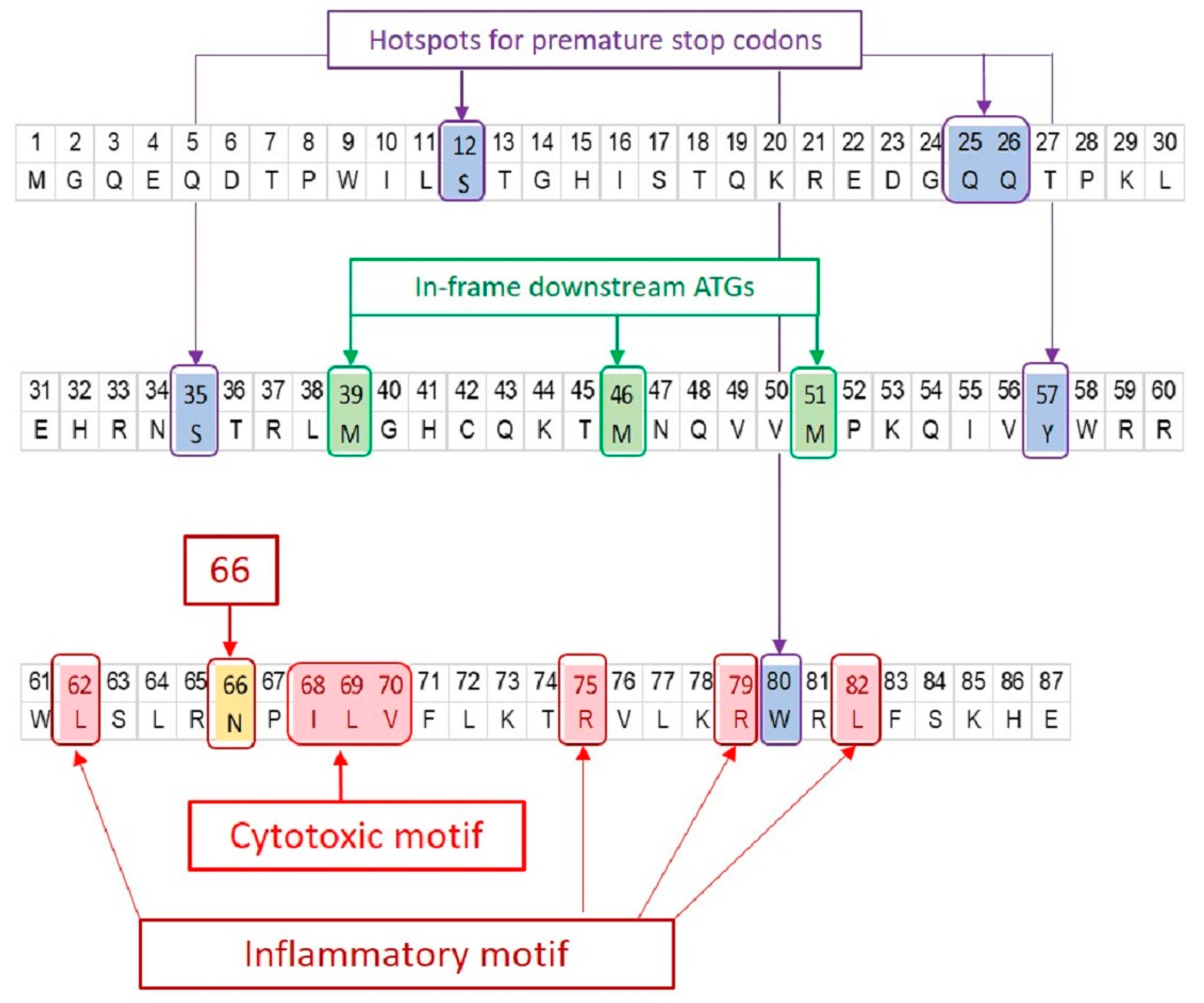
5.2.1. N66S
5.2.2. Inflammatory Motif
5.2.3. Cytotoxic Motif
5.2.4. Other Sequence Variants
5.3. Summary of Pathogenic Effects
6. Prevalence and Evolution of PB1-F2 Virulent Residues
7. Conclusions and Outstanding Questions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vasin, A.V.; Temkina, O.A.; Egorov, V.V.; Klotchenko, S.A.; Plotnikova, M.A.; Kiselev, O.I. Molecular mechanisms enhancing the proteome of influenza a viruses: An overview of recently discovered proteins. Virus Res. 2014, 185, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, X.; Liu, Q.; Bing, G.; Hu, Z.; Sun, H.; Xiong, X.; Jiang, M.; He, Q.; Wang, Y.; et al. PA-X protein contributes to virulence of triple-reassortant H1N2 influenza virus by suppressing early immune responses in swine. Virology 2017, 508, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; Maisonnasse, P.; Da Costa, B.; Delmas, B.; Chevalier, C.; Le Goffic, R. The influenza virus protein PB1-F2 increases viral pathogenesis through neutrophil recruitment and nk cells inhibition. PLoS ONE 2016, 11, e0165361. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Ortigoza, M.B.; Palese, P. Influenza a virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 2006, 80, 7976–7983. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza a virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel PB1-related protein translated from influenza a virus segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, G.W.; Wang, C.H.; Huang, C.H.; Wang, Y.C.; Shih, S.R. Differential localization and function of PB1-F2 derived from different strains of influenza a virus. J. Virol. 2010, 84, 10051–10062. [Google Scholar] [CrossRef] [PubMed]
- Deventhiran, J.; Kumar, S.R.; Raghunath, S.; Leroith, T.; Elankumaran, S. PB1-F2 protein does not impact the virulence of triple-reassortant H3N2 swine influenza virus in pigs but alters pathogenicity and transmission in turkeys. J. Virol. 2015, 90, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Hai, R.; Schmolke, M.; Varga, Z.T.; Manicassamy, B.; Wang, T.T.; Belser, J.A.; Pearce, M.B.; Garcia-Sastre, A.; Tumpey, T.M.; Palese, P. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J. Virol. 2010, 84, 4442–4450. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, O.; Embury-Hyatt, C.; Chevalier, C.; Jouneau, L.; Moroldo, M.; Da Costa, B.; Berhane, Y.; Delmas, B.; Weingartl, H.M.; Le Goffic, R. PB1-F2 attenuates virulence of highly pathogenic avian H5N1 influenza virus in chickens. PLoS ONE 2014, 9, e100679. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Chipuk, J.E.; Boyd, K.L.; Van De Velde, N.; Green, D.R.; McCullers, J.A. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010, 6, e1001014. [Google Scholar] [CrossRef] [PubMed]
- Bruns, K.; Studtrucker, N.; Sharma, A.; Fossen, T.; Mitzner, D.; Eissmann, A.; Tessmer, U.; Roder, R.; Henklein, P.; Wray, V.; et al. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza a virus protein. J. Biol. Chem. 2007, 282, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, C.; Al Bazzal, A.; Vidic, J.; Fevrier, V.; Bourdieu, C.; Bouguyon, E.; Le Goffic, R.; Vautherot, J.F.; Bernard, J.; Moudjou, M.; et al. PB1-F2 influenza a virus protein adopts a beta-sheet conformation and forms amyloid fibers in membrane environments. J. Biol. Chem. 2010, 285, 13233–13243. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The influenza a virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Chounan, R.; Higashi, Y.; Kurihara, N.; Kido, H. Mitochondrial targeting sequence of the influenza a virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004, 578, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Solbak, S.M.; Sharma, A.; Bruns, K.; Roder, R.; Mitzner, D.; Hahn, F.; Niebert, R.; Vedeler, A.; Henklein, P.; Henklein, P.; et al. Influenza a virus protein PB1-F2 from different strains shows distinct structural signatures. Biochim. Biophys. Acta 2013, 1834, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Henkel, M.; Mitzner, D.; Henklein, P.; Meyer-Almes, F.J.; Moroni, A.; Difrancesco, M.L.; Henkes, L.M.; Kreim, M.; Kast, S.M.; Schubert, U.; et al. The proapoptotic influenza a virus protein PB1-F2 forms a nonselective ion channel. PLoS ONE 2010, 5, e11112. [Google Scholar] [CrossRef] [PubMed]
- Vidic, J.; Richard, C.A.; Pechoux, C.; Da Costa, B.; Bertho, N.; Mazerat, S.; Delmas, B.; Chevalier, C. Amyloid assemblies of influenza a virus pb1-f2 protein damage membrane and induce cytotoxicity. J. Biol. Chem. 2016, 291, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Kamal, R.P.; Kumar, A.; Davis, C.T.; Tzeng, W.P.; Nguyen, T.; Donis, R.O.; Katz, J.M.; York, I.A. Emergence of highly pathogenic avian influenza a(H5N1) virus PB1-F2 variants and their virulence in BALB/c mice. J. Virol. 2015, 89, 5835–5846. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Krumbholz, A.; Eitner, A.; Krieg, R.; Halbhuber, K.J.; Wutzler, P. Prevalence of PB1-F2 of influenza a viruses. J. Gen. Virol. 2007, 88, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Tauber, S.; Ligertwood, Y.; Quigg-Nicol, M.; Dutia, B.M.; Elliott, R.M. Behaviour of influenza a viruses differentially expressing segment 2 gene products in vitro and in vivo. J. Gen. Virol. 2012, 93, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Praznovska, M.; Kosikova, M.; Bobisova, Z.; Holly, J.; Vareckova, E.; Kostolansky, F.; Russ, G. The ubiquitination of the influenza a virus PB1-F2 protein is crucial for its biological function. PLoS ONE 2015, 10, e0118477. [Google Scholar] [CrossRef] [PubMed]
- Buehler, J.; Navi, D.; Lorusso, A.; Vincent, A.; Lager, K.; Miller, C.L. Influenza a virus PB1-F2 protein expression is regulated in a strain-specific manner by sequences located downstream of the PB1-F2 initiation codon. J. Virol. 2013, 87, 10687–10699. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; Samarasinghe, A.; Vogel, P.; Green, A.M.; Weinlich, R.; McCullers, J.A. A novel cytotoxic sequence contributes to influenza a viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J. Virol. 2014, 88, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Schmolke, M.; Manicassamy, B.; Pena, L.; Sutton, T.; Hai, R.; Varga, Z.T.; Hale, B.G.; Steel, J.; Perez, D.R.; Garcia-Sastre, A. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza a virus in mammalian and avian species. PLoS Pathog. 2011, 7, e1002186. [Google Scholar] [CrossRef] [PubMed]
- Mazur, I.; Anhlan, D.; Mitzner, D.; Wixler, L.; Schubert, U.; Ludwig, S. The proapoptotic influenza a virus protein PB1-F2 regulates viral polymerase activity by interaction with the pb1 protein. Cell. Microbiol. 2008, 10, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Saitoh, T. Mitochondria-endoplasmic reticulum contact sites mediate innate immune responses. Adv. Exp. Med. Biol. 2017, 997, 187–197. [Google Scholar] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor mavs promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the mavs adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to mavs and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza a virus protein PB1-F2 translocates into mitochondria via tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Bouguyon, E.; Chevalier, C.; Vidic, J.; Da Costa, B.; Leymarie, O.; Bourdieu, C.; Decamps, L.; Dhorne-Pollet, S.; Delmas, B. Influenza a virus protein PB1-F2 exacerbates ifn-beta expression of human respiratory epithelial cells. J. Immunol. 2010, 185, 4812–4823. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Leymarie, O.; Chevalier, C.; Rebours, E.; Da Costa, B.; Vidic, J.; Descamps, D.; Sallenave, J.M.; Rauch, M.; Samson, M.; et al. Transcriptomic analysis of host immune and cell death responses associated with the influenza a virus PB1-F2 protein. PLoS Pathog. 2011, 7, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, O.; Meyer, L.; Tafforeau, L.; Lotteau, V.; Costa, B.D.; Delmas, B.; Chevalier, C.; Le Goffic, R. Influenza virus protein PB1-F2 interacts with CALCOCO2 (NDP52) to modulate innate immune response. J. Gen. Virol. 2017, 98, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Coulombe, F.; Downey, J.; Tzelepis, F.; Shalaby, K.; Tattoli, I.; Berube, J.; Rousseau, S.; Martin, J.G.; Girardin, S.E.; et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc. Natl. Acad. Sci. USA 2014, 111, E2110–E2119. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.E.; Wixler, L.; Nordhoff, C.; Nordmann, A.; Anhlan, D.; Wixler, V.; Ludwig, S. The influenza virus PB1-F2 protein has interferon antagonistic activity. Biol. Chem. 2011, 392, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Meunier, I.; von Messling, V. PB1-F2 modulates early host responses but does not affect the pathogenesis of H1N1 seasonal influenza virus. J. Virol. 2012, 86, 4271–4278. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by iav virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed]
- Pena, L.; Vincent, A.L.; Loving, C.L.; Henningson, J.N.; Lager, K.M.; Li, W.; Perez, D.R. Strain-dependent effects of PB1-F2 of triple-reassortant H3N2 influenza viruses in swine. J. Gen. Virol. 2012, 93, 2204–2214. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Henningson, J.; Ma, J.; Duff, M.; Lang, Y.; Li, Y.; Li, Y.; Nagy, A.; Sunwoo, S.; Bawa, B.; et al. Effects of PB1-F2 on the pathogenicity of H1N1 swine influenza virus in mice and pigs. J. Gen. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.L.; McCauley, J.W. The influenza virus protein PB1-F2 interacts with ikkbeta and modulates NF-kappab signalling. PLoS ONE 2013, 8, e63852. [Google Scholar] [CrossRef] [PubMed]
- Pinar, A.; Dowling, J.K.; Bitto, N.J.; Robertson, A.A.; Latz, E.; Stewart, C.R.; Drummond, G.R.; Cooper, M.A.; McAuley, J.L.; Tate, M.D.; et al. PB1-F2 peptide derived from avian influenza a virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J. Biol. Chem. 2017, 292, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Krejnusova, I.; Bystricka, M.; Polakova, K.; Russ, G. N-terminal region of the PB1-F2 protein is responsible for increased expression of influenza a viral protein PB1. Acta Virol. 2011, 55, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Basnet, S.; Burley, L.M.; Neumann, G.; Hatta, M.; Kawaoka, Y. Impact of amino acid mutations in PB2, PB1-F2, and NS1 on the replication and pathogenicity of pandemic (H1N1) 2009 influenza viruses. J. Virol. 2011, 85, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Zhang, K.; McCullers, J.A. The effects of influenza a virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J. Virol. 2010, 84, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, O.; Jouvion, G.; Herve, P.L.; Chevalier, C.; Lorin, V.; Lecardonnel, J.; Da Costa, B.; Delmas, B.; Escriou, N.; Le Goffic, R. Kinetic characterization of PB1-F2-mediated immunopathology during highly pathogenic avian H5N1 influenza virus infection. PLoS ONE 2013, 8, e57894. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Y.; Yang, S.R.; Wang, Y.T.; Lin, Y.H.; Chen, C.J. Amino acid residues 68–71 contribute to influenza a virus PB1-F2 protein stability and functions. Front. Microbiol. 2017, 8, 692. [Google Scholar] [CrossRef] [PubMed]
- Iverson, A.R.; Boyd, K.L.; McAuley, J.L.; Plano, L.R.; Hart, M.E.; McCullers, J.A. Influenza virus primes mice for pneumonia from staphylococcus aureus. J. Infect. Dis. 2011, 203, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Mitzner, D.; Dudek, S.E.; Studtrucker, N.; Anhlan, D.; Mazur, I.; Wissing, J.; Jansch, L.; Wixler, L.; Bruns, K.; Sharma, A.; et al. Phosphorylation of the influenza a virus protein PB1-F2 by PKC is crucial for apoptosis promoting functions in monocytes. Cell. Microbiol. 2009, 11, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Chanturiya, A.N.; Basanez, G.; Schubert, U.; Henklein, P.; Yewdell, J.W.; Zimmerberg, J. PB1-F2, an influenza a virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J. Virol. 2004, 78, 6304–6312. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Takaguchi, M.; Kawakami, T.; Suzuki, T. Sulfatide regulates caspase-3-independent apoptosis of influenza a virus through viral PB1-F2 protein. PLoS ONE 2013, 8, e61092. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza a virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Pena, L.; Vincent, A.L.; Loving, C.L.; Henningson, J.N.; Lager, K.M.; Lorusso, A.; Perez, D.R. Restored PB1-F2 in the 2009 pandemic H1N1 influenza virus has minimal effects in swine. J. Virol. 2012, 86, 5523–5532. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Howard, W.; Iqbal, M.; Nair, V.K.; Barclay, W.S.; Shelton, H. Influenza a virus PB1-F2 protein prolongs viral shedding in chickens lengthening the transmission window. J. Gen. Virol. 2016, 97, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; Green, A.M.; van de Velde, N.; McAuley, J.L.; Boyd, K.L.; Ghoneim, H.E.; McCullers, J.A. Immunopathogenic and antibacterial effects of H3N2 influenza a virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 2011, 85, 12324–12333. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; McCullers, J.A.; Kamal, R.P.; Vogel, P.; Green, A.M.; Ganesbom, S.; York, I.A. Dynamics of virulent PB1-F2 residues in human influenza a virus isolates and viral fitness in mice. Unpublished work. 2017. [Google Scholar]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza a viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Conenello, G.M.; Tisoncik, J.R.; Rosenzweig, E.; Varga, Z.T.; Palese, P.; Katze, M.G. A single n66s mutation in the PB1-F2 protein of influenza a virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 2011, 85, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Weeks-Gorospe, J.N.; Hurtig, H.R.; Iverson, A.R.; Schuneman, M.J.; Webby, R.J.; McCullers, J.A.; Huber, V.C. Naturally occurring swine influenza a virus PB1-F2 phenotypes that contribute to superinfection with gram-positive respiratory pathogens. J. Virol. 2012, 86, 9035–9043. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.; Deng, Y.M.; Gilbertson, B.; Mackenzie-Kludas, C.; Barr, I.; Brown, L. Rapid evolution of the PB1-F2 virulence protein expressed by human seasonal H3N2 influenza viruses reduces inflammatory responses to infection. Virol. J. 2017, 14, 162. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Scholtissek, C.; Franks, J.; Negovetich, N.J.; Aldridge, J.R.; Salomon, R.; Finkelstein, D.; Webster, R.G. Three amino acid changes in PB1-F2 of highly pathogenic H5N1 avian influenza virus affect pathogenicity in mallard ducks. Arch. Virol. 2010, 155, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Kuchipudi, S.V.; Mellits, K.H.; Sebastian, S.; James, J.; Liu, J.; Shelton, H.; Chang, K.C. Early apoptosis of porcine alveolar macrophages limits avian influenza virus replication and pro-inflammatory dysregulation. Sci. Rep. 2015, 5, 17999. [Google Scholar] [CrossRef] [PubMed]
- Betakova, T.; Kostrabova, A.; Lachova, V.; Turianova, L. Cytokines induced during influenza virus infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Henao, R.; Williams, J.; Nicholson, B.; Veldman, T.; Hudson, L.; Tsalik, E.L.; Lambkin-Williams, R.; Gilbert, A.; Mann, A.; et al. Differential evolution of peripheral cytokine levels in symptomatic and asymptomatic responses to experimental influenza virus challenge. Clin. Exp. Immunol. 2016, 183, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; York, I.A.; McCullers, J.A. Non-avian animal reservoirs present a source of influenza a PB1-F2 proteins with novel virulence-enhancing markers. PLoS ONE 2014, 9, e111603. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Han, G.Z.; Rambaut, A. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 2014, 508, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, Y.; Krauss, S.; Webster, R.G. Avian-to-human transmission of the PB1 gene of influenza a viruses in the 1957 and 1968 pandemics. J. Virol. 1989, 63, 4603–4608. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Host | IAV Subtype | % of Isolates with Full Length Protein (≥90aa) | Common Length Variants (% of Isolates) | Common Virulent Residues in Isolates Expressing PB1-F2 ≥62aa |
|---|---|---|---|---|
| Human | 1918pdmH1N1 | 100 | N/A | Complete inflammatory motif with 66S and 70V |
| Seasonal H1N1 | <1 | 57aa (96.8) | ||
| 2009pdmH1N1 | 0 | 11aa (100) | ||
| H1N1 SOIV a | 57 | 11aa (14.3) 57aa (14.3) 79aa (14.3) | 100% have 2 inflammatory residues (62L and 82L). | |
| H1N2 | 100 | N/A | No significant prevalence | |
| H1N2 SOIV a | 50 | 11aa (25) | 100% have 2 inflammatory residues (62L and 82L). | |
| H2N2 | >98 | N/A | Majority have complete inflammatory motif. | |
| Seasonal H3N2 | 91.3 | 34aa (6.7) | Majority have 1 cytotoxic residue (70V). 10% of recent (2015–2017) isolates have 70V and 79R. | |
| H3N2 SOIV a | >99 | N/A | Majority have 2 inflammatory residues (62L and 82L) | |
| H5N1 b | 94 | 23aa (3) | Majority have complete inflammatory motif. | |
| H5N6 b | 66 | 57aa (27) | 50% have complete inflammatory motif. | |
| H7N9 b | 86 | 57aa (5) | Majority have complete inflammatory motif. | |
| Swine | H1N1 | 42 | 11aa (29), 79aa (19) | 28.6% have complete inflammatory motif. |
| H1N2 | 82 | 79aa (5.7), 57aa (5) | 6.4% have complete inflammatory motif. Majority have 2 inflammatory residues (62L and 82L) | |
| H3N2 | 79 | 79aa (12), 35aa (3) | 2.4% have complete inflammatory motif. Majority have 2 inflammatory residues (62L and 82L) | |
| Equine | H3N8 | 50 | 81aa (50) | 85% have 3 inflammatory residues. |
| H7N7 | 75 | 34aa (25) | 100% have 3 inflammatory residues. | |
| Canine | H3N2 | 94.7 | 12aa (5.3) | 100% have 3 inflammatory residues. |
| H3N8 | 65.1 | 24aa (34.9) | 22% have complete inflammatory motif. | |
| Avian | H5N1 | 88.3 | 57aa (7.6), 24/25aa (2.03) | Majority have complete inflammatory motif. |
| H7N9 | 83 | 25aa (6), 34aa (5) | Majority have complete inflammatory motif. | |
| H9N2 | 81.6 | 79aa (12), 57 (6.4) | 50% have complete inflammatory motif. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamal, R.P.; Alymova, I.V.; York, I.A. Evolution and Virulence of Influenza A Virus Protein PB1-F2. Int. J. Mol. Sci. 2018, 19, 96. https://doi.org/10.3390/ijms19010096
Kamal RP, Alymova IV, York IA. Evolution and Virulence of Influenza A Virus Protein PB1-F2. International Journal of Molecular Sciences. 2018; 19(1):96. https://doi.org/10.3390/ijms19010096
Chicago/Turabian StyleKamal, Ram P., Irina V. Alymova, and Ian A. York. 2018. "Evolution and Virulence of Influenza A Virus Protein PB1-F2" International Journal of Molecular Sciences 19, no. 1: 96. https://doi.org/10.3390/ijms19010096
APA StyleKamal, R. P., Alymova, I. V., & York, I. A. (2018). Evolution and Virulence of Influenza A Virus Protein PB1-F2. International Journal of Molecular Sciences, 19(1), 96. https://doi.org/10.3390/ijms19010096