Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions


 , ,
, ,
Abstract
1. Introduction
2. Heat Shock Proteins
2.1. Heat Shock Proteins in Proteostasis
2.2. Heat Shock Factors Activate the Heat Shock Response
2.3. Hsp Activation by Membranes as Stress-Sensors
2.4. Cytoprotection by Hsps: Prevention of Apoptotic Cascade
3. ER-Stress, UPR, ERAD, Ubiquitination, and the Ubiquitin-Proteasome System (UPS)
3.1. Unfolded Protein Response
3.2. Endoplasmic Reticulum Associated Degradation
- (1)
- Recognition of misfolded or mutated proteins in the ER. This process involves detection of substructures within proteins, such as exposed large hydrophobic regions, unpaired cysteine residues, and immature glycans.
- (2)
- Retro-translocation of terminally misfolded proteins into the cytosol. The Hrd1E3 ubiquitin-protein ligase functions as a retrotranslocon (dislocon) to transport substrates to the cytosol. The direction of the transport is determined by the ubiquitin-binding factor Cdc48p in yeast and the valorin-containing protein (VCP/p97) in humans. The energy required for retro-translocation is provided by the ATPase activity of VCP/p97.
- (3)
- Ubiquitin-dependent degradation by the proteasome. Misfolded polypeptides are ubiquitinated by a cascade of enzymatic reactions within the ER membrane, such as the ubiquitin ligases Hrd1 and Doa10 [92]. Next, the polyubiquitinated polypeptide is recognized by specific subunits of the 26S proteasome (and thus ERAD is attached to UPS; Section 3.3) and translocates to the central chamber of the proteasome where the proteolytic active sites are located. ERAD has different branches for different misfolded domains [92].
3.3. Ubiquitination and UPS
3.4. Linkage between ERAD and UPR
4. Endo-Lysosomal System and Autophagy
4.1. Endo-Lysosomal and Autophagy Dysfuntion in NDDs
4.2. Autophagy
5. Prevention and Treatment of NDDs
5.1. Heat Shock Proteins in Neurodegenerative Disorders
5.2. Targeting Hsps for Treatment of NDDs
5.2.1. Therapeutic Potential of Small Molecule Hsp Co-Inducers
5.2.2. Natural Compounds that Induce/Co-Induce Chaperons and Are Applied for Treatment of NDDs
5.3. Targeting UPS, and Autophagy Dysfunction in NDDs
- Cathepsin activation, lipid clearance, lysosome membrane stabilization: by Hsp70, cholesterol modulation, and calpain inhibitors
- pH acidification: by GSK-3β inhibitors (valproate, lithium)
- Lysosomal exocytosis and exosome release by sphingomyelinase 2, phospholipase D and neuraminidase activation.
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Aβ | β-amyloid peptide |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| AMPK | 5′-AMP-activated protein kinase |
| APP | β-amyloid precursor protein |
| CMA | chaperon mediated autophagy |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum associated degradation |
| Hsp | heat shock protein |
| HSF | heat shock factor |
| HD | Huntington’s disease |
| LAMPA2A | lysosome-associated membrane protein 2 |
| LSD | lysosomal storage disorder |
| mTOR | mammalian target of rapamycin |
| NDD | neurodegenerative disease |
| PD | Parkinson’s disease |
| ULK | uncoordinated-51(unc-51) like kinase |
| UBD | ubiquitin binding domain |
| UPR | unfolded protein response |
| UPS | ubiquitin-proteasome system |
References
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yue, Z.Y. Neuronal aggregates: Formation, clearance, and spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.E.; Oddo, S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Esengul, Y.T.; Siddique, T. Protein recycling pathways in neurodegenerative diseases. Alzheimers Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Goloubinoff, P. Mechanisms of protein homeostatis in health, aging and disease. Swiss Med. Wkly. 2016, 146, w14306. [Google Scholar] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Cerebrovascular permeability to peptides—Manipulations of transport-systems at the blood-brain-barrier. Pharm. Res. 1995, 12, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Evidence for bulk flow of brain interstitial fluid: Significance for physiology and pathology. Neurochem. Int. 2004, 45, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.H.; Liao, Y.H.; Plogg, B.A.; Peng, W.G.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Sarge, K.D.; Abravaya, K. Transcriptional regulation of heat-shock genes—A paradigm for inducible genomic responses. J. Biol. Chem. 1992, 267, 21987–21990. [Google Scholar] [PubMed]
- Ellis, R.J. The molecular chaperone concept. Semin. Cell Biol. 1990, 1, 1–9. [Google Scholar] [PubMed]
- Horvath, I.; Multhoff, G.; Sonnleitner, A.; Vigh, L. Membrane-associated stress proteins: More than simply chaperones. BBA-Biomembranes 2008, 1778, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Meriin, A.B.; Yaglom, J.A.; Volloch, V.Z.; Sherman, M.Y. Role of Hsp70 in regulation of stress-kinase JNK: Implications in apoptosis and aging. FEBS Lett. 1998, 438, 1–4. [Google Scholar] [CrossRef]
- Garrido, C.; Schmitt, E.; Cande, C.; Vahsen, N.; Parcellier, A.; Kroemer, G. Hsp27 and Hsp70 potentially oncogenic apoptosis inhibitors. Cell Cycle 2003, 2, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Craig, E.A. Heat-shock proteins as molecular chaperones. Eur. J. Biochem. 1994, 219, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperon 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.W.; Caspers, G.J.; Leunissen, J.A. Genealogy of the alpha-crystallin—Small heat-shock protein superfamily. Int. J. Biol. Macromol. 1998, 22, 151–162. [Google Scholar] [CrossRef]
- Horwitz, J.; Bova, M.P.; Ding, L.L.; Haley, D.A.; Stewart, P.L. Lens alpha-crystallin: Function and structure. Eye 1999, 13, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Golenhofen, N.; Perng, M.D.; Quinlan, R.A.; Drenckhahn, D. Comparison of the small heat shock proteins alpha B-crystallin, MKBP, Hsp25, Hsp20, and cvhsp in heart and skeletal muscle. Histochem. Cell Biol. 2004, 122, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, H.; Vigh, L. The small heat shock proteins and their clients. Cell. Mol. Life Sci. 2007, 64, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B. Role of sHsps in organizing cytosolic protein aggregation and disaggregation. Cell Stress Chaperon 2017, 22, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; MacRae, T.H. The small heat shock proteins and their role in human disease. FEBS J. 2005, 272, 2613–2627. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Martin, E.; Gonzales, V.; Borchelt, D.R.; Lee, M.K. Differential regulation of small heat shock proteins in transgenic mouse models of neurodegenerative diseases. Neurobiol. Aging 2008, 29, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Gragerov, A.; Li, Z.; Xun, Z.; Burkholder, W.; Gottesman, M.E. Specificity of dnak peptide binding. J. Mol. Biol. 1994, 235, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, Z.; Broadley, S.A.; Shomura, Y.; Bracher, A.; Hartl, F.U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006, 25, 2519–2528. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Tse, E.; Freilich, R.; Mok, S.A.; Makley, L.N.; Southworth, D.R.; Gestwicki, J.E. Bag3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J. Mol. Biol. 2017, 429, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, C.A.; Connell, P.; Wu, Y.X.; Hu, Z.Y.; Thompson, L.J.; Yin, L.Y.; Patterson, C. Identification of chip, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [PubMed]
- McDonough, H.; Patterson, C. Chip: A link between the chaperone and proteasome systems. Cell Stress Chaperon 2003, 8, 303–308. [Google Scholar] [CrossRef]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational quality control: Folding, refolding, and degrading proteins. Science 1999, 286, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Min, J.N.; Whaley, R.A.; Sharpless, N.E.; Lockyer, P.; Portbury, A.L.; Patterson, C. Chip deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 2008, 28, 4018–4025. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Li, J.; Buchner, J. Structure, function and regulation of the hsp90 machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [PubMed]
- Balogi, Z.; Cheregi, O.; Giese, K.C.; Juhasz, K.; Vierling, E.; Vass, I.; Vigh, L.; Horvath, I. A mutant small heat shock protein with increased thylakoid association provides an elevated resistance against UV-B damage in synechocystis 6803. J. Biol. Chem. 2008, 283, 22983–22991. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Vigh, L. Cell biology stability in times of stress. Nature 2010, 463, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Glatz, A.; Nakamoto, H.; Mishkind, M.L.; Munnik, T.; Saidi, Y.; Goloubinoff, P.; Harwood, J.L.; Vigh, L. Heat shock response in photosynthetic organisms: Membrane and lipid connections. Prog. Lipid Res. 2012, 51, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Tsujimoto, N.; Nakagawa, H.; Iwaki, T.; Fukumaki, Y.; Iwaki, A. Association of HspB2, a member of the small heat shock protein family, with mitochondria. Exp. Cell Res. 2001, 271, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Torok, Z.; Pilbat, A.-M.; Gombos, I.; Hocsak, E.; Sümegi, B.; Horvath, I.; Vígh, L. Evidence on cholesterol-controlled lipif raft interaction of the small heat shock protein HspB11. In Cellular Trafficking of Cell Stress Proteins in Health and Disease; Henderson, B., Pockley, A.G., Eds.; Springer: Dordrecth, The Netherlands, 2012; Volume 6, pp. 75–86. ISBN 978-994-007-4739–4735. [Google Scholar]
- Bellyei, S.; Szigeti, A.; Boronkai, A.; Pozsgai, E.; Gomori, E.; Melegh, B.; Janaky, T.; Bognar, Z.; Hocsak, E.; Sumegi, B.; et al. Inhibition of cell death by a novel 16.2 kD heat shock protein predominantly via Hsp90 mediated lipid rafts stabilization and akt activation pathway. Apoptosis 2007, 12, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Torok, Z.; Goloubinoff, P.; Horvath, I.; Tsvetkova, N.M.; Glatz, A.; Balogh, G.; Varvasovszki, V.; Los, D.A.; Vierling, E.; Crowe, J.H.; et al. Synechocystis hsp17 is an amphitropic protein that stabilizes heat-stressed membranes and binds denatured proteins for subsequent chaperone-mediated refolding. Proc. Natl. Acad. Sci. USA 2001, 98, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed]
- Morano, K.A.; Thiele, D.J. Heat shock factor function and regulation in response to cellular stress, growth, and differentiation signals. Gene Expr. 1999, 7, 271–282. [Google Scholar] [PubMed]
- Shamovsky, I.; Gershon, D. Novel regulatory factors of HSF-1 activation: Facts and perspectives regarding their involvement in the age-associated attenuation of the heat shock response. Mech. Ageing Dev. 2004, 125, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Norman, S.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of Tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Q.; Wang, X.L.; Cao, X.H.; Ye, Z.Y.; Li, L.; Cai, W.Q. Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res. 2013, 1519, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.L. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Murshid, A. Molecular chaperone accumulation in cancer and decrease in Alzheimer’s disease: The potential roles of HSF1. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Hong, Y.; Yin, P.; Li, S.H.; Li, X.J. Differential HspBP1 expression accounts for the greater vulnerability of neurons than astrocytes to misfolded proteins. Proc. Natl. Acad. Sci. USA 2017, 114, E7803–E7811. [Google Scholar] [CrossRef] [PubMed]
- Prahlad, V.; Cornelius, T.; Morimoto, R.I. Regulation of the cellular heat shock response in caenorhabditis elegans by thermosensory neurons. Science 2008, 320, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Soti, C.; Csermely, P. Chaperones and aging: Role in neurodegeneration and in other civilizational diseases. Neurochem. Int. 2002, 41, 383–389. [Google Scholar] [CrossRef]
- Vigh, L.; Maresca, B.; Harwood, J.L. Does the membrane’s physical state control the expression of heat shock and other genes? Trends Biochem. Sci. 1998, 23, 369–374. [Google Scholar] [CrossRef]
- Balogh, G.; Peter, M.; Glatz, A.; Gombos, I.; Torok, Z.; Horvath, I.; Harwood, J.L.; Vigh, L. Key role of lipids in heat stress management. FEBS Lett. 2013, 587, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Balogi, Z.; Gombos, I.; Akerfelt, M.; Bjorkbom, A.; Balogh, G.; Torok, Z.; Maslyanko, A.; Fiszer-Kierzkowska, A.; Lisowska, K.; et al. Hyperfluidization-coupled membrane microdomain reorganization is linked to activation of the heat shock response in a murine melanoma cell line. Proc. Natl. Acad. Sci. USA 2007, 104, 7945–7950. [Google Scholar] [CrossRef] [PubMed]
- Crul, T.; Toth, N.; Piotto, S.; Literati-Nagy, P.; Tory, K.; Haldimann, P.; Kalmar, B.; Greensmith, L.; Torok, Z.; Balogh, G.; et al. Hydroximic acid derivatives: Pleiotropic Hsp co-inducers restoring homeostasis and robustness. Curr. Pharm. Des. 2013, 19, 309–346. [Google Scholar] [CrossRef] [PubMed]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef] [PubMed]
- Verdier, Y.; Zarandi, M.; Penke, B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimer’s disease. J. Pept. Sci. 2004, 10, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Chochina, S.V.; Avdulov, N.A.; Igbavboa, U.; Cleary, J.P.; O’Hare, E.O.; Wood, W.G. Amyloid beta-peptide(1-40) increases neuronal membrane fluidity: Role of cholesterol and brain region. J. Lipid Res. 2001, 42, 1292–1297. [Google Scholar] [PubMed]
- Kremer, J.J.; Pallitto, M.M.; Sklansky, D.J.; Murphy, R.M. Correlation of beta-amyloid aggregate size and hydrophobicity with decreased bilayer fluidity of model membranes. Biochemistry 2000, 39, 10309–10318. [Google Scholar] [CrossRef] [PubMed]
- Peters, I.; Igbavboa, U.; Schutt, T.; Haidari, S.; Hartig, U.; Rosello, X.; Bottner, S.; Copanaki, E.; Deller, T.; Kogel, D.; et al. The interaction of beta-amyloid protein with cellular membranes stimulates its own production. BBA-Biomembranes 2009, 1788, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Escriba, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Torok, Z.; Horvath, I.; Harwood, J.L.; Vigh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Mnich, K.; Samali, A. Heat shock preconditioning protects against ER stress-induced apoptosis through the regulation of the BH3-only protein BIM. FEBS Open Bio 2014, 4, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.T.; Copani, A.; Pike, C.J.; Whittemore, E.R.; Walencewicz, A.J.; Cotman, C.W. Apoptosis is induced by beta-amyloid in cultured central-nervous-system neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 7951–7955. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Anderson, A.J.; Cummings, B.J.; Cotman, C.W. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994, 5, 2529–2533. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; Tanaka, S.; Hansen, L.A. Caspase dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Guegan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [PubMed]
- Sreedhar, A.S.; Csermely, P. Heat shock proteins in the regulation of apoptosis: New strategies in tumor therapy—A comprehensive review. Pharmacol. Ther. 2004, 101, 227–257. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Reed, J.C.; Homma, S. Heat-shock proteins as regulators of apoptosis. Oncogene 2003, 22, 9041–9047. [Google Scholar] [CrossRef] [PubMed]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large potentials of small heat shock proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef] [PubMed]
- Voss, O.H.; Batra, S.; Kolattukudy, S.J.; Gonzalez-Mejia, M.E.; Smith, J.B.; Doseff, A.I. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J. Biol. Chem. 2007, 282, 25088–25099. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. Hsp70-DNAJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Saleh, A.; Nakazawa, A.; Kumar, S.; Srinivasula, S.M.; Kumar, V.; Weichselbaum, R.; Nalin, C.; Alnemri, E.S.; Kufe, D.; et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000, 19, 4310–4322. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER stress response: Getting the UPR hand on misfolded proteins. Curr. Biol. 2000, 10, R518–R521. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperons. BBA-Mol. Cell Res. 2013, 1833, 2410–2424. [Google Scholar]
- Graner, M.W.; Lillehei, K.O.; Katsanis, E. Endoplasmic reticulum chaperons and their roles in the immunogenicity of cancer vaccines. Front. Oncol. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.Z.; Tsai, J.; Xu, E.; Qiu, W.; Bereczki, E.; Santha, M.; Adeli, K. Apolipoprotein B100 acts as a molecular link between lipid-induced endoplasmic reticulum stress and hepatic insulin resistance. Hepatology 2009, 50, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Placido, A.I.; Pereira, C.M.F.; Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Oliveira, C.R.; Moreira, P.I. The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: Implications for Alzheimer’s disease. BBA-Mol. Basis Dis. 2014, 1842, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Area-Gomez, E. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell. Neurosci. 2013, 55, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Volgyi, K.; Juhasz, G.; Kovacs, Z.; Penke, B. Dysfunction of endoplasmic reticulum (ER) and mitochondria (MT) in Alzheimer’s disease: The role of the ER-MT cross-talk. Curr. Alzheimer Res. 2015, 12, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple mechanisms of unfolded protein response-induced cell death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Ben-Hamo, S.; Kenyon, C.; Henis-Korenblit, S. The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in c. Elegans. J. Cell Sci. 2013, 126, 4136–4146. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Mallucci, G.R. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology 2014, 76, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Foresti, O.; Carvalho, P. ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Jiang, L.L.; Huang, T.; Zhao, Y.J.; Liu, T.F.; Zhong, Y.W.; Li, X.G.; Campos, A.; Pomeroy, K.; Masliah, E.; et al. ER-associated degradation regulates Alzheimer’s amyloid pathology and memory function by modulating gamma-secretase activity. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Maldonado, M.A. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell. Mol. Immunol. 2006, 3, 255–261. [Google Scholar] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell. Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar] [PubMed]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Wu, Y.M.; Ferguson, S.M. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol. 2017, 216, 3291–3305. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Yuan, P.; Wu, Y.M.; Schrag, M.; Paradise, S.; Grutzendler, J.; Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. A beta localization in abnormal endosomes: Association with earliest a beta elevations in ad and down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Almeida, C.G.; Kearney, P.F.; Yu, F.M.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 2004, 24, 3592–3599. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.M.; Frechilla, D.; del Rio, J. Perspectives on the amyloid cascade hypothesis of Alzheimer’s disease. Rev. Neurol. 2010, 50, 667–675. [Google Scholar] [PubMed]
- Wang, X.; Zhou, X.; Li, G.Y.; Zhang, Y.; Wu, Y.L.; Song, W.H. Modifications and trafficking of APP in the pathogenesis of Alzheimer’s disease. Front. Mol. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Norstrom, E. Metabolic processing of the amyloid precursor protein—New pieces of the Alzheimer’s puzzle. Discov. Med. 2017, 23, 269–276. [Google Scholar] [PubMed]
- Peric, A.; Annaert, W. Early etiology of Alzheimer’s disease: Tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015, 129, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Ditaranto, K.; Tekirian, T.L.; Yang, A.J. Lysosomal membrane damage in soluble a beta-mediated cell death in Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T. Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’—A perspective review. Prog. Neurobiol. 2013, 105, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T. Can ‘calpain-cathepsin hypothesis’ explain Alzheimer neuronal death? Ageing Res. Rev. 2016, 32, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncosco, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and down syndrome—Differential effects of apoe genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology—Accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Barnett, J.L.; Mann, D.M.A.; Nixon, R.A. Colocalization of lysosomal hydrolase and beta-amyloid in diffuse plaques of the cerebellum and striatum in Alzheimer’s disease and down’s syndrome. J. Neuropathol. Exp. Neurol. 1996, 55, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? J. Neurochem. 2017, 140, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Telpoukhovskaia, M.A.; Bahr, B.A.; Chen, X.; Gan, L. Endo-lysosomal dysfunction: A converging mechanism in neurodegenerative diseases. Curr. Opin. Neurobiol. 2018, 48, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of down syndrome and Alzheimer’s disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperon-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The autophagy-lysosomal pathway in neurodegeneration: A TFEB perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jing, Y.Y.; Kang, X.; Yang, L.; Wang, D.L.; Zhang, W.; Zhang, L.; Chen, P.; Chang, J.F.; Yang, X.M.; et al. Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Res. 2017, 45, 1144–1158. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y. Regulation and function of uncoordinated-51 like kinase proteins. Antioxid. Redox Signal. 2012, 17, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Toth, A.M.; Foldi, I.; Szucs, M.; Janaky, T. Intraneuronal ss-amyloid and its interactions with proteins and subcellular organelles. Electrophoresis 2012, 33, 3608–3616. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Herman, A.M.; Moussa, C.E.H. Inflammation in the early stages of neurodegenerative pathology. J. Neuroimmunol. 2011, 238, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Caroni, P. Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Renkawek, K.; Bosman, G.J.C.G.M.; Dejong, W.W. Expression of small heat-shock protein Hsp-27 in reactive gliosis in Alzheimer-disease and other types of dementia. Acta Neuropathol. 1994, 87, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Renkawek, K.; Voorter, C.E.; Bosman, G.J.; van Workum, F.P.; de Jong, W.W. Expression of alpha B-crystallin in Alzheimer’s disease. Acta Neuropathol. 1994, 87, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.E.; Santha, M.; Penke, B.; Vígh, L. How to stabilize both the proteins and the membranes: Diverse effects of sHsps in neuroprotection. In The Big Book on Small Heat Shock Proteins; Tanguay, R.M., Hightower, L.E., Eds.; Springer: New York City, NY, USA, 2015; Volume 8, pp. 527–562. ISBN 978-523-319-16076-16074. [Google Scholar]
- Wilhelmus, M.M.M.; Otte-Holler, I.; Wesseling, P.; de Waal, R.M.W.; Boelens, W.C.; Verbeek, M.M. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2006, 32, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Inaguma, Y.; Goto, S.; Inagaki, T.; Kato, K. Alpha-B-crystallin and Hsp28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J. Neurol. Sci. 1993, 119, 203–208. [Google Scholar] [CrossRef]
- Jellinger, K.A. Cell death mechanisms in Parkinson’s disease. J. Neural Transm. 2000, 107, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Wisniewski, T.; Iwaki, A.; Corbin, E.; Tomokane, N.; Tateishi, J.; Goldman, J.E. Accumulation of alpha-B-crystallin in central-nervous-system glia and neurons in pathological conditions. Am. J. Pathol. 1992, 140, 345–356. [Google Scholar] [PubMed]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Selig, E.; Griffin, M.D.W.; Carver, J.A.; Ecroyd, H. Small heat-shock proteins prevent α-synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation. J. Biol. Chem. 2016, 291, 22618–22629. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase l1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, V.; Meister-Broekema, M.; Minoia, M.; Carra, S.; Kampinga, H.H. Barcoding heat shock proteins to human diseases: Looking beyond the heat shock response. Dis. Model Mech. 2014, 7, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.S.; Akbar, M.T.; Bouri, S.; Senda, A.; Joshi, K.; Chen, H.J.; Latchman, D.S.; WellSa, D.J.; de Belleroche, J. Protective effects of heat shock protein 27 in a model of ALS occur in the early stages of disease progression. Neurobiol. Dis. 2008, 30, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.E.; Szegedi, V.; Varga, E.; Juhasz, G.; Horvath, J.; Borbely, E.; Csibrany, B.; Alfoldi, R.; Lenart, N.; Penke, B.; et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/ps1 mice. Cell Stress Chaperon 2013, 18, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.M.; Chan, H.Y.E.; Gray-Board, G.L.; Chai, Y.H.; Paulson, H.L.; Bonini, N.M. Suppression of polyglutamine-mediated neurodegeneration in drosophila by the molecular chaperone Hsp70. Nat. Genet. 1999, 23, 425–428. [Google Scholar] [PubMed]
- Cummings, C.J.; Sun, Y.; Opal, P.; Antalffy, B.; Mestril, R.; Orr, H.T.; Dillmann, W.H.; Zoghbi, H.Y. Over-expression of inducible Hsp70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum. Mol. Genet. 2001, 10, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Murao, N.; Namba, T.; Takehara, M.; Adachi, H.; Katsuno, M.; Sobue, G.; Matsushima, T.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J. Neurosci. 2011, 31, 5225–5234. [Google Scholar] [CrossRef] [PubMed]
- Kudva, Y.C.; Hiddinga, H.J.; Butler, P.C.; Mueske, C.S.; Eberhardt, N.L. Small heat shock proteins inhibit in vitro a beta(1-42) amyloidogenesis. FEBS Lett. 1997, 416, 117–121. [Google Scholar] [CrossRef]
- Bruinsma, I.B.; Bruggink, K.A.; Kinast, K.; Versleijen, A.A.M.; Segers-Nolten, I.M.J.; Subramaniam, V.; Kuiperij, H.B.; Boelens, W.; de Waal, R.M.W.; Verbeek, M.M. Inhibition of alpha-synuclein aggregation by small heat shock proteins. Proteins Struct. Funct. Bioinform. 2011, 79, 2956–2967. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.; Ban, T.; Sakai, M.; Pasta, S.Y.; Ramakrishna, T.; Naiki, H.; Goto, Y.; Rao, C.M. Alpha B-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid beta-peptide and beta 2-microglobulin. Biochem. J. 2005, 392, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.H.; Koppenhafer, S.L.; Bonini, N.M.; Paulson, H.L. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J. Neurosci. 1999, 19, 10338–10347. [Google Scholar] [PubMed]
- Wilhelmus, M.M.M.; Boelens, W.C.; Otte-Holler, I.; Kamps, B.; de Waal, R.M.W.; Verbeek, M.M. Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res. 2006, 1089, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Klucken, J.; Strathearn, K.E.; Liu, F.; Nguyen, P.; Rochet, J.C.; Hyman, B.T.; McLean, P.J. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem. Biophys. Res. Commun. 2006, 351, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Zourlidou, A.; Smith, M.D.P.; Latchman, D.S. Hsp27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J. Neurochem. 2004, 88, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Yurinskaya, M.M.; Mit’kevich, V.A.; Barykin, E.P.; Garbuz, D.G.; Evgen’ev, M.B.; Makarov, A.A.; Vinokurov, M.G. Heat-shock protein Hsp70 protects neuroblastoma cells SK-N-SH from the neurotoxic effects of hydrogen peroxide and the beta-amyloid peptide. Mol. Biol. 2015, 49, 924–927. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell. Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Arrigo, A. The role of heat shock proteins during neurodegeneration in Alzheimer’s, Parkinson’s and Huntington’s disease. In Heat Shock Proteins in Neural Cells. Neuroscience Intelligence Unit; Richter-Landsberg, C., Ed.; Springer: New York, NY, USA, 2009; pp. 81–99. ISBN 978-970-387-39954-39956. [Google Scholar]
- Friedlander, R.M. Mechanisms of disease: Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- De Ruiter, J.P.; Uylings, H.B. Morphometric and dendritic analysis of fascia dentata granule cells in human aging and senile dementia. Brain Res. 1987, 402, 217–229. [Google Scholar] [CrossRef]
- Hanks, S.D.; Flood, D.G. Region-specific stability of dendritic extent in normal human aging and regression in alzheimers disease. I. Ca1 of hippocampus. Brain Res. 1991, 540, 63–82. [Google Scholar] [CrossRef]
- Bartelt-Kirbach, B.; Moron, M.; Glomb, M.; Beck, C.M.; Weller, M.P.; Golenhofen, N. HspB5/alphaB-crystallin increases dendritic complexity and protects the dendritic arbor during heat shock in cultured rat hippocampal neurons. Cell. Mol. Life Sci. 2016, 73, 3761–3775. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. The protective role of plant biophenols in mechanisms of Alzheimer’s disease. J. Nutr. Biochem. 2017, 47, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vigh, L.; Literati, P.N.; Horvath, I.; Torok, Z.; Balogh, G.; Glatz, A.; Kovacs, E.; Boros, I.; Ferdinandy, P.; Farkas, B.; et al. Bimoclomol: A nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat. Med. 1997, 3, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Biro, K.; Jednakovits, A.; Kukorelli, T.; Hegedus, E.; Koranyi, L. Bimoclomol (BRLP-42) ameliorates peripheral neuropathy in streptozotocin-induced diabetic rats. Brain Res. Bull. 1997, 44, 259–263. [Google Scholar] [CrossRef]
- Erdo, F.; Erdo, S.L. Bimoclomol protects against vascular consequences of experimental subarachnoid hemorrhage in rats. Brain Res. Bull. 1998, 45, 163–166. [Google Scholar] [CrossRef]
- Jednakovits, A.; Kurucz, I.; Nanasi, P.P. Effect of subchronic bimoclomol treatment on vascular responsiveness and heat shock protein production in spontaneously hypertensive rats. Life Sci. 2000, 67, 1791–1797. [Google Scholar] [CrossRef]
- Lubbers, N.L.; Polakowski, J.S.; Wegner, C.D.; Burke, S.E.; Diaz, G.J.; Daniell, K.M.; Cox, B.F. Oral bimoclomol elevates heat shock protein 70 and reduces myocardial infarct size in rats. Eur. J. Pharmacol. 2002, 435, 79–83. [Google Scholar] [CrossRef]
- Polakowski, J.S.; Wegner, C.D.; Cox, B.F. Bimoclomol elevates heat shock protein 70 and cytoprotects rat neonatal cardiomyocytes. Eur. J. Pharmacol. 2002, 435, 73–77. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Gray, J.; Priestman, D.A.; Wallom, K.L.; Atkins, J.; Olsen, O.D.; Klein, A.; Drndarski, S.; Petersen, N.H.; Ingemann, L.; et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 2016, 8, 355ra118. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Aguila, M.; McCulley, C.H.; Bevilacqua, D.; Mendes, H.F.; Athanasiou, D.; Novoselov, S.S.; Kanuga, N.; Munro, P.M.; Coffey, P.J.; et al. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.; Nirmalananthan, N.; Gray, A.L.; La Spada, A.R.; Hanna, M.G.; Greensmith, L. Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: Implications for therapy. Brain 2013, 136, 926–943. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Edet-Amana, E.; Greensmith, L. Treatment with a coinducer of the heat shock response delays muscle denervation in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Novoselov, S.; Gray, A.; Cheetham, M.E.; Margulis, B.; Greensmith, L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J. Neurochem. 2008, 107, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kieran, D.; Kalmar, B.; Dick, J.R.; Riddoch-Contreras, J.; Burnstock, G.; Greensmith, L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004, 10, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Shefner, J.M.; Simpson, E.; Grasso, D.; Yu, H.; Zhang, H.; Shui, A.; Schoenfeld, D.; Brown, R.H.; Wieland, S.; et al. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 2008, 38, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Greensmith, L. Cellular chaperones as therapeutic targets in ALS to restore protein homeostasis and improve cellular function. Front. Mol. Neurosci. 2017, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Haldimann, P.; Muriset, M.; Vigh, L.; Goloubinoff, P. The novel hydroxylamine derivative NG-094 suppresses polyglutamine protein toxicity in caenorhabditis elegans. J. Biol. Chem. 2011, 286, 18784–18794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ke, L.; Mackovicova, K.; Der Want, J.J.; Sibon, O.C.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Effects of different small HSPB members on contractile dysfunction and structural changes in a drosophila melanogaster model for atrial fibrillation. J. Mol. Cell. Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, D.C.; Bruce, C.R.; Drew, B.G.; Tory, K.; Kolonics, A.; Estevez, E.; Chung, J.; Watson, N.; Gardner, T.; Lee-Young, R.S.; et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014, 63, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, B.; Peterfai, E.; Kulcsar, E.; Literati-Nagy, Z.; Buday, B.; Tory, K.; Mandl, J.; Sumegi, B.; Fleming, A.; Roth, J.; et al. Beneficial effect of the insulin sensitizer (hsp inducer) BGP-15 on olanzapine-induced metabolic disorders. Brain Res. Bull. 2010, 83, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, B.; Tory, K.; Peitl, B.; Bajza, A.; Koranyi, L.; Literati-Nagy, Z.; Hooper, P.L.; Vigh, L.; Szilvassy, Z. Improvement of insulin sensitivity by a novel drug candidate, BGP-15, in different animal studies. Metab. Syndr. Relat. Disord. 2014, 12, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, Z.; Tory, K.; Literati-Nagy, B.; Bajza, A.; Vigh, L., Jr.; Vigh, L.; Mandl, J.; Szilvassy, Z. Synergic insulin sensitizing effect of rimonabant and BGP-15 in zucker-obese rats. Pathol. Oncol. Res. 2013, 19, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, Z.; Tory, K.; Literati-Nagy, B.; Kolonics, A.; Torok, Z.; Gombos, I.; Balogh, G.; Vigh, L., Jr.; Horvath, I.; Mandl, J.; et al. The hsp co-inducer BGP-15 can prevent the metabolic side effects of the atypical antipsychotics. Cell Stress Chaperon 2012, 17, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, Z.; Tory, K.; Literati-Nagy, B.; Kolonics, A.; Vigh, L., Jr.; Vigh, L.; Mandl, J.; Szilvassy, Z. A novel insulin sensitizer drug candidate BGP-15 can prevent metabolic side effects of atypical antipsychotics. Pathol. Oncol. Res. 2012, 18, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Wang, L.; Li, Q.; Cao, Y.; Dong, X.; Liang, J.; Wu, X. Hsp70 plays an important role in high-fat diet induced gestational hyperglycemia in mice. J. Physiol. Biochem. 2015, 71, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, B.; Kulcsar, E.; Literati-Nagy, Z.; Buday, B.; Peterfai, E.; Horvath, T.; Tory, K.; Kolonics, A.; Fleming, A.; Mandl, J.; et al. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: A proof of concept randomized double-blind clinical trial. Horm. Metab. Res. 2009, 41, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, S.M.; van der, P.C.; Sayer, T.A.; Schertzer, J.D.; Henstridge, D.C.; Church, J.E.; Lamon, S.; Russell, A.P.; Davies, K.E.; Febbraio, M.A.; Lynch, G.S. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 2012, 484, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Sumegi, K.; Fekete, K.; Antus, C.; Debreceni, B.; Hocsak, E.; Gallyas, F., Jr.; Sumegi, B.; Szabo, A. BGP-15 protects against oxidative stress- or lipopolysaccharide-induced mitochondrial destabilization and reduces mitochondrial production of reactive oxygen species. PLoS ONE 2017, 12, e0169372. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, B.; Kimbler, D.E.; Pang, J.; Choi, J.; Moskophidis, D.; Yanasak, N.; Dhandapani, K.M.; Mivechi, N.F. Therapeutic inducers of the HSP70/hsp110 protect mice against traumatic brain injury. J. Neurochem. 2014, 130, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Torok, Z.; Tsvetkova, N.M.; Balogh, G.; Horvath, I.; Nagy, E.; Penzes, Z.; Hargitai, J.; Bensaude, O.; Csermely, P.; Crowe, J.H.; et al. Heat shock protein coinducers with no effect on protein denaturation specifically modulate the membrane lipid phase. Proc. Natl. Acad. Sci. USA 2003, 100, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Gungor, B.; Gombos, I.; Crul, T.; Ayaydin, F.; Szabo, L.; Torok, Z.; Mates, L.; Vigh, L.; Horvath, I. Rac1 participates in thermally induced alterations of the cytoskeleton, cell morphology and lipid rafts, and regulates the expression of heat shock proteins in B16F10 melanoma cells. PLoS ONE 2014, 9, e89136. [Google Scholar] [CrossRef] [PubMed]
- Torok, Z.; Crul, T.; Maresca, B.; Schutz, G.J.; Viana, F.; Dindia, L.; Piotto, S.; Brameshuber, M.; Balogh, G.; Peter, M.; et al. Plasma membranes as heat stress sensors: From lipid-controlled molecular switches to therapeutic applications. Biochim. Biophys. Acta 2014, 1838, 1594–1618. [Google Scholar] [CrossRef] [PubMed]
- Gombos, I.; Crul, T.; Piotto, S.; Gungor, B.; Torok, Z.; Balogh, G.; Peter, M.; Slotte, J.P.; Campana, F.; Pilbat, A.M.; et al. Membrane-lipid therapy in operation: The hsp co-inducer BGP-15 activates stress signal transduction pathways by remodeling plasma membrane rafts. PLoS ONE 2011, 6, e28818. [Google Scholar] [CrossRef] [PubMed]
- Sapra, G.; Tham, Y.K.; Cemerlang, N.; Matsumoto, A.; Kiriazis, H.; Bernardo, B.C.; Henstridge, D.C.; Ooi, J.Y.; Pretorius, L.; Boey, E.J.; et al. The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat. Commun. 2014, 5, 5705. [Google Scholar] [CrossRef] [PubMed]
- Hargitai, J.; Lewis, H.; Boros, I.; Racz, T.; Fiser, A.; Kurucz, I.; Benjamin, I.; Vigh, L.; Penzes, Z.; Csermely, P.; et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 2003, 307, 689–695. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M., Jr.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Budzynski, M.A.; Crul, T.; Himanen, S.V.; Toth, N.; Otvos, F.; Sistonen, L.; Vigh, L. Chaperone co-inducer BGP-15 inhibits histone deacetylases and enhances the heat shock response through increased chromatin accessibility. Cell Stress Chaperon 2017, 22, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmaros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Horvath, I.; Nagy, E.; Hoyk, Z.; Benko, S.; Bensaude, O.; Vigh, L. The hyperfluidization of mammalian cell membranes acts as a signal to initiate the heat shock protein response. FEBS J. 2005, 272, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Maulucci, G.; Gombos, I.; Horvath, I.; Torok, Z.; Peter, M.; Fodor, E.; Pali, T.; Benko, S.; Parasassi, T.; et al. Heat stress causes spatially-distinct membrane re-modelling in K562 leukemia cells. PLoS ONE 2011, 6, e21182. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Peter, M.; Liebisch, G.; Horvath, I.; Torok, Z.; Nagy, E.; Maslyanko, A.; Benko, S.; Schmitz, G.; Harwood, J.L.; et al. Lipidomics reveals membrane lipid remodelling and release of potential lipid mediators during early stress responses in a murine melanoma cell line. Biochim. Biophys. Acta 2010, 1801, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Csoboz, B.; Balogh, G.E.; Kusz, E.; Gombos, I.; Peter, M.; Crul, T.; Gungor, B.; Haracska, L.; Bogdanovics, G.; Torok, Z.; et al. Membrane fluidity matters: Hyperthermia from the aspects of lipids and membranes. Int. J. Hyperth. 2013, 29, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Glatz, A.; Pilbat, A.M.; Nemeth, G.L.; Vince-Kontar, K.; Josvay, K.; Hunya, A.; Udvardy, A.; Gombos, I.; Peter, M.; Balogh, G.; et al. Involvement of small heat shock proteins, trehalose, and lipids in the thermal stress management in schizosaccharomyces pombe. Cell Stress Chaperon 2016, 21, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Balogh, G.; Gombos, I.; Liebisch, G.; Horvath, I.; Torok, Z.; Nagy, E.; Maslyanko, A.; Benko, S.; Schmitz, G.; et al. Nutritional lipid supply can control the heat shock response of b16 melanoma cells in culture. Mol. Membr. Biol. 2012, 29, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Glatz, A.; Gudmann, P.; Gombos, I.; Torok, Z.; Horvath, I.; Vigh, L.; Balogh, G. Metabolic crosstalk between membrane and storage lipids facilitates heat stress management in schizosaccharomyces pombe. PLoS ONE 2017, 12, e0173739. [Google Scholar] [CrossRef] [PubMed]
- Saidi, Y.; Peter, M.; Finka, A.; Cicekli, C.; Vigh, L.; Goloubinoff, P. Membrane lipid composition affects plant heat sensing and modulates Ca2+-dependent heat shock response. Plant Signal. Behav. 2010, 5, 1530–1533. [Google Scholar] [CrossRef] [PubMed]
- Vigh, L.; Nakamoto, H.; Landry, J.; Gomez-Munoz, A.; Harwood, J.L.; Horvath, I. Membrane regulation of the stress response from prokaryotic models to mammalian cells. Ann. N. Y. Acad. Sci. 2007, 1113, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Vigh, L.; Torok, Z.; Balogh, G.; Glatz, A.; Piotto, S.; Horvath, I. Membrane-regulated stress response: A theoretical and practical approach. Adv. Exp. Med. Biol. 2007, 594, 114–131. [Google Scholar] [PubMed]
- Grimm, M.O.W.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Vigh, L.; Horvath, I.; Maresca, B.; Harwood, J.L. Can the stress protein response be controlled by ‘membrane-lipid therapy’? Trends Biochem. Sci. 2007, 32, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Kasza, A.; Hunya, A.; Frank, Z.; Fulop, F.; Torok, Z.; Balogh, G.; Santha, M.; Balind, A.; Bernath, S.; Blundell, K.L.; et al. Dihydropyridine derivatives modulate heat shock responses and have a neuroprotective effect in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2016, 53, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Grille, S.; Zaslawski, A.; Thiele, S.; Plat, J.; Warnecke, D. The functions of steryl glycosides come to those who wait: Recent advances in plants, fungi, bacteria and animals. Prog. Lipid Res. 2010, 49, 262–288. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, S.; Kobayashi, T.; Kobayashi, S.; Murakami-Murofushi, K. Expression of cholesteryl glucoside by heat shock in human fibroblasts. Cell Stress Chaperon 2000, 5, 3–7. [Google Scholar] [CrossRef]
- Kunimoto, S.; Murofushi, W.; Kai, H.; Ishida, Y.; Uchiyama, A.; Kobayashi, T.; Kobayashi, S.; Murofushi, H.; Murakami-Murofushi, K. Steryl glucoside is a lipid mediator in stress-responsive signal transduction. Cell Struct. Funct. 2002, 27, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Sasaki, N.; Hanazawa, S.; Gotoh, M.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Novel sterol glucosyltransferase in the animal tissue and cultured cells: Evidence that glucosylceramide as glucose donor. Biochim. Biophys. Acta 2011, 1811, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Halling, K.K.; Ramstedt, B.; Slotte, J.P. Glycosylation induces shifts in the lateral distribution of cholesterol from ordered towards less ordered domains. Biochim. Biophys. Acta 2008, 1778, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Alarcon, S.V.; Lee, S.; Lee, M.J.; Giaccone, G.; Neckers, L.; Trepel, J.B. Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 2009, 9, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Paine-Murrieta, G.D.; Taylor, C.W.; Pulcini, E.J.; Akinaga, S.; Benjamin, I.J.; Whitesell, L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer Res. 2000, 6, 3312–3318. [Google Scholar] [PubMed]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of heat shock transcription factor HSF1 activation by hsp90 (hsp90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar] [CrossRef]
- Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Pagoulatos, G.; Angelidis, C.; Kusakabe, M.; Yoshiki, A.; Kobayashi, Y.; Doyu, M.; et al. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J. Neurosci. 2003, 23, 2203–2211. [Google Scholar] [PubMed]
- Tokui, K.; Adachi, H.; Waza, M.; Katsuno, M.; Minamiyama, M.; Doi, H.; Tanaka, K.; Hamazaki, J.; Murata, S.; Tanaka, F.; et al. 17-dmag ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an sbma model mouse. Hum. Mol. Genet. 2009, 18, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-aag, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Huang, L.; Chen, J.; Li, J.J.; Wang, R.; Kim, E.; Chen, Y.; Justicia, C.; Sakata, K.; et al. A cns-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol. Psychiatry 2017, 22, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Vega, F.V. Resveratrol protects primary rat hepatocytes against necrosis induced by reactive oxygen species. Biomed. Pharmacother. 2008, 62, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yang, J.Y.; Mou, Y.H.; Wang, L.H.; Zhou, Y.N.; Wu, C.F. Differences in the activities of resveratrol and ascorbic acid in protection of ethanol-induced oxidative DNA damage in human peripheral lymphocytes. Food Chem. Toxicol. 2012, 50, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; He, J.H.; Xie, H.B.; Yang, Y.S.; Li, J.C.; Zou, Y. Resveratrol induces antioxidant and heat shock protein mRNA expression in response to heat stress in black-boned chickens. Poult. Sci. 2014, 93, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.A.; Andrade, S.; Duarte, A.; Neves, A.R.; Queiroz, J.F.; Nunes, C.; Sevin, E.; Fenart, L.; Gosselet, F.; Coelho, M.A.N.; et al. Resveratrol and grape extract-loaded solid lipid nanoparticles for the treatment of Alzheimer’s disease. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.M.; Wang, N.; Liu, X.W. Resveratrol and amyloid-beta: Mechanistic insights. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- He, X.P.; Li, Z.H.; Rizak, J.D.; Wu, S.H.; Wang, Z.B.; He, R.Q.; Su, M.; Qin, D.D.; Wang, J.K.; Hu, X.T. Resveratrol attenuates formaldehyde induced hyperphosphorylation of Tau protein and cytotoxicity in N2a cells. Front. Neurosci. 2017, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Resveratrol ameliorates Tau hyperphosphorylation at Ser396 site and oxidative damage in rat hippocampal slices exposed to vanadate: Implication of ERK1/2 and GSK-3/beta signaling cascades. J. Agric. Food Chem. 2017, 65, 9626–9634. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauss, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7, 13753. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H. Biophenols pharmacology against the amyloidogenic activity in Alzheimer’s disease. Biomed. Pharmacother. 2017, 89, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.B.; Rui, Y.H.; Qin, L.Q.; Xu, J.Y.; Han, S.F.; Yuan, L.X.; Yin, X.B.; Wan, Z.X. Vitamin D combined with resveratrol prevents cognitive decline in SAMP8 mice. Curr. Alzheimer Res. 2017, 14, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Koyani, R.D.; Vazquez-Duhalt, R. Enzymatic activation of the emerging drug resveratrol. Appl. Biochem. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cordova-Gomez, M.; Galano, A.; Alvarez-Idaboy, J.R. Piceatannol, a better peroxyl radical scavenger than resveratrol. RSA Adv. 2013, 3, 20209–20218. [Google Scholar] [CrossRef]
- Yang, T.; Wang, L.; Zhu, M.; Zhang, L.; Yan, L. Properties and molecular mechanisms of resveratrol: A review. Pharmazie 2015, 70, 501–506. [Google Scholar] [PubMed]
- Kosuru, R.; Rai, U.; Prakash, S.; Singh, A.; Singh, S. Promising therapeutic potential of pterostilbene and its mechanistic insight based on preclinical evidence. Eur. J. Pharmacol. 2016, 789, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, W.; Zhou, Z.; Deng, S.; Ma, X.; Li, C.; Shu, X. Therapeutic versatility of resveratrol derivatives. Nutrients 2017, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Kasiotis, K.M.; Pratsinis, H.; Kletsas, D.; Haroutounian, S.A. Resveratrol and related stilbenes: Their anti-aging and anti-angiogenic properties. Food Chem. Toxicol. 2013, 61, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Rimando, A.; Pallas, M.; Camins, A.; Porquet, D.; Reeves, J.; Shukitt-Hale, B.; Smith, M.A.; Joseph, J.A.; Casadesus, G. Low-dose pterostilbene, but not resveratrol, is a potent neuromodulator in aging and Alzheimer’s disease. Neurobiol. Aging 2012, 33, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.A.; Fisher, D.R.; Cheng, V.; Rimando, A.M.; Shukitt-Hale, B. Cellular and behavioral effects of stilbene resveratrol analogues: Implications for reducing the deleterious effects of aging. J. Agric. Food Chem. 2008, 56, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- Jurenka, J.S. Anti-inflammatory properties of curcumin, a major constituent of curcuma longa: A review of preclinical and clinical research. Altern. Med. Rev. 2009, 14, 277. [Google Scholar]
- Randino, R.; Grimaldi, M.; Persico, M.; De Santis, A.; Cini, E.; Cabri, W.; Riva, A.; D’Errico, G.; Fattorusso, C.; D’Ursi, A.M.; et al. Investigating the neuroprotective effects of turmeric extract: Structural interactions of beta-amyloid peptide with single curcuminoids. Sci. Rep. 2016, 6, 38846. [Google Scholar] [CrossRef] [PubMed]
- Mathuranath, P.S.; George, A.; Ranjith, N.; Justus, S.; Kumar, M.S.; Menon, R.; Sarma, P.S.; Verghese, J. Incidence of Alzheimer’s disease in india: A 10 years follow-up study. Neurol. India 2012, 60, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Gounden, S.; Chuturgoon, A. Curcumin upregulates antioxidant defense, lon protease, and heat-shock protein 70 under hyperglycemic conditions in human hepatoma cells. J. Med. Food 2017, 20, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Kalaycioglu, Z.; Gazioglu, I.; Erim, F.B. Comparison of antioxidant, anticholinesterase, and antidiabetic activities of three curcuminoids isolated from curcuma longa L. Nat. Prod. Res. 2017, 31, 2914–2917. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Li, Z.; Qiu, D.H.; Gu, Q.O.; Lei, Q.F.; Mao, L. The inhibitory effects of different curcuminoids on beta-amyloid protein, beta-amyloid precursor protein and beta-site amyloid precursor protein cleaving enzyme 1 in swapp HEK293 cells. Neurosci. Lett. 2010, 485, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.P.N.; Mohamed, T.; Teckwani, K.; Tin, G. Curcumin binding to beta amyloid: A computational study. Chem. Biol. Drug Des. 2015, 86, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, X.; Wang, C.; Teng, Z.; Li, Y. Curcumin decreases hyperphosphorylation of Tau by down-regulating Caveolin-1/GSK-3beta in N2a/app695swe cells and APP/ps1 double transgenic Alzheimer’s disease mice. Am. J. Chin. Med. 2017, 45, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, J.R.; Poore, C.P.; Bin Sulaimee, N.H.; Pareek, T.; Cheong, W.F.; Wenk, M.R.; Pant, H.C.; Frautschy, S.A.; Low, C.M.; Kesavapany, S. Curcumin ameliorates neuroinflammation, neurodegeneration, and memory deficits in p25 transgenic mouse model that bears hallmarks of Alzheimer’s disease. J. Alzheimers Dis. 2017, 60, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.W.; Su, C.X.; Feng, H.L.; Chen, X.P.; Dong, Y.F.; Rao, Y.X.; Ren, Y.; Yang, J.D.; Shi, J.; Tian, J.Z.; et al. Curcumin regulates insulin pathways and glucose metabolism in the brains of APPswe/PS1dE9 mice. Int. J. Immunopathol. Pharmacol. 2017, 30, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Garcea, G.; Jones, D.J.L.; Singh, R.; Dennison, A.R.; Farmer, P.B.; Sharma, R.A.; Steward, W.P.; Gescher, A.J.; Berry, D.P. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br. J. Cancer 2004, 90, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S.S.R. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.; Fritz, M.; Brender, J.R.; Smith, P.E.S.; Lee, D.K.; Ramamoorthy, A. Determining the effects of lipophilic drugs on membrane structure by solid-state nmr spectroscopy: The case of the antioxidant curcumin. J. Am. Chem. Soc. 2009, 131, 4490–4498. [Google Scholar] [CrossRef] [PubMed]
- Semalty, A.; Semalty, M.; Rawat, M.S.M.; Franceschi, F. Supramolecular phospholipids-polyphenolics interactions: The PHYTOSOME (R) strategy to improve the bioavailability of phytochemicals. Fitoterapia 2010, 81, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, J.; Appendino, G.; Dern, A.S.; Schneider, E.; McKinnon, T.P.; Brown, M.J.; Togni, S.; Dixon, B.M. Comparative absorption of a standardized curcuminoid mixture and its lecithin formulation. J. Nat. Prod. 2011, 74, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, F.; Zacconi, P.; Bertuccioli, A.; Togni, S.; Eggenhoffner, R.; Giacomelli, L.; Scaltrini, S. A naturally-inspired, curcumin-based lecithin formulation (Meriva(R) formulated as the finished product Algocur(R)) alleviates the osteo-muscular pain conditions in rugby players. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4935–4940. [Google Scholar] [PubMed]
- Giori, A.; Franceschi, F. Phospholipid complexes of curcumin having improved bioavailability. EP 060078201, 9 March 2006. [Google Scholar]
- Tian, C.; Asghar, S.; Wu, Y.; Kambere Amerigos, D.; Chen, Z.; Zhang, M.; Yin, L.; Huang, L.; Ping, Q.; Xiao, Y. N-acetyl-L-cysteine functionalized nanostructured lipid carrier for improving oral bioavailability of curcumin: Preparation, in vitro and in vivo evaluations. Drug Deliv. 2017, 24, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Agrawal, A.K.; Gupta, R. Exosomes for the enhanced tissue bioavailability and efficacy of curcumin. AAPS J. 2017, 19, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Gera, M.; Sharma, N.; Ghosh, M.; Huynh, D.L.; Lee, S.J.; Min, T.; Kwon, T.; Jeong, D.K. Nanoformulations of curcumin: An emerging paradigm for improved remedial application. Oncotarget 2017, 8, 66680–66698. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Veena, M.S.; Stevenson, K.; Tang, C.; Ho, B.; Suh, J.D.; Duarte, V.M.; Faull, K.F.; Mehta, K.; Srivatsan, E.S.; et al. Liposome-encapsulated curcumin suppresses growth of head and neck squamous cell carcinoma in vitro and in xenografts through the inhibition of nuclear factor kappa B by an AKT-Independent pathway. Clin. Cancer Res. 2008, 14, 6228–6236. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Kasoju, N.; Bora, U. Encapsulation of curcumin in alginate-chitosan-pluronic composite nanoparticles for delivery to cancer cells. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Aseh, A.; Rios, C.N.; Aggarwal, B.B.; Mathur, A.B. Fabrication and characterization of silk fibroin-derived curcumin nanoparticles for cancer therapy. Int. J. Nanomed. 2009, 4, 115–122. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Metusalach; Brown, J.A. Carotenoid pigments in seafoods and aquaculture. Crit. Rev. Food Sci. Nutr. 1998, 38, 1–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Osawa, T. Cis astaxanthin and especially 9-cis astaxanthin exhibits a higher antioxidant activity in vitro compared to the all-trans isomer. Biochem. Biophys. Res. Commun. 2007, 357, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Grimmig, B.; Kim, S.H.; Nash, K.; Bickford, P.C.; Shytle, R.D. Neuroprotective mechanisms of astaxanthin: A potential therapeutic role in preserving cognitive function in age and neurodegeneration. Geroscience 2017, 39, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.P.; Poppe, S.C.; Bondan, E.F. Neuroprotective properties of the marine carotenoid astaxanthin and omega-3 fatty acids, and perspectives for the natural combination of both in krill oil. Nutrients 2014, 6, 1293–1317. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Olanow, C.W. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 1996, 47, 161S–170S. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, Y.J.; Kwon, K.H. Neuroprotective effects of astaxanthin in oxygen-glucose deprivation in SH-SY5Y cells and global cerebral ischemia in rat. J. Clin. Biochem. Nutr. 2010, 47, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Shi, H.Z.; Guo, Q.S.; Yu, Y.B.; Wang, A.M.; Lv, F.; Shen, W.B. Effects of astaxanthin and emodin on the growth, stress resistance and disease resistance of yellow catfish (pelteobagrus fulvidraco). Fish Shellfish Immunol. 2016, 51, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chen, C.Y.; Chiou, J.Y.; Peng, R.Y.; Peng, C.H. Astaxanthine secured apoptotic death of PC12 cells induced by beta-amyloid peptide 25–35: Its molecular action targets. J. Med. Food 2010, 13, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Polich, J.; Ehlers, C.L.; Otis, S.; Mandell, A.J.; Bloom, F.E. P300 latency reflects the degree of cognitive decline in dementing illness. Electroencephalogr. Clin. Neurophysiol. 1986, 63, 138–144. [Google Scholar] [CrossRef]
- Satoh, A.; Tsuji, S.; Okada, Y.; Murakami, N.; Urami, M.; Nakagawa, K.; Ishikura, M.; Katagiri, M.; Koga, Y.; Shirasawa, T. Preliminary clinical evaluation of toxicity and efficacy of a new astaxanthin-rich haematococcus pluvialis extract. J. Clin. Biochem. Nutr. 2009, 44, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, M.; Satoh, A.; Tsuji, S.; Shirasawa, T. Effects of astaxanthin-rich haematococcus pluvialis extract on cognitive function: A randomised, double-blind, placebo-controlled study. J. Clin. Biochem. Nutr. 2012, 51, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Nalawade, P.; Gajjar, A. Optimization of astaxanthin microencapsulation in hydrophilic carriers using response surface methodology. Arch. Pharm. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Suzuki, R.; Nara, E.; Yokoyama, A.; Miyashita, K. Antioxidant activity of polar carotenoids including astaxanthin-beta-glucoside from marine bacterium on PC liposomes. Fish Sci. 2000, 66, 980–985. [Google Scholar] [CrossRef]
- Chen, C.S.; Wu, S.H.; Wu, Y.Y.; Fang, J.M.; Wu, T.H. Properties of Astaxanthin/Ca2+ complex formation in the deceleration of Cis/Trans isomerization. Org. Lett. 2007, 9, 2985–2988. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Chen, R.; Guo, Z.Y.; Li, C.P.; Li, P.C. The preparation and stability of the inclusion complex of astaxanthin with beta-cyclodextrin. Food Chem. 2007, 101, 1580–1584. [Google Scholar] [CrossRef]
- De Paiva Lacerda, S.; Espitalier, F.; Hoffart, V.; Re, M.I. Liquid anti-solvent recrystallization to enhance dissolution of CRS74, a new antiretroviral drug. Drug Dev. Ind. Pharm. 2015, 41, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Ciapara, I.; Felix-Valenzuela, L.; Goycoolea, F.M.; Arguelles-Monal, W. Microencapsulation of astaxanthin in a chitosan matrix. Carbohydr. Polym. 2004, 56, 41–45. [Google Scholar] [CrossRef]
- Kittikaiwan, P.; Powthongsook, S.; Pavasant, P.; Shotipruk, A. Encapsulation of haematococcus pluvialis using chitosan for astaxanthin stability enhancement. Carbohydr. Polym. 2007, 70, 378–385. [Google Scholar] [CrossRef]
- Acevedo, F.; Rubilar, M.; Jofre, I.; Villarroel, M.; Navarrete, P.; Esparza, M.; Romero, F.; Vilches, E.A.; Acevedo, V.; Shene, C. Oil bodies as a potential microencapsulation carrier for astaxanthin stabilisation and safe delivery. J. Microencapsul. 2014, 31, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Jiao, Y.P.; Wang, Y.F.; Zhou, C.R.; Zhang, Z.Y. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Del. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Pestieau, A.; Krier, F.; Lebrun, P.; Brouwers, A.; Streel, B.; Evrard, B. Optimization of a PGSS (particles from gas saturated solutions) process for a fenofibrate lipid-based solid dispersion formulation. Int. J. Pharm. 2015, 485, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Meeus, J.; Lenaerts, M.; Scurr, D.J.; Amssoms, K.; Davies, M.C.; Roberts, C.J.; Van Den Mooter, G. The influence of spray-drying parameters on phase behavior, drug distribution, and in vitro release of injectable microspheres for sustained release. J. Pharm. Sci. 2015, 104, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Paidi, S.K.; Jena, S.K.; Ahuja, B.K.; Devasari, N.; Suresh, S. Preparation, in vitro and in vivo evaluation of spray-dried ternary solid dispersion of biopharmaceutics classification system class II model drug. J. Pharm. Pharmacol. 2015, 67, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Gerding, D.; Oomah, B.D.; Acevedo, F.; Morales, E.; Bustamante, M.; Shene, C.; Rubilar, M. High carotenoid bioaccessibility through linseed oil nanoemulsions with enhanced physical and oxidative stability. Food Chem. 2016, 199, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Deane, C.A.S.; Brown, I.R. Knockdown of heat shock proteins HSPA6 (Hsp70B’) and HSPA1A (Hsp70-1) sensitizes differentiated human neuronal cells to cellular stress. Neurochem. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cleren, C.; Calingasan, N.Y.; Chen, J.; Beal, M.F. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005, 94, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Sassa, H.; Takaishi, Y.; Terada, H. The triterpene celastrol as a very potent inhibitor of lipid-peroxidation in mitochondria. Biochem. Biophys. Res. Commun. 1990, 172, 890–897. [Google Scholar] [CrossRef]
- Sassa, H.; Kogure, K.; Takaishi, Y.; Terada, H. Structural basis of potent antiperoxidation activity of the triterpene celastrol in mitochondria—Effect of negative membrane-surface charge on lipid-peroxidation. Free Radic. Biol. Med. 1994, 17, 201–207. [Google Scholar] [CrossRef]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.M.; Alvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Tao, X.L.; Sun, Y.; Dong, Y.; Xiao, Y.L.; Hu, D.W.; Shi, Y.P.; Zhu, Q.L.; Dai, H.; Zhang, N.Z. A prospective, controlled, double-blind, cross over study of tripterygium-wilfodii hook-F in treatment of rheumatoid-arthritis. Chin. Med. J. 1989, 102, 327–332. [Google Scholar] [PubMed]
- Freag, M.S.; Saleh, W.M.; Abdallah, O.Y. Self-assembled phospholipid-based phytosomal nanocarriers as promising platforms for improving oral bioavailability of the anticancer celastrol. Int. J. Pharm. 2017, 535, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Khodagholi, F.; Eftekharzadeh, B.; Maghsoudi, N.; Rezaei, P.F. Chitosan prevents oxidative stress-induced amyloid beta formation and cytotoxicity in NT2 neurons: Involvement of transcription factors nrf2 and nf-kappa B. Mol. Cell. Biochem. 2010, 337, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Anissian, D.; Ghasemi-Kasman, M.; Khalili-Fomeshi, M.; Akbari, A.; Hashemian, M.; Kazemi, S.; Moghadamnia, A.A. Piperine-loaded chitosan-STPP nanoparticles reduce neuronal loss and astrocytes activation in chemical kindling model of epilepsy. Int. J. Biol. Macromol. 2017, 107, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, T.; Rokutan, K.; Nikawa, T.; Kishi, K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 1996, 111, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Marunouchi, T.; Inomata, S.; Sanbe, A.; Takagi, N.; Tanonaka, K. Protective effect of geranylgeranylacetone via enhanced induction of hspb1 and hspb8 in mitochondria of the failing heart following myocardial infarction in rats. Eur. J. Pharmacol. 2014, 730, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Fujibayashi, T.; Hashimoto, N.; Jijiwa, M.; Hasegawa, Y.; Kojima, T.; Ishiguro, N. Protective effect of geranylgeranylacetone, an inducer of heat shock protein 70, against drug-induced lung injury/fibrosis in an animal model. BMC Pulm. Med. 2009, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, J.Y.; Kang, S.W.; Chun, G.S.; Ban, J.Y. Protective effect of geranylgeranylacetone against hydrogen peroxide-induced oxidative stress in human neuroblastoma cells. Life Sci. 2015, 131, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Ogai, K.; Sugitani, K.; Hisano, S.; Kato, S. Geranylgeranylacetone suppresses N-Methyl-N-nitrosourea-induced photoreceptor cell loss in mice. Adv. Exp. Med. Biol. 2016, 854, 237–243. [Google Scholar] [PubMed]
- Dong, Z.; Shinmei, Y.; Dong, Y.; Inafuku, S.; Fukuhara, J.; Ando, R.; Kitaichi, N.; Kanda, A.; Tanaka, K.; Noda, K.; et al. Effect of geranylgeranylacetone on the protection of retinal ganglion cells in a mouse model of normal tension glaucoma. Heliyon 2016, 2, e00191. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Kwong, J.M.K.; Caprioli, J. Retinal ganglion cell protection with geranylgeranylacetone, a heat shock protein inducer, in a rat glaucoma model. Investing. Ophthalmol. Vis. Sci. 2003, 44, 1982–1992. [Google Scholar] [CrossRef]
- Fujiki, M.; Furukawa, Y.; Kobayashi, H.; Abe, T.; Ishii, K.; Uchida, S.; Kamida, T. Geranylgeranylacetone limits secondary injury, neuronal death, and progressive necrosis and cavitation after spinal cord injury. Brain Res. 2005, 1053, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Abe, E.; Fujiki, M.; Nagai, Y.; Shiqi, K.; Kubo, T.; Ishii, K.; Abe, T.; Kobayashi, H. The phosphatidylinositol-3 kinase/akt pathway mediates geranylgeranylacetone-induced neuroprotection against cerebral infarction in rats. Brain Res. 2010, 1330, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Fujiki, M.; Uchida, S.; Morishige, M.; Momii, Y.; Ishii, K. A single oral dose of geranylgeranylacetone upregulates vascular endothelial growth factor and protects against kainic acid-induced neuronal cell death: Involvement of the phosphatidylinositol-3 kinase/akt pathway. Pathobiology 2017, 84, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Fujiki, M.; Nagai, Y.; Abe, T.; Kobayashi, H. Geranylgeranylacetone, a noninvasive heat shock protein inducer, induces protein kinase C and leads to neuroprotection against cerebral infarction in rats. Neurosci. Lett. 2006, 396, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.D.; Dixit, V.M. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Elia, A.E.H.; Xu, Q.K.; Thoma, C.R.; Izhar, L.; Leng, Y.M.; Guo, A.L.; Chen, Y.N.; Rush, J.; Hsu, P.W.C.; et al. Global identification of modular cullin-ring ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaeid, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014, 35, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. (Warsz) 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Rubinsztein, D.C. Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet. 2006, 15, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.; Menzies, F.M.; Renna, M.; Acevedo-Arozena, A.; Corrochano, S.; Sadiq, O.; Brown, S.D.; Rubinsztein, D.C. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Papp, D.; Kovacs, T.; Billes, V.; Varga, M.; Tarnoci, A.; Hackler, L.; Puskas, L.G.; Liliom, H.; Tarnok, K.; Schlett, K.; et al. AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy 2016, 12, 273–286. [Google Scholar] [CrossRef] [PubMed]






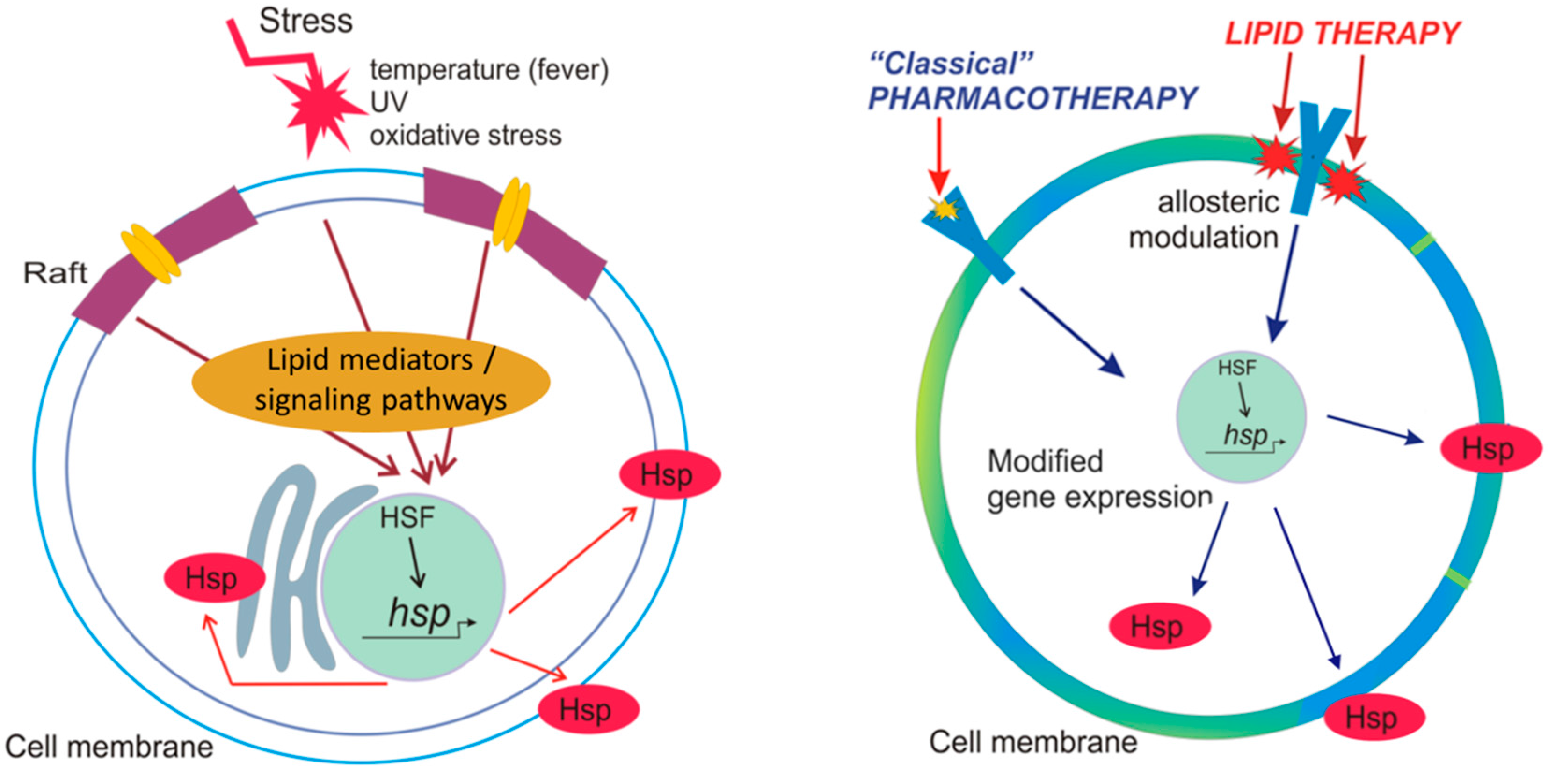{kind=link}
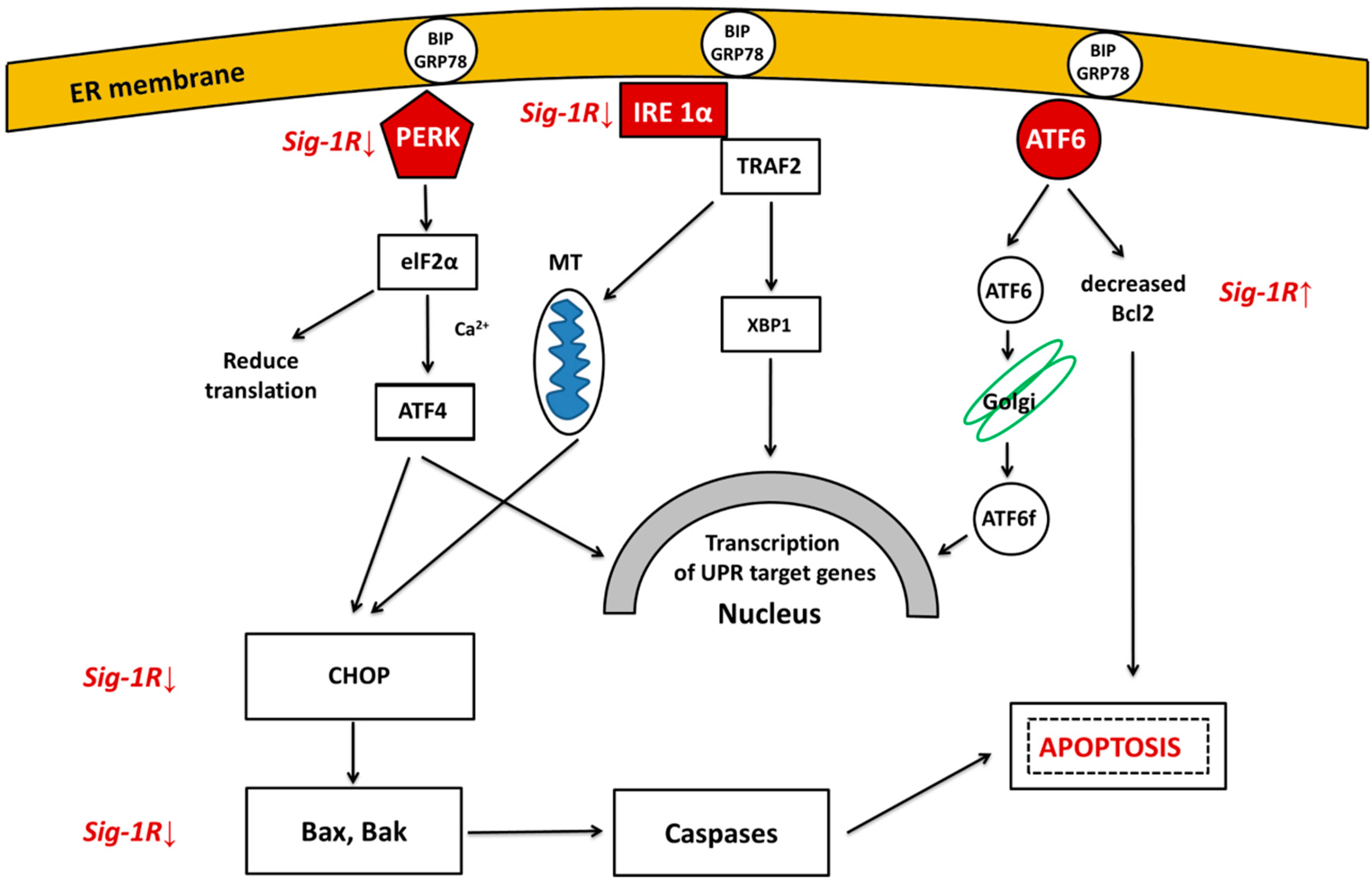{kind=link}
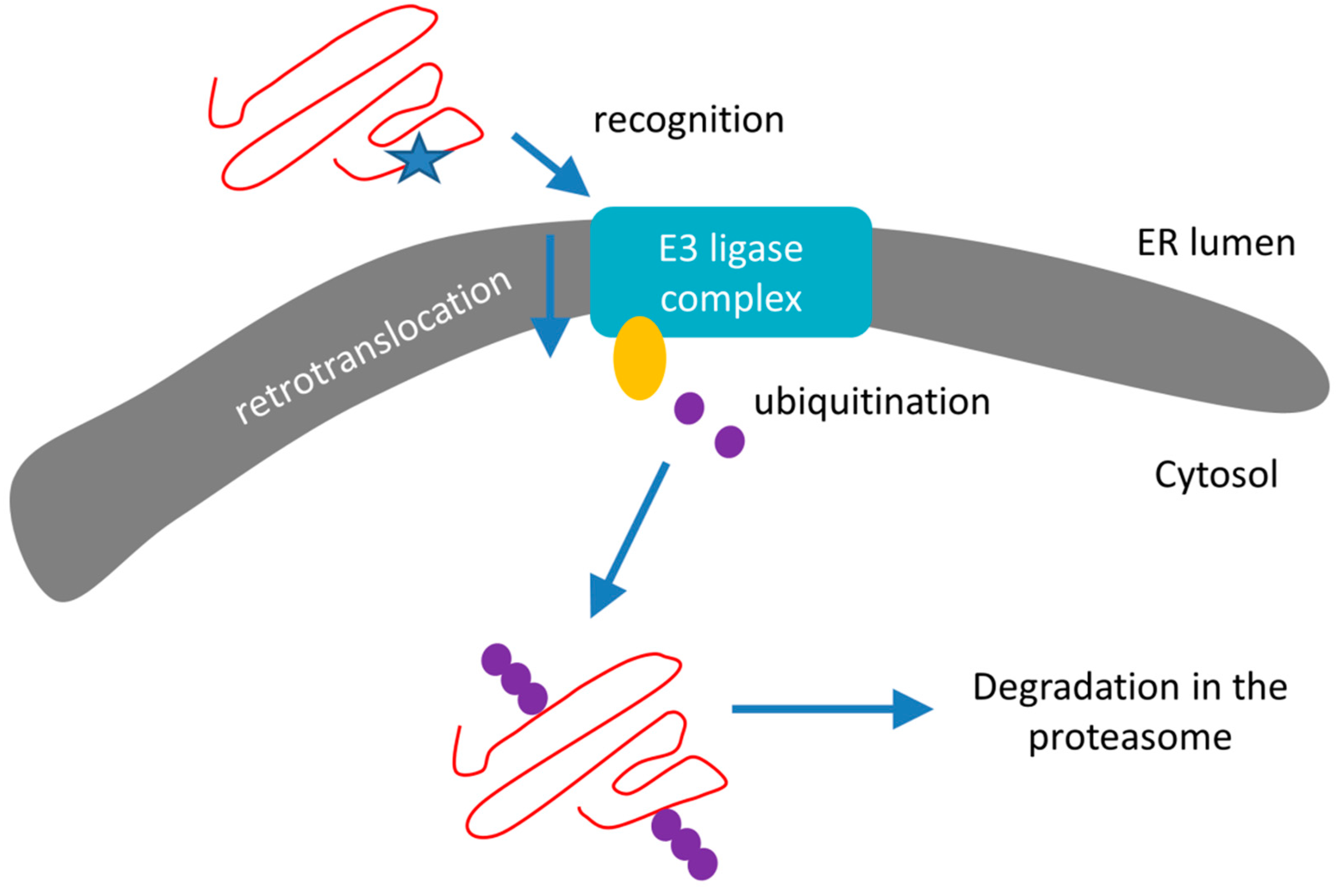{kind=link}
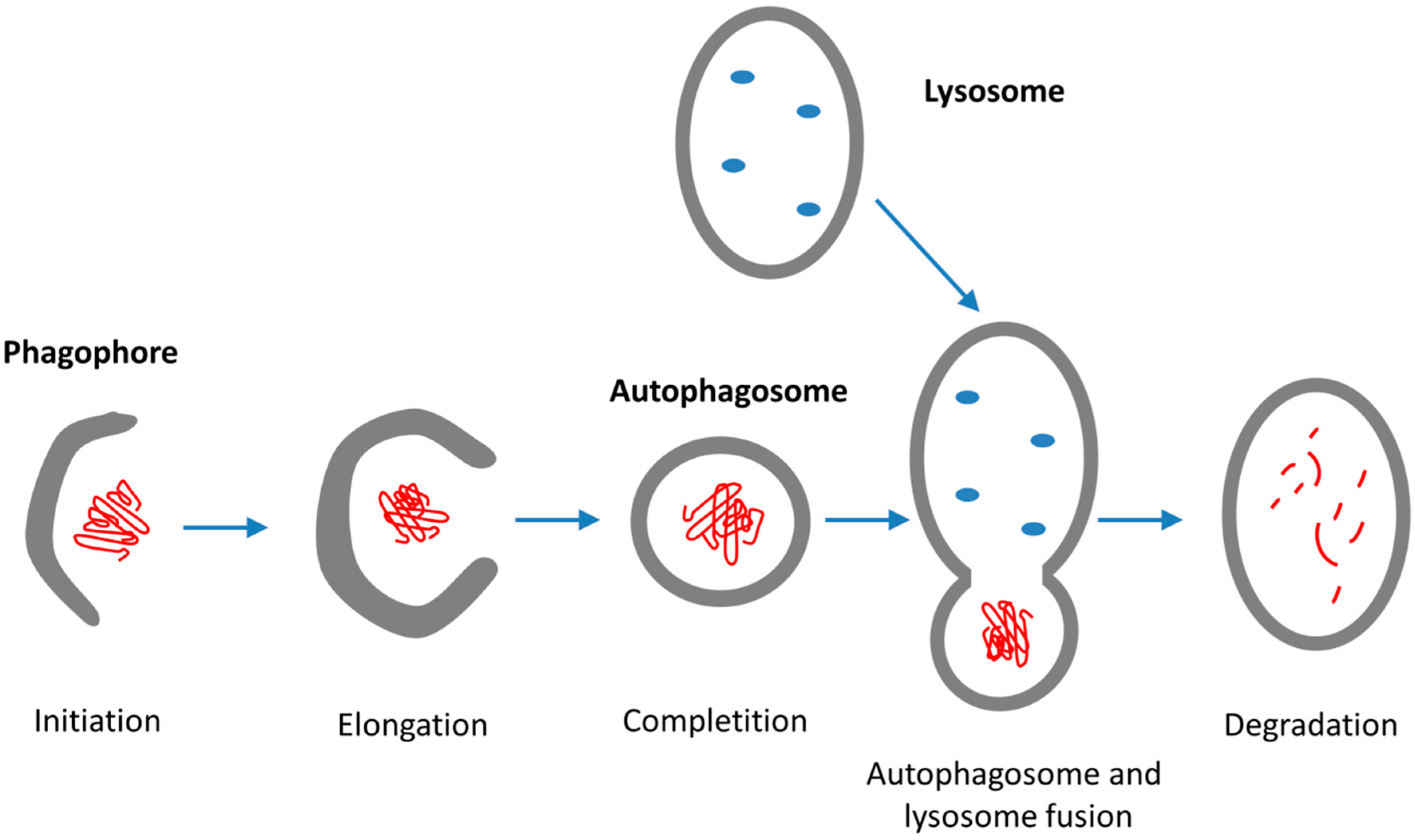{kind=link}
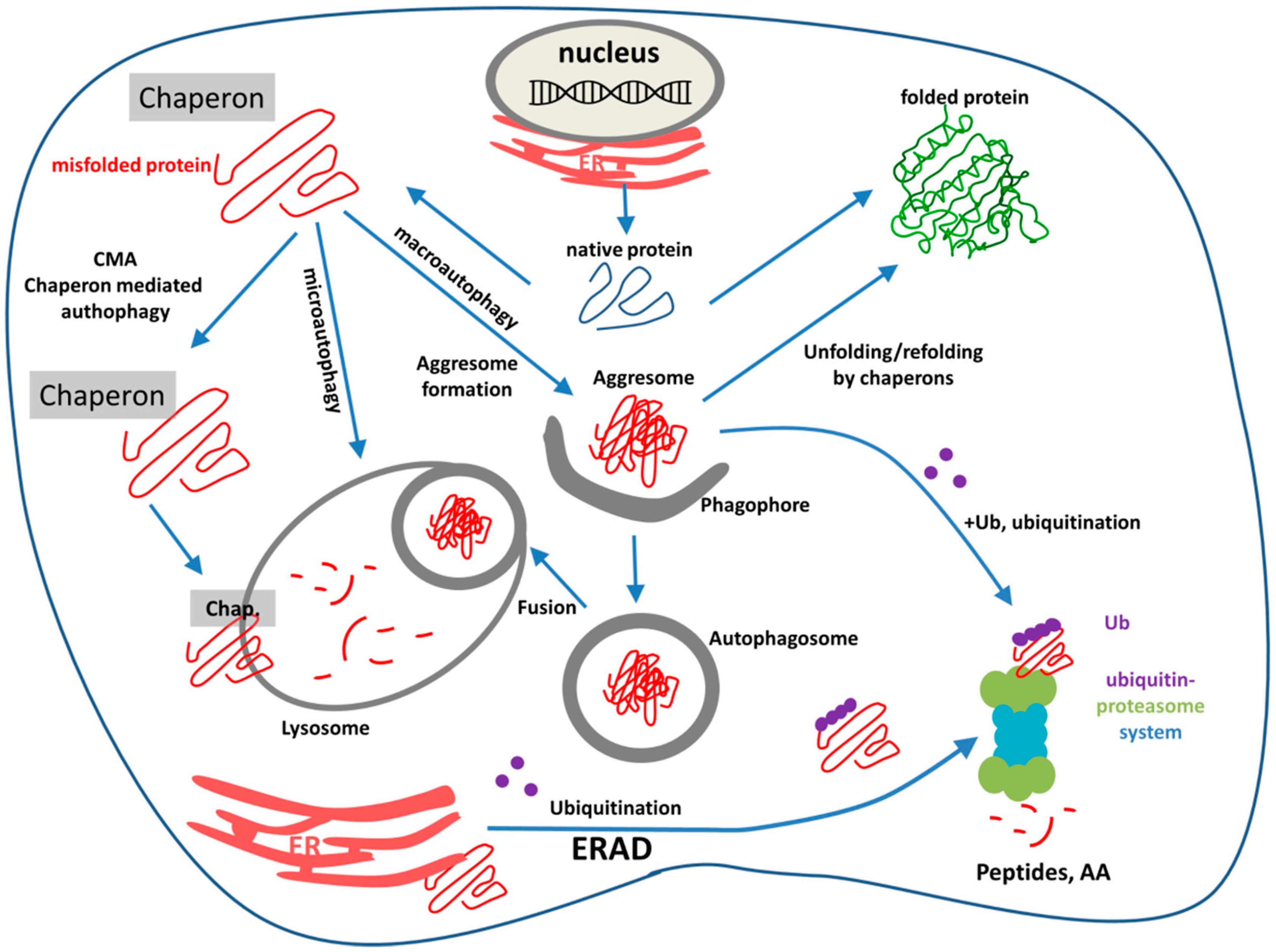{kind=link}
| List of Diseases | Misfolded Proteins |
|---|---|
| Alzheimer’s disease | β-amyloid (Aβ) |
| hyperphosphorylated Tau (pTau) | |
| α-synuclein | |
| Parkinson’s disease | α-synuclein |
| Huntington’s disease | huntingtin |
| Lewy-body dementia | α-synuclein |
| Amyotrophic lateral sclerosis | TDP-43 |
| Prion diseases | superoxide dismutase (SOD) |
| prion protein (PrPsc) |
| Name of the Process/Pathway | Localization | Participating Players, Structures |
|---|---|---|
| Ubiquitin-proteasome system (UPS) | cytoplasm, proteasome | E1, E2, E3 enzymes, ubiquitin, UBD adaptors, proteasome |
| ER-associated degradation (ERAD) | ER, cytoplasm, proteasome | Recognition proteins, E3 ligase complex, Doa10 and Hrd1 complexes, ubiquitin, proteasome |
| Autophagy | ||
| Chaperon-mediated autophagy (CMA) | cytoplasm, lysosome | Cytosolic chaperon, protein-translocation complex LAMP2A, Hsp90AA1, GFAP, lysosomal enzymes |
| Macroautophagy | cytoplasm, lysosome | Aggresome, phagophor, autophagophor, lysosomal hydrolyses |
| Microautophagy | cytoplasm, lysosome | HspA8, late endosome, ESCRT for transport, lysosomal hydrolyses |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.E.; Vígh, L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. Int. J. Mol. Sci. 2018, 19, 325. https://doi.org/10.3390/ijms19010325
Penke B, Bogár F, Crul T, Sántha M, Tóth ME, Vígh L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. International Journal of Molecular Sciences. 2018; 19(1):325. https://doi.org/10.3390/ijms19010325
Chicago/Turabian StylePenke, Botond, Ferenc Bogár, Tim Crul, Miklós Sántha, Melinda E. Tóth, and László Vígh. 2018. "Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions" International Journal of Molecular Sciences 19, no. 1: 325. https://doi.org/10.3390/ijms19010325
APA StylePenke, B., Bogár, F., Crul, T., Sántha, M., Tóth, M. E., & Vígh, L. (2018). Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. International Journal of Molecular Sciences, 19(1), 325. https://doi.org/10.3390/ijms19010325