Role of 3-Hydroxy Fatty Acid-Induced Hepatic Lipotoxicity in Acute Fatty Liver of Pregnancy



Abstract
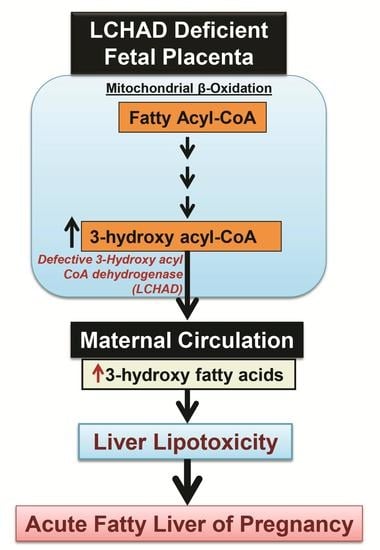
:
{kind=link}
{kind=link}
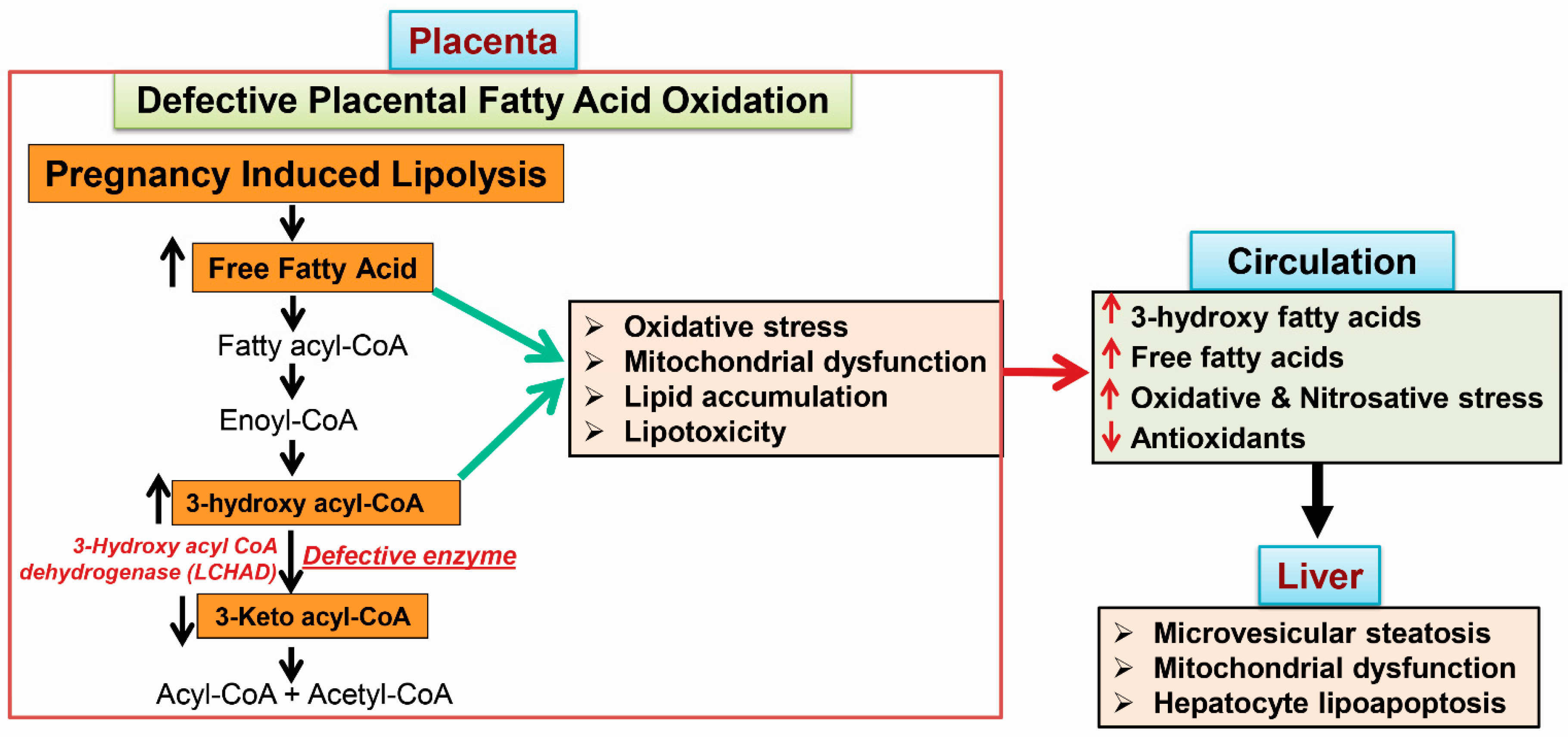{kind=link}
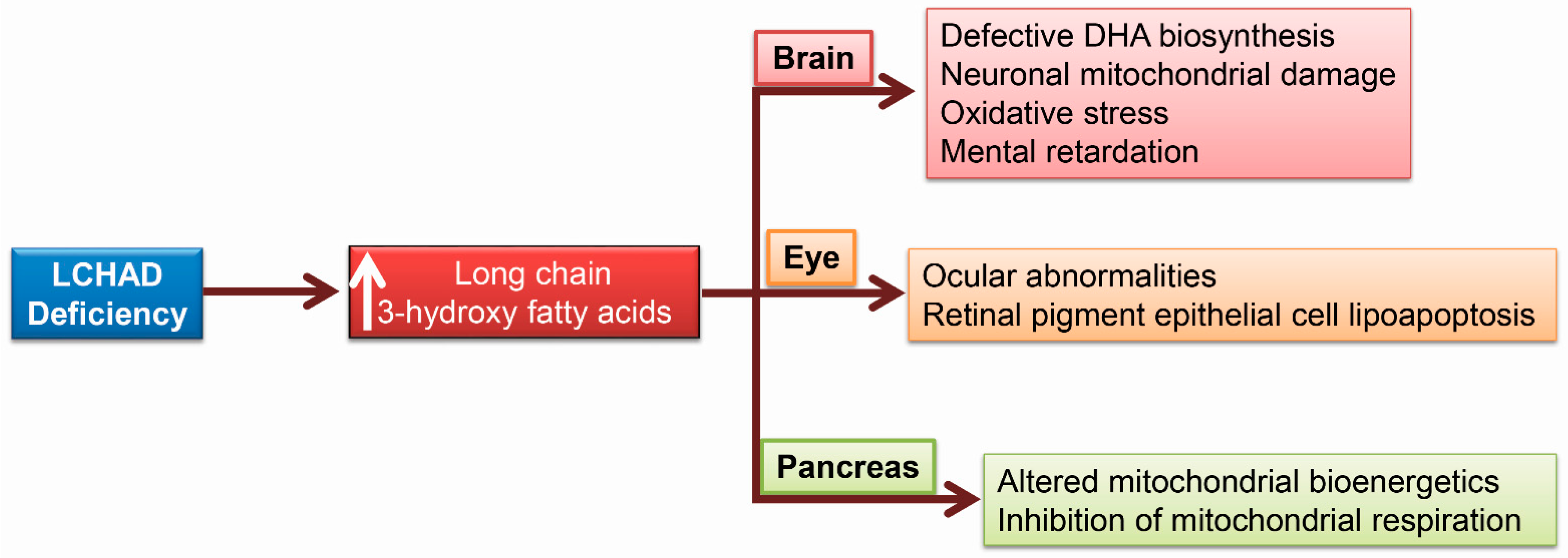{kind=link}
1. Fatty Acid Oxidation
1.1. Mitochondrial Fatty Acid Oxidation
1.2. Peroxisomal and Microsomal Fatty Acid Oxidation
2. Maternal Liver Disease Associated with Fatty Acid Oxidation Defects
2.1. Acute Fatty Liver of Pregnancy
2.2. The Incidence of AFLP and LCHAD Mutations
2.3. 3-Hydroxy Fatty Acid Accumulation
3. Metabolic Phenotypes Associated with 3-Hydroxy Fatty Acid Accumulation
3.1. Hypoglycemia in AFLP and Hormonal Regulation
3.2. Mitochondrial Trifunctional Protein (MTP)-Deficient Mice Develop Intra Uterine Growth Retardation (IUGR), Neonatal Hypoglycemia, and Sudden Death
3.3. MTP Heterozygous Mice Develop Hepatic Insulin Resistance
4. Mechanisms of 3-Hydroxy Fatty Acid-Induced Lipotoxicity
4.1. Placental Damage in AFLP Patients
4.2. Subcellular Damage and Oxidative Injury
4.3. 3-Hydroxy Fatty Acid-Induced Hepatocyte Lipoapoptosis
5. 3-HFA and Pediatric Complications Unique to LCHAD Deficiency
5.1. Brain Damage Due to 3-Hydroxy Fatty Acids
5.2. Ocular Abnormalities in Patients with LCHAD Deficiency
5.3. 3-Hydroxy Fatty Acids Alter Pancreatic Islet β-Cell Bioenergetics
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AFLP | acute fatty liver of pregnancy |
| ALT | alanine amino transferase |
| AST | aspartate amino transferase |
| ALP | alkaline phosphatase |
| CPT2 | carnitine palmitoyl transferase 2 |
| DHA | docosa hexaenoic acid |
| FoxO | forkhead family of transcription factor class O |
| GGT | γ-glutamyl transpeptidase |
| HELLP | hemolysis elevated liver enzymes with low platelet count |
| iPSC | induced-pluripotent stem cells |
| LCHAD | long chain hydroxy acyl-CoA dehydrogenease |
| MAPK | mitogen activated protein kinases |
| MTP | mitochondiral trifunctional protien |
| MCHAD | medium chain hydroxy acyl-CoA dehydrognease |
| PGC1α | peroxisome proliferator-activated receptor-γ coactivator-1α |
| PUMA | p35-upregulated modulator of apoptosis |
| RPE | retinal pigment epithelial cells |
| TCA cycle | tricarboxylic acid cycle |
| ZO-1 | zona occludens-1 |
| 3-HFA | 3-hydroxy fatty acid |
| 3-HMA | 3-hydroxy myristic acid |
| 3-HOA | 3-hydroxy octanoic acid |
| 3-HPA | 3-hydroxy palmitic acid |
References
- Natarajan, S.K.; Eapen, C.E.; Pullimood, A.B.; Balasubramanian, K.A. Oxidative stress in experimental liver microvesicular steatosis: Role of mitochondria and peroxisomes. J. Gastroenterol. Hepatol. 2006, 21, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Thangaraj, K.R.; Eapen, C.E.; Ramachandran, A.; Balasubramanian, K.A. Acute fatty liver of pregnancy: An update on mechanism. Obstet. Med. 2011, 4, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A. Acute fatty liver of pregnancy: An update on pathogenesis and clinical implications. World J. Gastroenterol. 2006, 12, 7397–7404. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.M.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Mitochondria and redox signaling in steatohepatitis. Antioxid. Redox Signal. 2011, 15, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Dhar, M.; Sepkovic, D.W.; Hirani, V.; Magnusson, R.P.; Lasker, J.M. Omega oxidation of 3-hydroxy fatty acids by the human CYP4F gene subfamily enzyme CYP4F11. J. Lipid Res. 2008, 49, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P.; Osei-Hyiaman, D.; Wiland, H.; Abdelmegeed, M.A.; Song, B.J. PPAR/RXR regulation of fatty acid metabolism and fatty acid ω-hydroxylase (CYP4) isozymes: Implications for prevention of lipotoxicity in fatty liver disease. PPAR Res. 2009, 2009, 952734. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sun, X.; Sun, Y.; Hou, L.; Yao, M.; Lian, K.; Li, J.; Lu, X.; Jiang, L. Elevation of cortical C26:0 due to the decline of peroxisomal β-oxidation potentiates amyloid β generation and spatial memory deficits via oxidative stress in diabetic rats. Neuroscience 2016, 315, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Thangaraj, K.R.; Eapen, C.E.; Ramachandran, A.; Mukhopadhya, A.; Mathai, M.; Seshadri, L.; Peedikayil, A.; Ramakrishna, B.; Balasubramanian, K.A. Liver injury in acute fatty liver of pregnancy: Possible link to placental mitochondrial dysfunction and oxidative stress. Hepatology 2010, 51, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Sykut-Cegielska, J.; Gradowska, W.; Piekutowska-Abramczuk, D.; Andresen, B.S.; Olsen, R.K.; Oltarzewski, M.; Pronicki, M.; Pajdowska, M.; Bogdanska, A.; Jablonska, E.; et al. Urgent metabolic service improves survival in long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency detected by symptomatic identification and pilot newborn screening. J. Inherit. Metab. Dis. 2011, 34, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Bennett, M.J.; Rogers, B.B. Long-chain l-3-hydroxyacyl-coenzyme a dehydrogenase deficiency: A molecular and biochemical review. Lab. Investig. 2002, 82, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, M.; Van Calcar, S.; Ney, D.; Wolff, J.; Harding, C. Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). A case report and survey. J. Inherit. Metab. Dis. 1999, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, M.B.; Purnell, J.Q.; Jordan, J.; Stadler, D.; Haqq, A.M.; Harding, C.O. Effects of higher dietary protein intake on energy balance and metabolic control in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol. Genet. Metab. 2007, 90, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Tserng, K.Y.; Jin, S.J.; Kerr, D.S.; Hoppel, C.L. Abnormal urinary excretion of unsaturated dicarboxylic acids in patients with medium-chain acyl-CoA dehydrogenase deficiency. J. Lipid Res. 1990, 31, 763–771. [Google Scholar] [PubMed]
- Tserng, K.Y.; Jin, S.J.; Kerr, D.S.; Hoppel, C.L. Urinary 3-hydroxydicarboxylic acids in pathophysiology of metabolic disorders with dicarboxylic aciduria. Metabolism 1991, 40, 676–682. [Google Scholar] [CrossRef]
- Divry, P.; David, M.; Gregersen, N.; Kolvraa, S.; Christensen, E.; Collet, J.P.; Dellamonica, C.; Cotte, J. Dicarboxylic aciduria due to medium chain acyl CoA dehydrogenase defect. A cause of hypoglycemia in childhood. Acta Paediatr. Scand. 1983, 72, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Kolvraa, S.; Rasmussen, K.; Mortensen, P.B.; Divry, P.; David, M.; Hobolth, N. General (medium-chain) acyl-CoA dehydrogenase deficiency (non-ketotic dicarboxylic aciduria): Quantitative urinary excretion pattern of 23 biologically significant organic acids in three cases. Clin. Chim. Acta 1983, 132, 181–191. [Google Scholar] [CrossRef]
- Yang, Z.; Yamada, J.; Zhao, Y.; Strauss, A.W.; Ibdah, J.A. Prospective screening for pediatric mitochondrial trifunctional protein defects in pregnancies complicated by liver disease. JAMA 2002, 288, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Jamwal, K.D.; Ramachandran, A.; Balasubramanian, K.A.; Eapen, C.E. Pregnancy-related liver disorders. J. Clin. Exp. Hepatol. 2014, 4, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A. Role of genetic screening in identifying susceptibility to acute fatty liver of pregnancy. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 494–495. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.T.; Almashhrawi, A.A.; Rahman, R.N.; Hammoud, G.M.; Ibdah, J.A. Liver diseases in pregnancy: Diseases unique to pregnancy. World J. Gastroenterol. 2013, 19, 7639–7646. [Google Scholar] [CrossRef] [PubMed]
- London, V.; Grube, S.; Sherer, D.M.; Abulafia, O. Hyperemesis gravidarum: A review of recent literature. Pharmacology 2017, 100, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Outlaw, W.M.; Ibdah, J.A. Impaired fatty acid oxidation as a cause of liver disease associated with hyperemesis gravidarum. Med. Hypotheses 2005, 65, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aranguren, L.C.; Prada, C.E.; Riano-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Dasouki, M.J.; Strauss, A.W. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Variable expressivity of maternal illness during pregnancy and unusual presentation with infantile cholestasis and hypocalcaemia. J. Inherit. Metab. Dis. 1999, 22, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Bartha, J.L.; Visiedo, F.; Fernandez-Deudero, A.; Bugatto, F.; Perdomo, G. Decreased mitochondrial fatty acid oxidation in placentas from women with preeclampsia. Placenta 2012, 33, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yang, Z.; Han, Y.; Yu, H. Fatty acid oxidation changes and the correlation with oxidative stress in different preeclampsia-like mouse models. PLoS ONE 2014, 9, e109554. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.W.; Yang, Z.; Ding, X.Y.; Yu, H. Differences in liver injury and trophoblastic mitochondrial damage in different preeclampsia-like mouse models. Chin. Med. J(Engl). 2015, 128, 1627–1635. [Google Scholar] [PubMed]
- Yang, Z.; Lantz, P.E.; Ibdah, J.A. Post-mortem analysis for two prevalent β-oxidation mutations in sudden infant death. Pediatr. Int. 2007, 49, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, Y.; Bennett, M.J.; Strauss, A.W.; Ibdah, J.A. Fetal genotypes and pregnancy outcomes in 35 families with mitochondrial trifunctional protein mutations. Am. J. Obstet. Gynecol. 2002, 187, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Ramakrishna, B.; Zachariah, U.; Ramachandran, J.; Eapen, C.E.; Kurian, G.; Chandy, G. How accurate are the swansea criteria to diagnose acute fatty liver of pregnancy in predicting hepatic microvesicular steatosis? Gut 2011, 60, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Kingham, J.G. Swansea criteria for diagnosis of acute fatty liver of pregnancy. Gut 2010. [Google Scholar] [CrossRef]
- Goel, A.; Nair, S.C.; Viswabandya, A.; Masilamani, V.P.; Rao, S.V.; George, A.; Regi, A.; Jose, R.; Zachariah, U.; Subramani, K.; et al. Preliminary experience with use of recombinant activated factor VII to control postpartum hemorrhage in acute fatty liver of pregnancy and other pregnancy-related liver disorders. Indian J. Gastroenterol. 2013, 32, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Bennett, M.J.; Rinaldo, P.; Zhao, Y.; Gibson, B.; Sims, H.F.; Strauss, A.W. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999, 340, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Yang, Z.; Bennett, M.J. Liver disease in pregnancy and fetal fatty acid oxidation defects. Mol. Genet. Metab. 2000, 71, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Zhao, Y.; Viola, J.; Gibson, B.; Bennett, M.J.; Strauss, A.W. Molecular prenatal diagnosis in families with fetal mitochondrial trifunctional protein mutations. J. Pediatr. 2001, 138, 396–399. [Google Scholar] [CrossRef] [PubMed]
- van Eerd, D.C.; Brusse, I.A.; Adriaens, V.F.; Mankowski, R.T.; Praet, S.F.; Michels, M.; Langeveld, M. Management of an LCHADD patient during pregnancy and high intensity exercise. JIMD Rep. 2017, 32, 95–100. [Google Scholar] [PubMed]
- Haglind, C.B.; Stenlid, M.H.; Ask, S.; Alm, J.; Nemeth, A.; Dobeln, U.; Nordenstrom, A. Growth in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. JIMD Rep. 2013, 8, 81–90. [Google Scholar] [PubMed]
- Shekhawat, P.; Bennett, M.J.; Sadovsky, Y.; Nelson, D.M.; Rakheja, D.; Strauss, A.W. Human placenta metabolizes fatty acids: Implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1098–E1105. [Google Scholar] [CrossRef] [PubMed]
- Ch’ng, C.L.; Morgan, M.; Hainsworth, I.; Kingham, J.G. Prospective study of liver dysfunction in pregnancy in southwest wales. Gut 2002, 51, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Rathi, U.; Bapat, M.; Rathi, P.; Abraham, P. Effect of liver disease on maternal and fetal outcome—A prospective study. Indian J. Gastroenterol. 2007, 26, 59–63. [Google Scholar] [PubMed]
- Dani, R.; Mendes, G.S.; Medeiros, J.L.; Peret, F.J.; Nunes, A. Study of the liver changes occurring in preeclampsia and their possible pathogenetic connection with acute fatty liver of pregnancy. Am. J. Gastroenterol. 1996, 91, 292–294. [Google Scholar] [PubMed]
- Nedoszytko, B.; Sieminska, A.; Strapagiel, D.; Dabrowski, S.; Slomka, M.; Sobalska-Kwapis, M.; Marciniak, B.; Wierzba, J.; Skokowski, J.; Fijalkowski, M.; et al. High prevalence of carriers of variant c.1528G>C of hadha gene causing long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) in the population of adult kashubians from north poland. PLoS ONE 2017, 12, e0187365. [Google Scholar] [CrossRef] [PubMed]
- Barycki, J.J.; O’Brien, L.K.; Strauss, A.W.; Banaszak, L.J. Glutamate 170 of human l-3-hydroxyacyl-CoA dehydrogenase is required for proper orientation of the catalytic histidine and structural integrity of the enzyme. J. Biol. Chem. 2001, 276, 36718–36726. [Google Scholar] [CrossRef] [PubMed]
- Joost, K.; Ounap, K.; Zordania, R.; Uudelepp, M.L.; Olsen, R.K.; Kall, K.; Kilk, K.; Soomets, U.; Kahre, T. Prevalence of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency in estonia. JIMD Rep. 2012, 2, 79–85. [Google Scholar] [PubMed]
- den Boer, M.E.; Wanders, R.J.; Morris, A.A.; Ijist, L.; Heymans, H.S.; Wijburg, F.A. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Clinical presentation and follow-up of 50 patients. Pediatrics 2002, 109, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Baudach, S.; Schnackenberg, U.; Vreken, P.; Wanders, R.J. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Neonatal manifestation at the first day of life presenting with tachypnoea. J. Inherit. Metab. Dis. 1999, 22, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Tuuli, I.; Emilia, A.; Jussi, T.; Risto, L.; Tiina, T.; Leena, L. Peripheral neuropathy in patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency—A follow-up emg study of 12 patients. Eur. J. Paediatr. Neurol. 2016, 20, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Kivela, T.; Lappi, M.; Summanen, P.; Nikoskelainen, E.; Pihko, H. Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: A new type of hereditary metabolic chorioretinopathy. Ophthalmology 1998, 105, 810–824. [Google Scholar] [CrossRef]
- Tyni, T.; Majander, A.; Kalimo, H.; Rapola, J.; Pihko, H. Pathology of skeletal muscle and impaired respiratory chain function in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency with the G1528C mutation. Neuromuscul. Disord. 1996, 6, 327–337. [Google Scholar] [CrossRef]
- Tyni, T.; Paetau, A.; Strauss, A.W.; Middleton, B.; Kivela, T. Mitochondrial fatty acid β-oxidation in the human eye and brain: Implications for the retinopathy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr. Res. 2004, 56, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Pihko, H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 1999, 88, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Tonin, A.M.; Amaral, A.U.; Busanello, E.N.; Gasparotto, J.; Gelain, D.P.; Gregersen, N.; Wajner, M. Mitochondrial bioenergetics deregulation caused by long-chain 3-hydroxy fatty acids accumulating in lchad and MTP deficiencies in rat brain: A possible role of mptp opening as a pathomechanism in these disorders? Biochim. Biophys. Acta 2014, 1842, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Tonin, A.M.; Ferreira, G.C.; Grings, M.; Viegas, C.M.; Busanello, E.N.; Amaral, A.U.; Zanatta, A.; Schuck, P.F.; Wajner, M. Disturbance of mitochondrial energy homeostasis caused by the metabolites accumulating in LCHAD and MTP deficiencies in rat brain. Life Sci. 2010, 86, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Tonin, A.M.; Grings, M.; Busanello, E.N.; Moura, A.P.; Ferreira, G.C.; Viegas, C.M.; Fernandes, C.G.; Schuck, P.F.; Wajner, M. Long-chain 3-hydroxy fatty acids accumulating in LCHAD and MTP deficiencies induce oxidative stress in rat brain. Neurochem. Int. 2010, 56, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; Moffitt, M.; Joseph, D.; Harthcock, P.A.; Boriack, R.L.; Ibdah, J.A.; Strauss, A.W.; Bennett, M.J. Accumulation of free 3-hydroxy fatty acids in the culture media of fibroblasts from patients deficient in long-chain l-3-hydroxyacyl-CoA dehydrogenase: A useful diagnostic aid. Clin. Chem. 2001, 47, 1190–1194. [Google Scholar] [PubMed]
- Cecatto, C.; Godoy Kdos, S.; da Silva, J.C.; Amaral, A.U.; Wajner, M. Disturbance of mitochondrial functions provoked by the major long-chain 3-hydroxylated fatty acids accumulating in MTP and LCHAD deficiencies in skeletal muscle. Toxicol. In Vitro 2016, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cecatto, C.; Hickmann, F.H.; Rodrigues, M.D.; Amaral, A.U.; Wajner, M. Deregulation of mitochondrial functions provoked by LCHFA accumulating in LCHAD and MTP deficiencies in rat heart: Mpt pore opening as a potential contributing pathomechanism of cardiac alterations in these disorders. FEBS J. 2015, 282, 4714–4726. [Google Scholar] [CrossRef] [PubMed]
- Eskelin, P.M.; Laitinen, K.A.; Tyni, T.A. Elevated hydroxyacylcarnitines in a carrier of lchad deficiency during acute liver disease of pregnancy—A common feature of the pregnancy complication? Mol. Genet. Metab. 2010, 100, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez Junquera, C.; Balmaseda, E.; Gil, E.; Martinez, A.; Sorli, M.; Cuartero, I.; Merinero, B.; Ugarte, M. Acute fatty liver of pregnancy and neonatal long-chain 3-hydroxyacyl-coenzyme a dehydrogenase (LCHAD) deficiency. Eur. J. Pediatr. 2009, 168, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; Butt, Y.; Bennett, M.J. Accumulation of 3-hydroxy-fatty acids in the culture medium of long-chain l-3-hydroxyacyl CoA dehydrogenase (LCHAD) and mitochondrial trifunctional protein-deficient skin fibroblasts: Implications for medium chain triglyceride dietary treatment of LCHAD deficiency. Pediatr. Res. 2003, 53, 783–787. [Google Scholar] [PubMed]
- Neuman-Laniec, M.; Wierzba, J.; Irga, N.; Zaborowska-Soltys, M.; Balcerska, A. LCHAD (long-chain 3-hydroxyacyl-CoA dehydrogenase) deficiency as a cause of sudden death of a three months old infant. Med. Wieku Rozwoj. 2002, 6, 221–226. [Google Scholar] [PubMed]
- Bellig, L.L. Maternal acute fatty liver of pregnancy and the associated risk for long-chain 3-hydroxyacyl-coenzyme a dehydrogenase (LCHAD) deficiency in infants. Adv. Neonatal. Care 2004, 4, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, N.; Seaquist, E.R. Neuroendocrine responses to hypoglycemia. Ann. N. Y. Acad. Sci. 2010, 1212, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Halldin, M.U.; Forslund, A.; Von Dobeln, U.; Eklund, C.; Gustafsson, J. Increased lipolysis in LCHAD deficiency. J. Inherit. Metab. Dis. 2007, 30, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Haglind, C.B.; Nordenstrom, A.; Ask, S.; von Dobeln, U.; Gustafsson, J.; Stenlid, M.H. Increased and early lipolysis in children with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency during fast. J. Inherit. Metab. Dis. 2015, 38, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Ingham, S.A.; Mohr, A.M.; Wehrkamp, C.J.; Ray, A.; Roy, S.; Cazanave, S.C.; Phillippi, M.A.; Mott, J.L. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 2014, 60, 1942–1956. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, J.A.; Paul, H.; Zhao, Y.; Binford, S.; Salleng, K.; Cline, M.; Matern, D.; Bennett, M.J.; Rinaldo, P.; Strauss, A.W. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Investig. 2001, 107, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Martin, D.; Pascale De, L.; Villain, E.; Jouvet, P.; Rabier, D.; Brivet, M.; Saudubray, J.M. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation 1999, 100, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Morris, E.M.; Ridenhour, S.; Meers, G.M.; Hsu, F.F.; Turk, J.; Ibdah, J.A. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology 2013, 57, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Payne, R.M.; Ibdah, J.A. Mitochondrial trifunctional protein defects: Clinical implications and therapeutic approaches. Adv. Drug Deliv. Rev. 2008, 60, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in otsuka long-evans tokushima fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G619–G626. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Wei, Y.; Ibdah, J.A. Non-alcoholic fatty liver disease and the metabolic syndrome: An update. World J. Gastroenterol. 2008, 14, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Oey, N.A.; den Boer, M.E.; Ruiter, J.P.; Wanders, R.J.; Duran, M.; Waterham, H.R.; Boer, K.; Van der Post, J.A.; Wijburg, F.A. High activity of fatty acid oxidation enzymes in human placenta: Implications for fetal-maternal disease. J. Inherit. Metab. Dis. 2003, 26, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Oey, N.A.; den Boer, M.E.; Wijburg, F.A.; Vekemans, M.; Auge, J.; Steiner, C.; Wanders, R.J.; Waterham, H.R.; Ruiter, J.P.; Attie-Bitach, T. Long-chain fatty acid oxidation during early human development. Pediatr. Res. 2005, 57, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Oey, N.A.; Ruiter, J.P.; Attie-Bitach, T.; Ijlst, L.; Wanders, R.J.; Wijburg, F.A. Fatty acid oxidation in the human fetus: Implications for fetal and adult disease. J. Inherit. Metab. Dis. 2006, 29, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.; McDermott, L. Long chain pufa transport in human term placenta. J. Nutr. 2009, 139, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.M.; Phillips, M.; Wendon, J. Rare fatal complications of acute fatty liver of pregnancy. Crit. Care 1999, 3, P186. [Google Scholar] [CrossRef]
- Matern, D.; Schehata, B.M.; Shekhawa, P.; Strauss, A.W.; Bennett, M.J.; Rinaldo, P. Placental floor infarction complicating the pregnancy of a fetus with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Mol. Genet. Metab. 2001, 72, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Runmei, M. Retrospective study of seven cases with acute fatty liver of pregnancy. ISRN Obstet. Gynecol. 2013, 2013, 730569. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.G.; Dorland, L.; Holwerda, U.; de Almeida, I.T.; Poll-The, B.T.; Jakobs, C.; Duran, M. Simultaneous analysis of plasma free fatty acids and their 3-hydroxy analogs in fatty acid β-oxidation disorders. Clin. Chem. 1998, 44, 463–471. [Google Scholar] [PubMed]
- Steinmann, D.; Knab, J.; Priebe, H.J. Perioperative management of a child with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Paediatr. Anaesth. 2010, 20, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.H.; Yang, B.Z.; Nada, M.A.; Roe, C.R. Improved detection of the G1528C mutation in LCHAD deficiency. Biochem. Mol. Med. 1996, 58, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Clark, S.; Andresen, B.S.; Bischoff, C.; Olsen, R.K.; Gregersen, N.; Chakrapani, A.; Downing, M.; Manning, N.J.; Sharrard, M.; et al. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J. Inherit. Metab. Dis. 2005, 28, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Innes, A.M.; Seargeant, L.E.; Balachandra, K.; Roe, C.R.; Wanders, R.J.; Ruiter, J.P.; Casiro, O.; Grewar, D.A.; Greenberg, C.R. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr. Res. 2000, 47, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Minami, S.; Mitani, A.; Tanizaki, Y.; Booka, M.; Okutani, T.; Yamaguchi, S.; Ino, K. Acute fatty liver of pregnancy associated with fetal mitochondrial trifunctional protein deficiency. J. Obstet. Gynaecol. Res. 2015, 41, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Thomas, S.; Ramamoorthy, P.; Basivireddy, J.; Pulimood, A.B.; Ramachandran, A.; Balasubramanian, K.A. Oxidative stress in the development of liver cirrhosis: A comparison of two different experimental models. J. Gastroenterol. Hepatol. 2006, 21, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, D.Z.; Li, Y.M.; Zhang, X.Q.; Zhou, X.X.; Jin, X. Proteomic analysis of liver mitochondria from rats with nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 4778–4786. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Jin, X.; Xia, L.; Shen, S.M.; Wang, X.L.; Cai, J.; Chen, G.Q.; Wang, L.S.; Fang, N.Y. Alterations of mitochondrial enzymes contribute to cardiac hypertrophy before hypertension development in spontaneously hypertensive rats. J. Proteome Res. 2009, 8, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Ucar, F.; Sezer, S.; Erdogan, S.; Akyol, S.; Armutcu, F.; Akyol, O. The relationship between oxidative stress and nonalcoholic fatty liver disease: Its effects on the development of nonalcoholic steatohepatitis. Redox Rep. 2013, 18, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yusoff, N.S.N.; Mustapha, Z.; Sharif, S.E.T.; Govindasamy, C.; Sirajudeen, K.N.S. Effect of antihypertensive drug treatment on oxidative stress markers in heart of spontaneously hypertensive rat models. J. Environ. Pathol. Toxicol. Oncol. 2017, 36, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate attenuates palmitate-induced bim and puma up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Mott, J.L.; Elmi, N.A.; Bronk, S.F.; Werneburg, N.W.; Akazawa, Y.; Kahraman, A.; Garrison, S.P.; Zambetti, G.P.; Charlton, M.R.; et al. JNK1-dependent puma expression contributes to hepatocyte lipoapoptosis. J. Biol. Chem. 2009, 284, 26591–26602. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Wang, X.; Zhou, H.; Rahmani, M.; Grant, S.; Durrant, D.E.; Klaassen, C.D.; Yamamoto, M.; Sanyal, A.J. Degradation of keap1 activates BH3-only proteins bim and puma during hepatocyte lipoapoptosis. Cell Death Differ. 2014, 21, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.K.; Glaser, S.S. Cholangiocyte lipoapoptosis: Implications for biliary damage during nonalcoholic fatty liver disease. Hepatology 2014, 60, 1809–1811. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Stringham, B.A.; Mohr, A.M.; Wehrkamp, C.J.; Lu, S.; Phillippi, M.A.; Harrison-Findik, D.; Mott, J.L. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J. Lipid Res. 2017, 58, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.Y.; Pan, J.; Usuda, N.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor α natural ligand metabolism. J. Biol. Chem. 1998, 273, 15639–15645. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.O.; Gillingham, M.B.; van Calcar, S.C.; Wolff, J.A.; Verhoeve, J.N.; Mills, M.D. Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Kivela, T.T. Early dietary therapy for long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency can maintain vision despite subnormal retinal function. Acta Paediatr. 2016, 105, 1461. [Google Scholar] [CrossRef] [PubMed]
- Fahnehjelm, K.T.; Holmstrom, G.; Ying, L.; Haglind, C.B.; Nordenstrom, A.; Halldin, M.; Alm, J.; Nemeth, A.; von Dobeln, U. Ocular characteristics in 10 children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: A cross-sectional study with long-term follow-up. Acta Ophthalmol. 2008, 86, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Fahnehjelm, K.T.; Liu, Y.; Olsson, D.; Amren, U.; Haglind, C.B.; Holmstrom, G.; Halldin, M.; Andreasson, S.; Nordenstrom, A. Most patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency develop pathological or subnormal retinal function. Acta Paediatr. 2016, 105, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Boese, E.A.; Jain, N.; Jia, Y.; Schlechter, C.L.; Harding, C.O.; Gao, S.S.; Patel, R.C.; Huang, D.; Weleber, R.G.; Gillingham, M.B.; et al. Characterization of chorioretinopathy associated with mitochondrial trifunctional protein disorders: Long-term follow-up of 21 cases. Ophthalmology 2016, 123, 2183–2195. [Google Scholar] [CrossRef] [PubMed]
- Polinati, P.P.; Ilmarinen, T.; Trokovic, R.; Hyotylainen, T.; Otonkoski, T.; Suomalainen, A.; Skottman, H.; Tyni, T. Patient-specific induced pluripotent stem cell-derived RPE cells: Understanding the pathogenesis of retinopathy in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3371–3382. [Google Scholar] [CrossRef] [PubMed]
- Apiratpracha, W.; Yoshida, E.M.; Scudamore, C.H.; Weiss, A.A.; Byrne, M.F. Chronic pancreatitis: A sequela of acute fatty liver of pregnancy. Hepatobiliary Pancreat. Dis. Int. 2008, 7, 101–104. [Google Scholar] [PubMed]
- Castanon-Gonzalez, J.A.; Vazquez-de Anda, G.F.; Gallegos-Perez, H.; Hernandez-Lopez, G.; Eid-Lidt, G.; Miranda-Ruiz, R. Acute fatty liver of pregnancy complicated by pancreatitis. Gac. Med. Mex. 1997, 133, 253–258. [Google Scholar] [PubMed]
- De Oliveira, C.V.; Moreira, A.; Baima, J.P.; Franzoni Lde, C.; Lima, T.B.; Yamashiro Fda, S.; Coelho, K.Y.; Sassaki, L.Y.; Caramori, C.A.; Romeiro, F.G.; et al. Acute fatty liver of pregnancy associated with severe acute pancreatitis: A case report. World J. Hepatol. 2014, 6, 527–531. [Google Scholar] [CrossRef] [PubMed]
- English, N.; Rao, J. Acute fatty liver of pregnancy with hypoglycaemia, diabetes insipidus and pancreatitis, preceded by intrahepatic cholestasis of pregnancy. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Minakami, H.; Kimura, K.; Kanazawa, T.; Tamada, T.; Kaneko, K. Acute fatty liver of pregnancy with hyperlipidemia, acute hemorrhagic pancreatitis and disseminated intravascular Coagulation. Asia Ocean. J. Obstet. Gynaecol. 1985, 11, 371–376. [Google Scholar] [CrossRef]
- Minakami, H.; Kimura, K.; Tamada, T.; Matsuda, A.; Kobayashi, S.; Matsumoto, S. Acute fatty liver of pregnancy: Report of a case complicating DIC and acute pancreatitis (author’s transl). Nihon Sanka Fujinka Gakkai Zasshi 1982, 34, 637–640. [Google Scholar] [PubMed]
- Moldenhauer, J.S.; O’Brien, J.M.; Barton, J.R.; Sibai, B. Acute fatty liver of pregnancy associated with pancreatitis: A life-threatening complication. Am. J. Obstet. Gynecol. 2004, 190, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Doliba, N.M.; Liu, Q.; Li, C.; Chen, J.; Chen, P.; Liu, C.; Frederick, D.W.; Baur, J.A.; Bennett, M.J.; Naji, A.; et al. Accumulation of 3-hydroxytetradecenoic acid: Cause or corollary of glucolipotoxic impairment of pancreatic β-cell bioenergetics? Mol. Metab. 2015, 4, 926–939. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, S.K.; Ibdah, J.A. Role of 3-Hydroxy Fatty Acid-Induced Hepatic Lipotoxicity in Acute Fatty Liver of Pregnancy. Int. J. Mol. Sci. 2018, 19, 322. https://doi.org/10.3390/ijms19010322
Natarajan SK, Ibdah JA. Role of 3-Hydroxy Fatty Acid-Induced Hepatic Lipotoxicity in Acute Fatty Liver of Pregnancy. International Journal of Molecular Sciences. 2018; 19(1):322. https://doi.org/10.3390/ijms19010322
Chicago/Turabian StyleNatarajan, Sathish Kumar, and Jamal A. Ibdah. 2018. "Role of 3-Hydroxy Fatty Acid-Induced Hepatic Lipotoxicity in Acute Fatty Liver of Pregnancy" International Journal of Molecular Sciences 19, no. 1: 322. https://doi.org/10.3390/ijms19010322
APA StyleNatarajan, S. K., & Ibdah, J. A. (2018). Role of 3-Hydroxy Fatty Acid-Induced Hepatic Lipotoxicity in Acute Fatty Liver of Pregnancy. International Journal of Molecular Sciences, 19(1), 322. https://doi.org/10.3390/ijms19010322