Identification of a Rare Germline Heterozygous Deletion Involving the Polycistronic miR-17–92 Cluster in Two First-Degree Relatives from a BRCA 1/2 Negative Chilean Family with Familial Breast Cancer: Possible Functional Implications


Abstract
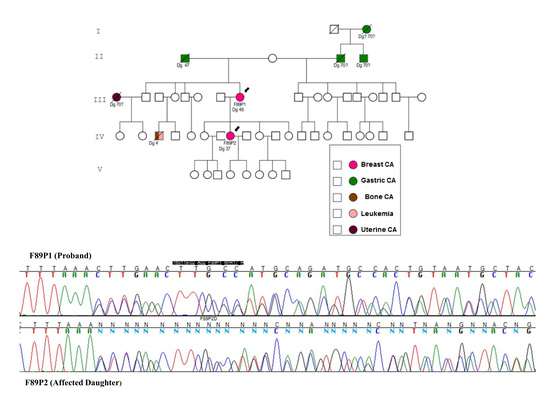
:
1. Introduction
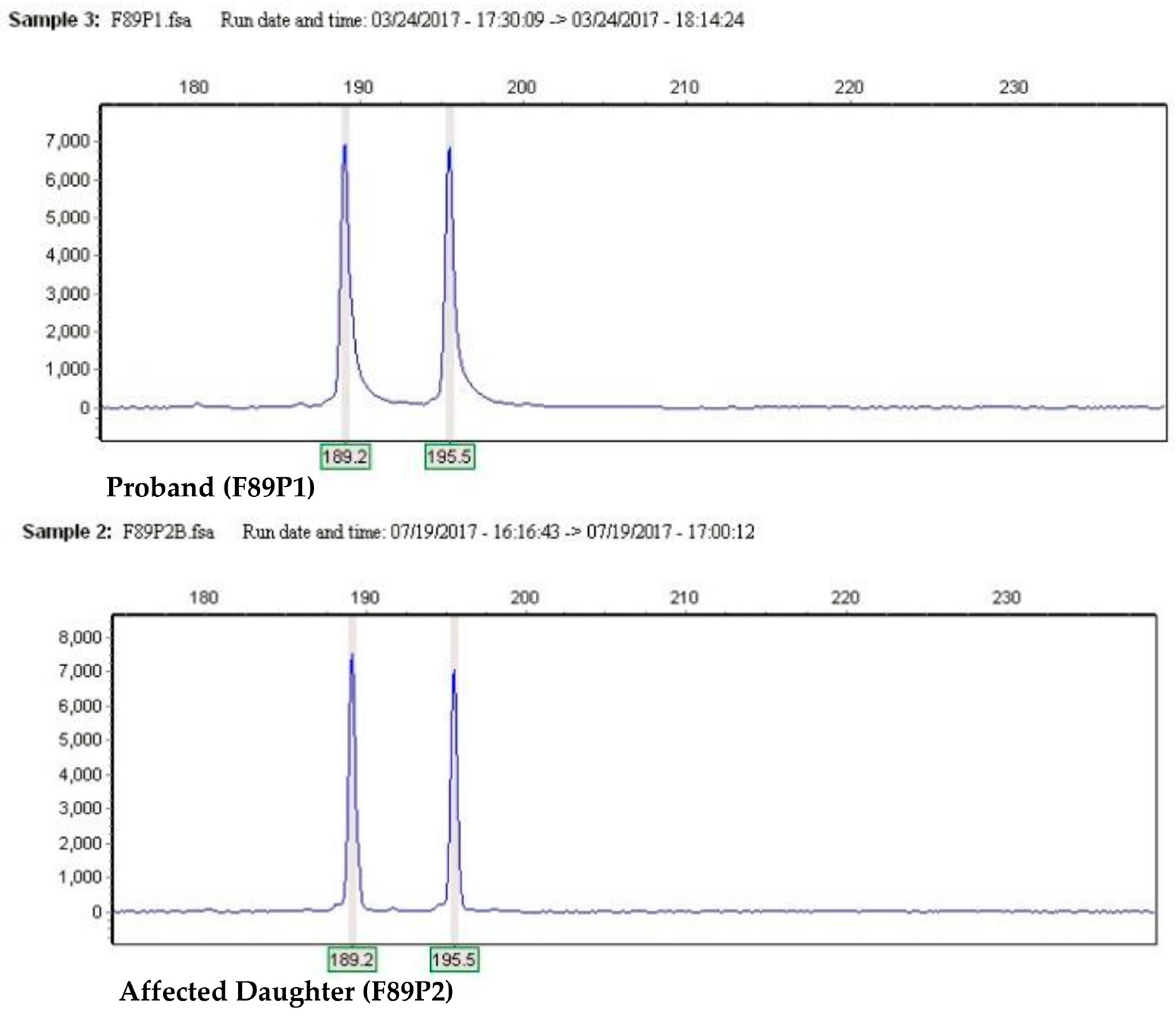
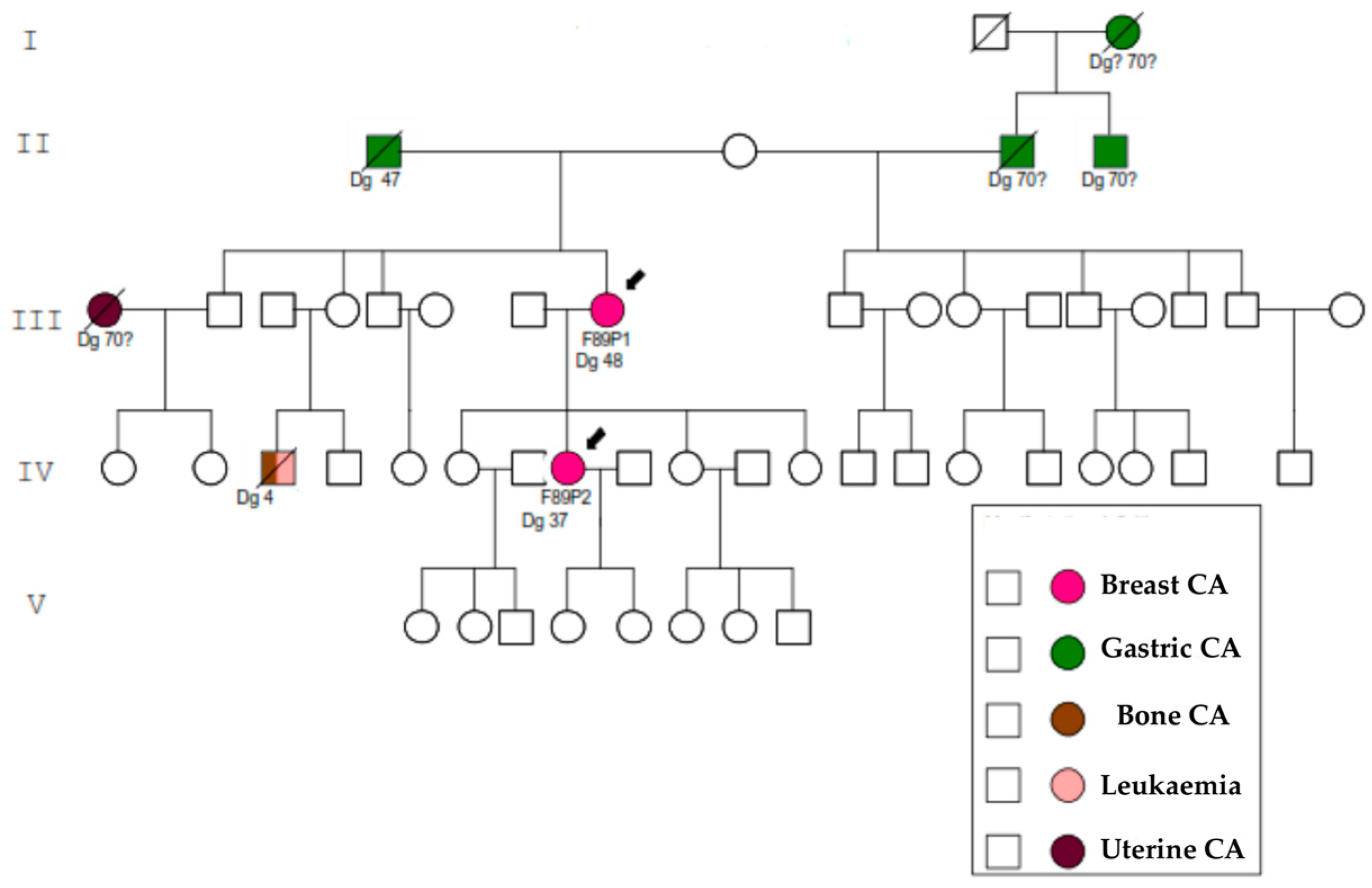
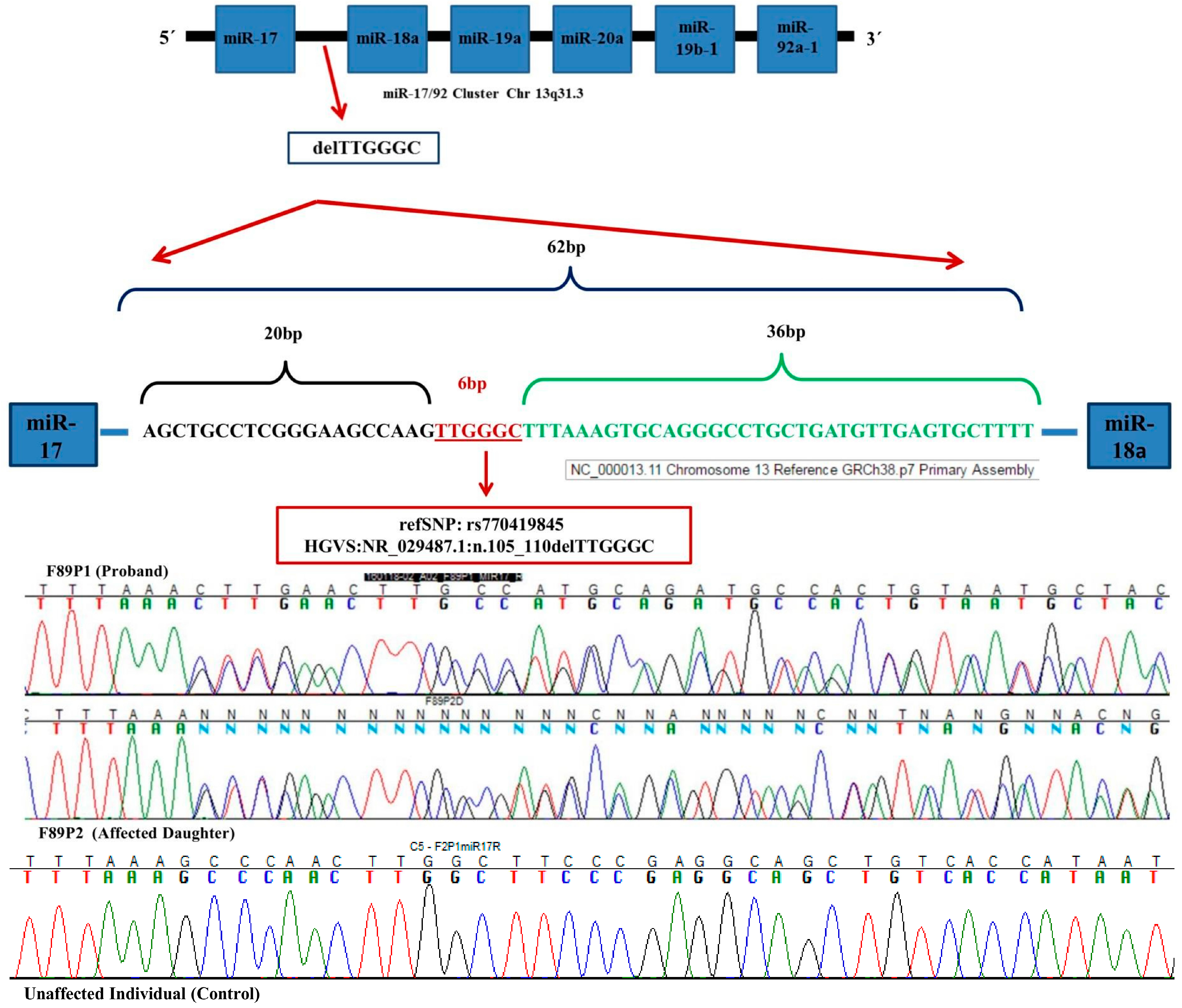
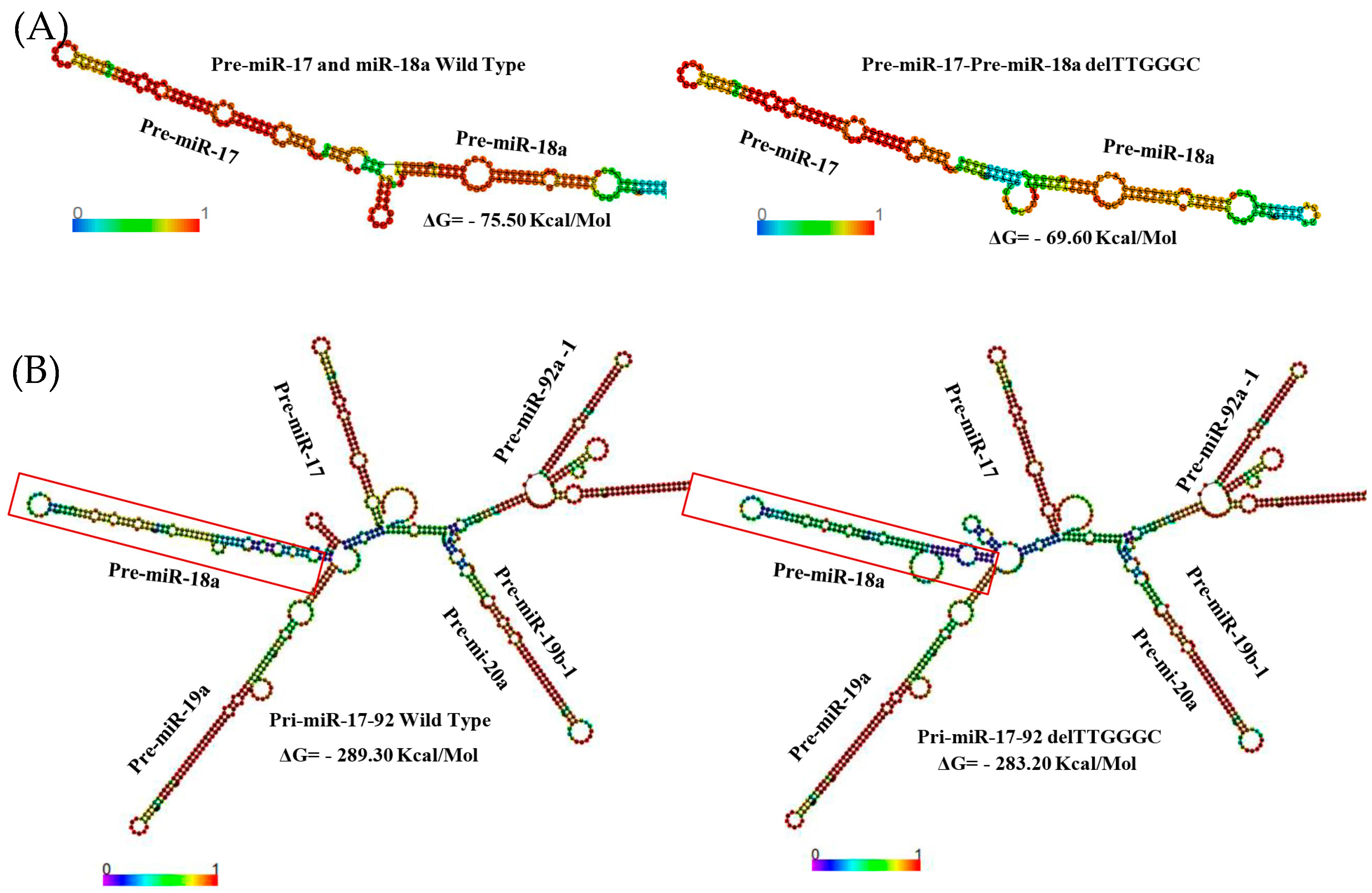
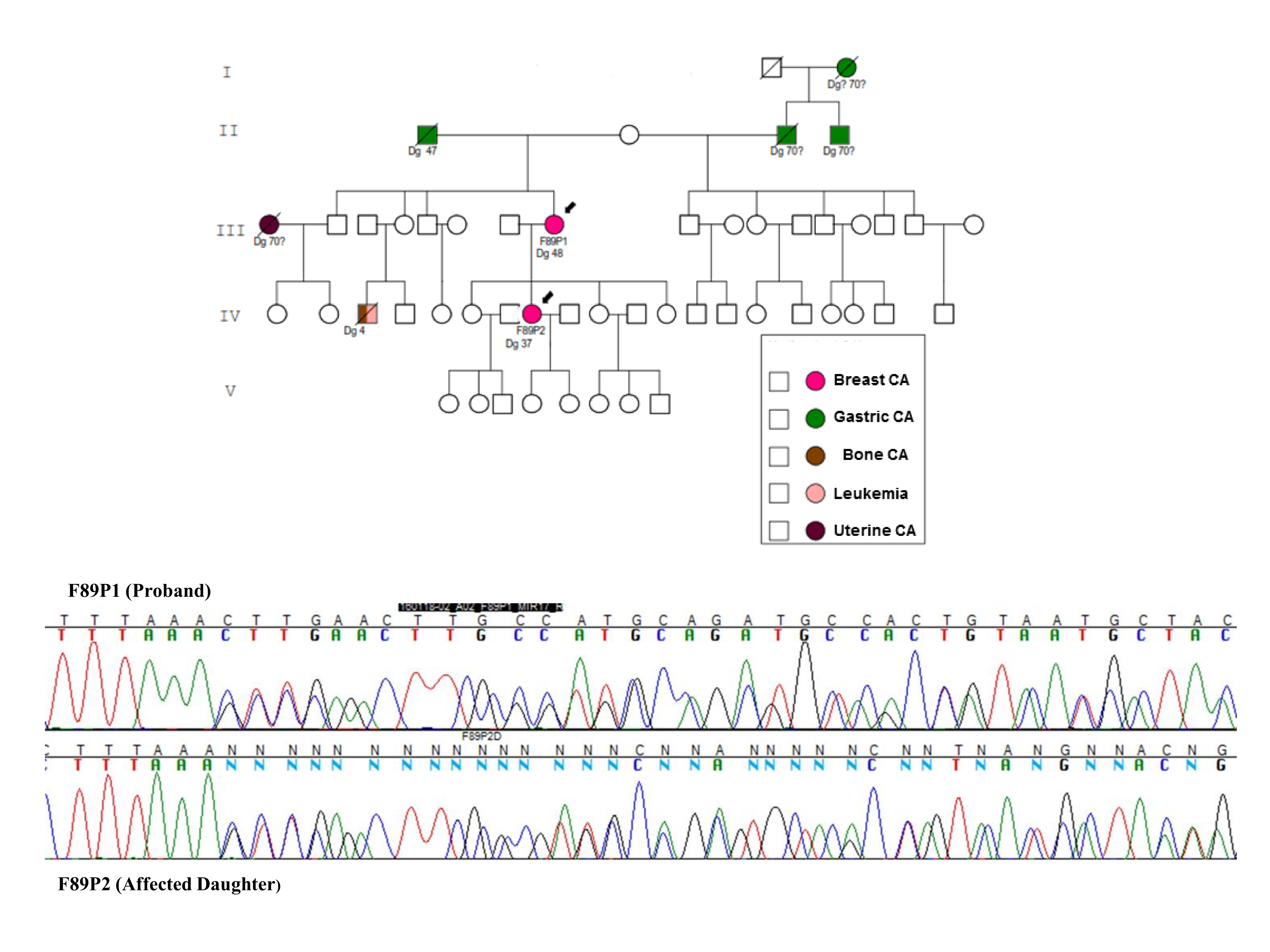
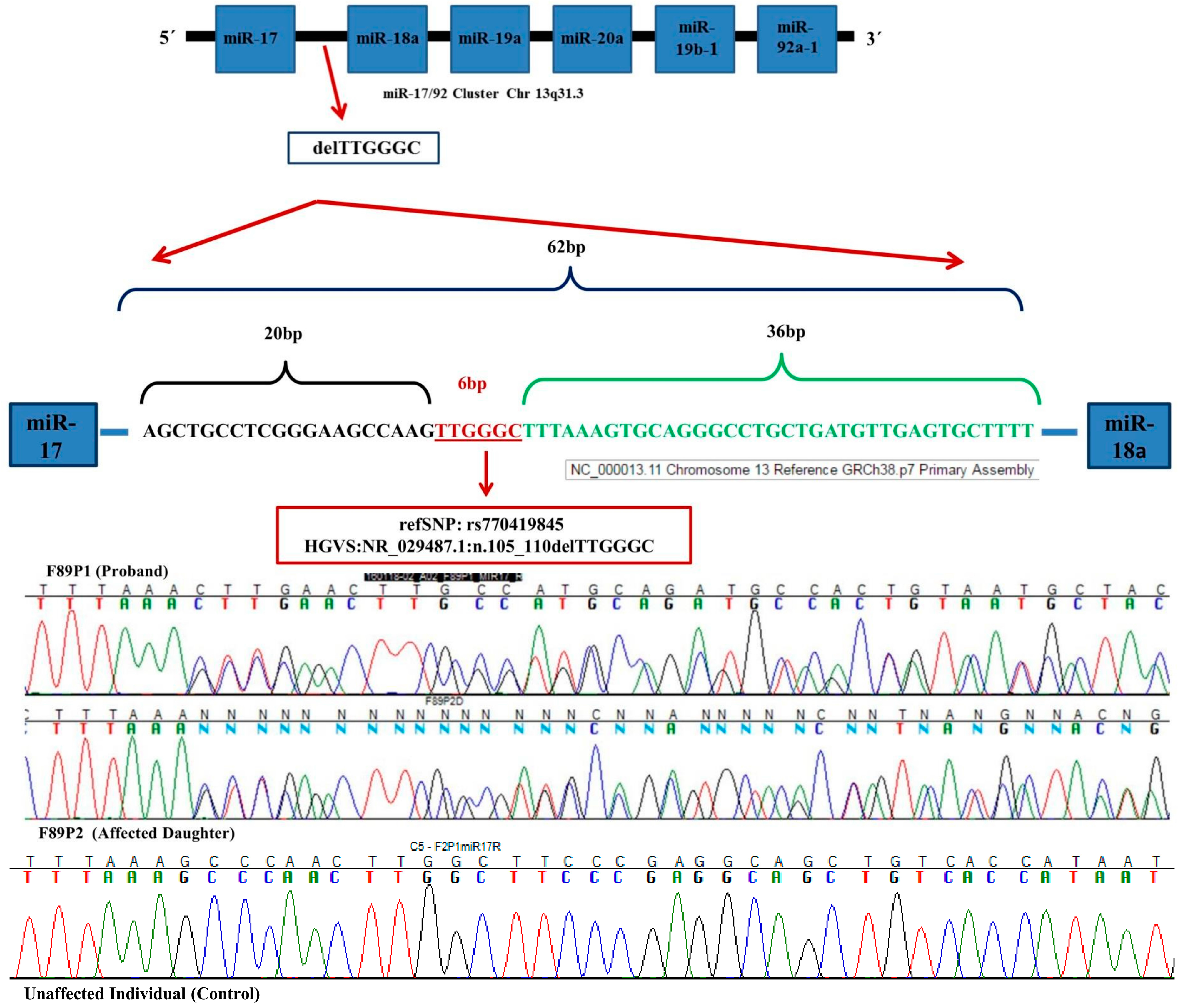
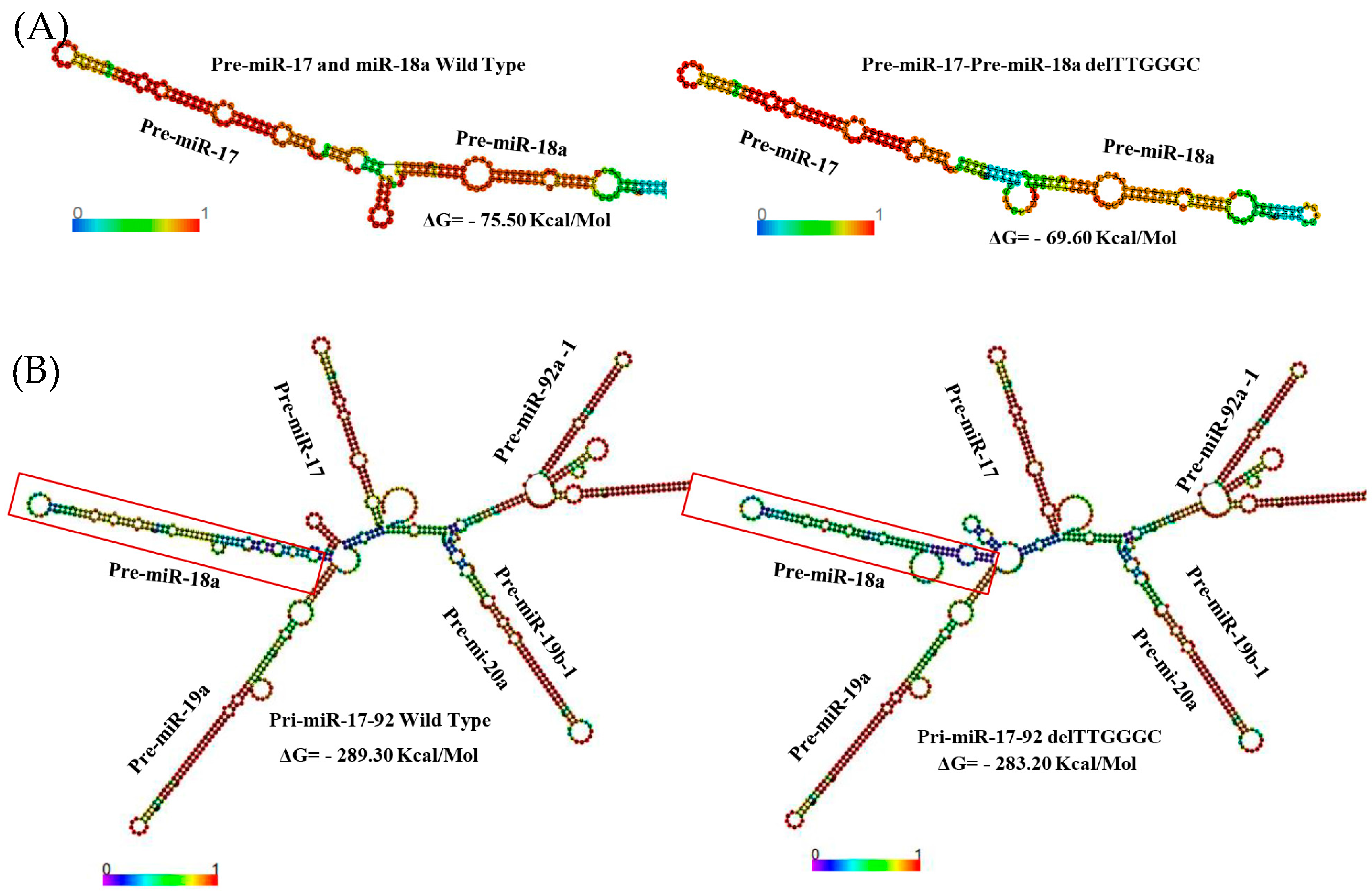
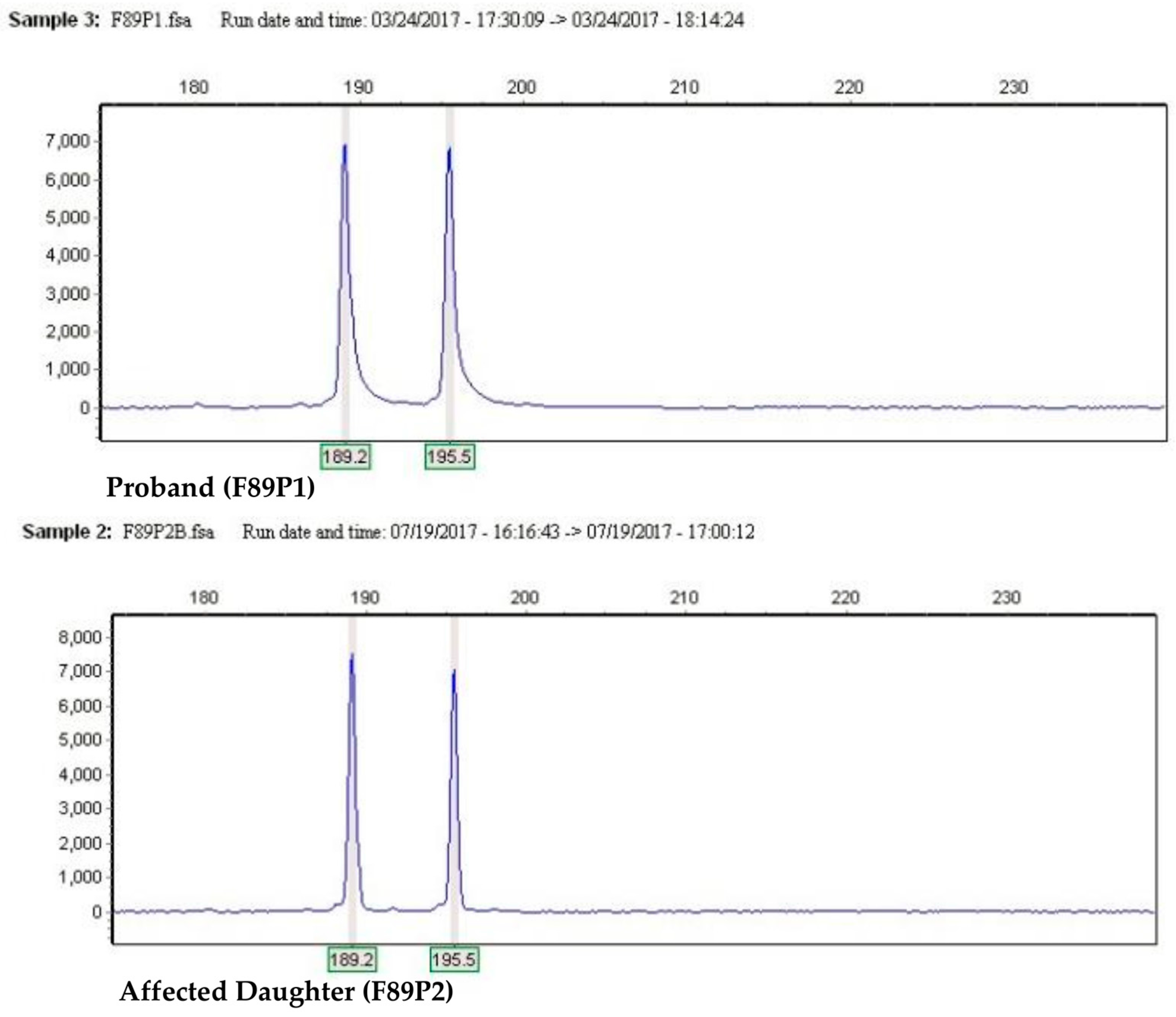
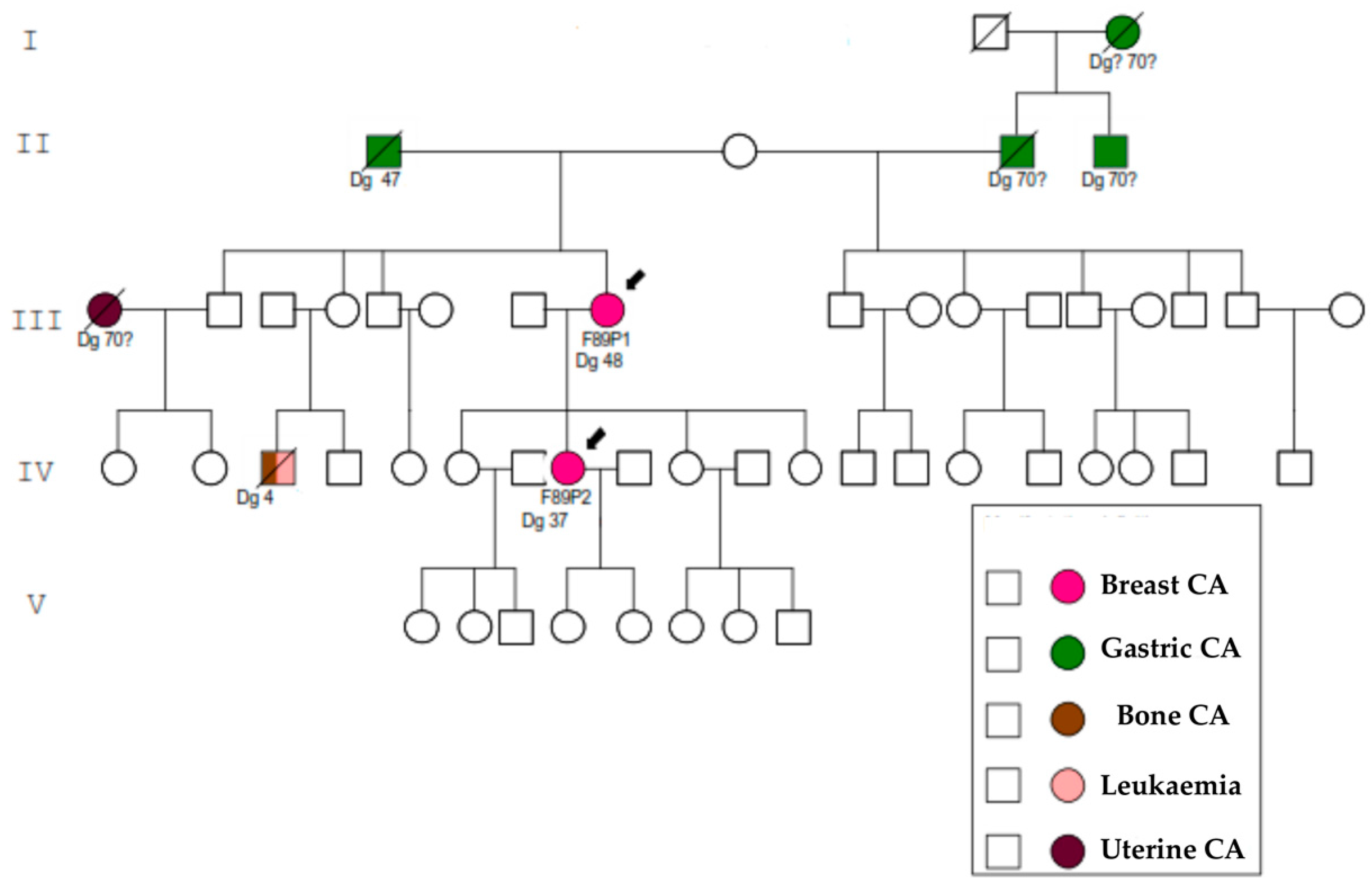
2. Results
3. Discussion
4. Materials and Methods
4.1. Families
4.2. Controls
4.3. Mutation Analysis
4.4. Pre-miR-17 Complete Sequence Analysis
4.5. Pre-miR-17 rs770419845 Analysis
4.6. Bioinformatic Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Health Ministry of Chile. Mortality Rates in Chile; Department of Health Statistics and Information (DEIS): Santiago, Chile, 2011; Available online: http://www.deis.cl/ (accessed on 8 November 2017).
- Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br. J. Cancer 2000, 83, 1301–1308. [Google Scholar] [CrossRef]
- Stratton, M.R.; Rahman, N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008, 40, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, J.; Siepel, A.; Lai, E.C. Diverse modes of evolutionary emergence and flux of conserved microRNA clusters. RNA 2014, 20, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, J.; Zhang, H.; Lu, J. microRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol. 2016, 33, 2232–2247. [Google Scholar] [CrossRef] [PubMed]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005, 33, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The microRNA-17-92 family of microRNA clusters in development and disease. Cancer J. 2012, 18, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004, 64, 3087–3095. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel regulators in the hallmarks of human cancer. Cancer Lett. 2009, 285, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Robles, A.I.; Harris, C.C. Genetic variation in microRNA networks: The implications for cancer research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Coke, R. Ethnic origin and evolution of the Chilean population. Rev. Med. Chile 1976, 104, 365–368. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, H.; Rogers, K.M. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered. Cancer Clin. Pract. 2015, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and function. Thromb. Haemost. 2012, 107, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Chaulk, S.G.; Thede, G.L.; Kent, O.A.; Xu, Z.; Gesner, E.M.; Veldhoen, R.A.; Khanna, S.K.; Goping, I.S.; MacMillan, A.M.; Mendell, J.T.; et al. Role of pri-miRNA tertiary structure in miR-17~92 miRNA biogenesis. RNA Biol. 2011, 8, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Mehtab, S.; Patwardhan, A.; Krishnan, Y. Pri-miR-17-92a transcript folds into a tertiary structure and autoregulates its processing. RNA 2012, 18, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hormazabal, P.; Gutierrez-Enriquez, S.; Gaete, D.; Reyes, J.M.; Peralta, O.; Waugh, E.; Gomez, F.; Margarit, S.; Bravo, T.; Blanco, R.; et al. Spectrum of BRCA1/2 point mutations and genomic rearrangements in high-risk breast/ovarian cancer Chilean families. Breast Cancer Res. Treat. 2011, 126, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Families: n (%) |
|---|---|
| ≥3 family members with breast and/or ovarian cancer | 139 (30.4%) |
| 2 family members with breast and/or ovarian cancer | 155 (33.8%) |
| Single affected individual with breast cancer, diagnosed at ≤35 years of age | 81 (17.7%) |
| Single affected individual with breast cancer, diagnosed at 36–50 years of age | 83 (18.1%) |
| Total | 458 (100%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Mayo, T.; Ziegler, A.; Morales, S.; Jara, L. Identification of a Rare Germline Heterozygous Deletion Involving the Polycistronic miR-17–92 Cluster in Two First-Degree Relatives from a BRCA 1/2 Negative Chilean Family with Familial Breast Cancer: Possible Functional Implications. Int. J. Mol. Sci. 2018, 19, 321. https://doi.org/10.3390/ijms19010321
De Mayo T, Ziegler A, Morales S, Jara L. Identification of a Rare Germline Heterozygous Deletion Involving the Polycistronic miR-17–92 Cluster in Two First-Degree Relatives from a BRCA 1/2 Negative Chilean Family with Familial Breast Cancer: Possible Functional Implications. International Journal of Molecular Sciences. 2018; 19(1):321. https://doi.org/10.3390/ijms19010321
Chicago/Turabian StyleDe Mayo, Tomás, Annemarie Ziegler, Sebastián Morales, and Lilian Jara. 2018. "Identification of a Rare Germline Heterozygous Deletion Involving the Polycistronic miR-17–92 Cluster in Two First-Degree Relatives from a BRCA 1/2 Negative Chilean Family with Familial Breast Cancer: Possible Functional Implications" International Journal of Molecular Sciences 19, no. 1: 321. https://doi.org/10.3390/ijms19010321
APA StyleDe Mayo, T., Ziegler, A., Morales, S., & Jara, L. (2018). Identification of a Rare Germline Heterozygous Deletion Involving the Polycistronic miR-17–92 Cluster in Two First-Degree Relatives from a BRCA 1/2 Negative Chilean Family with Familial Breast Cancer: Possible Functional Implications. International Journal of Molecular Sciences, 19(1), 321. https://doi.org/10.3390/ijms19010321